

Abstract
SHANK3 (also known as ProSAP2) regulates the structural organization of dendritic spines and is a binding partner of neuroligins; genes encoding neuroligins are mutated in autism and Asperger syndrome. Here, we report that a mutation of a single copy of SHANK3 on chromosome 22q13 can result in language and/or social communication disorders. These mutations concern only a small number of individuals, but they shed light on one gene dosage-sensitive synaptic pathway that is involved in autism spectrum disorders.
Keywords: Autistic Disorder; genetics; Base Sequence; Carrier Proteins; genetics; DNA Mutational Analysis; Female; Genetic Screening; Humans; In Situ Hybridization, Fluorescence; Male; Molecular Sequence Data; Mutation; Pedigree
Autism spectrum disorders (ASD) affect about 6 of every 1000 children and are characterized by impairments in reciprocal social interaction and communication as well as restricted and stereotyped patterns of interests and activities1. ASD ranges from severe (in the case of autistic disorder with moderate or severe cognitive impairment) to a milder variant (Asperger syndrome with higher cognitive ability). Although the causative genes remain largely unknown2, familial and twin studies indicate that ASD is one of the most genetic neuropsychiatric disorders. Standard karyotype analyses show chromosomal rearrangements in 3%–6% of cases, the most common being deletions and duplications on chromosomes 15q, 22q and 7q3. One of the most frequent rearrangements associated with cognitive deficits, the 22q13.3 microdeletion syndrome is characterized by neonatal hypotonia, global developmental delay, normal to accelerated growth, absent to severely delayed speech, autistic behavior, and minor dysmorphic features4. The loss of terminal 22q13.3 can be subtle and can go undetected by routine chromosome analysis; FISH is often required to confirm the presence of this deletion.
Among the three genes (ACR, RABL2B, SHANK3) located in the minimal telomeric region5, SHANK3 (also known as ProSAP2) is the strongest candidate for the neurobehavioral symptoms observed in patients with 22q13 deletions. SHANK3 is a scaffolding protein found in excitatory synapses directly opposite to the presynaptic active zone. Shank proteins are believed to function as master organizers of the postsynaptic density (PSD), owing to their ability to form multimeric complexes with postsynaptic receptors, signaling molecules and cytoskeletal proteins present in dendritic spines and PSDs6,7. SHANK3 can bind to the cell adhesion proteins neuroligins8; we have previously found genes encoding neuroligins (NLGN3 and NLGN4) to be mutated in individuals with autism and Asperger syndrome9. SHANK3 was disrupted by a de novo balanced translocation in a child with all the features of the 22q13.3 deletion syndrome10. In this paper, we report evidence showing that abnormal gene dosage of SHANK3 is associated with severe cognitive deficits, including language and speech disorder and ASD.
We used FISH analysis (n=97) and/or direct sequencing (n=227) to investigate chromosome 22q13 and SHANK3 in patients with ASD (Supplementary Methods). We also sequenced all SHANK3 exons in a minimum of 190 controls to ascertain the diversity of SHANK3 nonsynonymous variations in the general population. SHANK3 spans 57 kb and contains 24 exons. Seven exons are alternatively spliced, including exon 18, which is detected mostly in the brain (Supplementary Fig. 1). During our screening, three families with ASD showed unambiguous alteration of 22q13 or SHANK3. In family ASD 1, the proband with autism, absent language and moderate mental retardation carried a de novo deletion of 22q13 (the clinical description of all patients is provided in the Supplementary Note). The deletion breakpoint was located in intron 8 of SHANK3 and removed 142 kb of the terminal 22q13 (Fig. 1a). This deletion had been “repaired” by addition of telomeric repeats and was similar to the minimum deleted region described previously5. The recurrent deletions in this region may be due to the quadruplex-forming G-rich sequence (QGRS) surrounding the breakpoint (Supplementary Fig. 2), which provides a structural substrate for inappropriate telomere formation.
Figure 1.
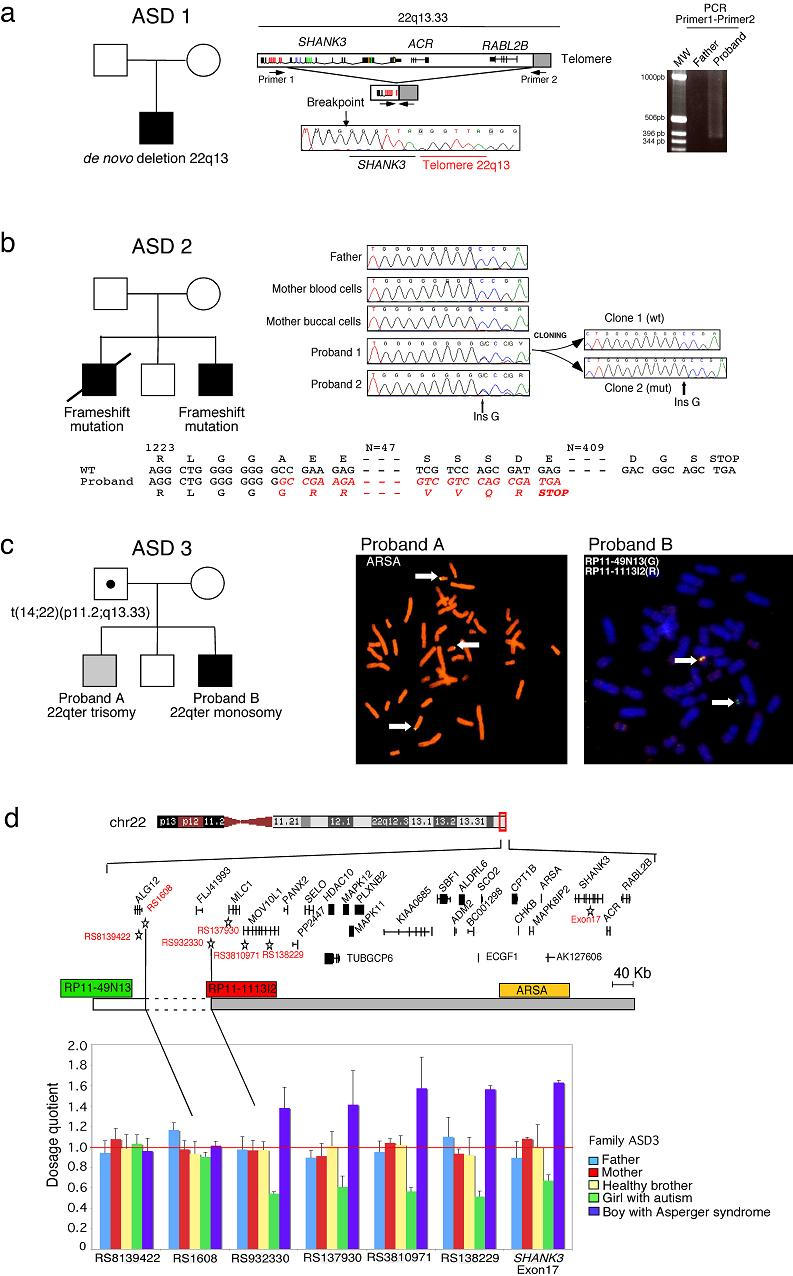
Genetic analyses of three families with ASD and SHANK3 mutations, (a) In family ASD 1, the proband carries a de novo terminal deletion of the paternal chromosome 22q13. The deletion breakpoint is located in intron 8 of SHANK3. The breakpoint was sequenced after amplification of the proband DNA using primer 1 in SHANK3 and primer 2 in the telomeric repeats. The heterogenous smear in the proband is likely due to the difference in telomere length from chromosome to chromosome and/or priming at different locations by the telomeric primer, (b) In family ASD 2, the two probands carry the same de novo SHANK3 frame-shift mutation on the maternal chromosome 22q13. The mutation is absent from the mother blood and buccal cells, suggesting a germinal mosaicism. The guanine insertion is located in exon 21 of SHANK3, leading to a premature truncated protein, (c) In family ASD 3, the father carries a balanced translocation t(14,22)(p11.2;q13.33), proband A (Asperger syndrome) presents a partial 22qter trisomy and proband B (autism) has a 22qter deletion, (d) Using quantitative fluorescent PCR, we mapped the breakpoint between the genes ALG12 and MLC1. The dosage quotient has a theoretical value of 0.5 for a deletion and 1.5 for a duplication.
In family ASD 2, two brothers with autism were heterozygous for an insertion of a guanine nucleotide in exon 21 (Fig. 1b). Both brothers had severely impaired speech and severe mental retardation. The mutation was absent in an unaffected brother and the unaffected parents. Using 14 informative SNPs, we found that the mutation was located on the same maternal haplotype in the two affected brothers and that the unaffected brother did not have this haplotype (Supplementary Fig. 3). The mutation was absent in the DNA isolated from blood leukocytes and mouth cells of the mother. These results strongly suggest a germinal mosaicism in the mother. The guanine insertion creates a frameshift at nucleotide 3680, modifying the C-terminal sequence of the protein (Fig. 1b). This putative truncated protein lacks several crucial domains involved in mGluR and actin binding (Homer, AbPl, cortactin) and in the synaptic targeting and postsynaptic assembly of SHANK3 multimers11,12. Consistent with the loss of these domains, when we over-expressed the truncated protein in rat hippocampal neuronal cells, we did not observe any synaptic localization compared with the wild-type sequence (Supplementary Fig. 4).
In family ASD 3, we identified a terminal 22q deletion in a girl with autism and severe language delay and a 22qter partial trisomy in her brother with Asperger syndrome, who demonstrated precocious language development and fluent speech (Fig. 1c). We found that these unbalanced cytogenetic abnormalities were inherited from a paternal translocation, t(14;22)(p11.2;q13.33). The chromosome 14p11.2 breakpoint fell within the heterochromatic DNA sequence characteristic of acrocentric chromosomes and did not contain any putative transcripts or genes. On chromosome 22q13.33, using informative SNPs and quantitative PCR, we mapped the breakpoint between ALG12 and MLC1 (Fig. 1d). The deletion and duplication rearrangement observed in both siblings involved 25 genes, including SHANK3, located in the 800-kb terminal sequence of 22q13. No other SHANK3 deletions or duplications were observed after screening 155 individuals by quantitative PCR (58 with autism, 38 with Asperger syndrome and 59 controls).
In the remaining individuals with ASD, we identified seven who had rare nonsynonymous variations, which were not observed in controls (n=270–333; Fig. 2 and Supplementary Table 1). However, all these variations were inherited from healthy parents, ruling out their direct involvement as dominant mutations in the disorder. Notably, for two substitutions modifying highly conserved amino acids (R12C and R300C; Supplementary Fig. 3), we observed that the overexpressed mutated GFP Shank3 fusion proteins clustered but showed significantly less colocalization with the presynaptic marker protein Bassoon, suggesting nonsynaptic clustering (Supplementary Fig. 4). These observations might reflect posttranslational modifications or abnormal folding of the protein. Thus, although these genetic variations cannot be considered as causal mutations, they might nevertheless modify the synaptic scaffolding and represent risk factors for ASD in interaction with other susceptibility genes.
Figure 2.
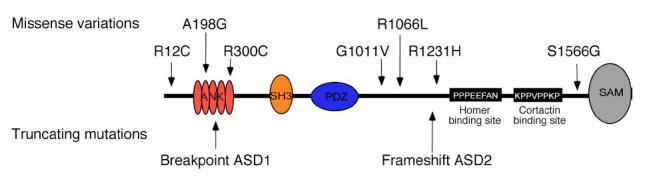
Localization of rare nonsynonymous variations or truncating SHANK3 mutations identified in families with ASD. ANK: ankyrin repeats; SH3: Src homology 3 domain; PDZ: postsynaptic density 95/Discs large/zona occludens-1 homology domain; SAM: sterile alpha motif domain.
In this study, we show that a SHANK3 heterozygous mutation can cause ASD. Notably, in the boy with Asperger syndrome in family ASD 3, the presence of an additional copy of 22q13/SHANK3 did not impair his language ability but seems to have led to a severe impairment in social communication. These results, together with previous reports13,14, highlight the importance of a fine gene dosage for the development of speech/language and/or social communication in humans.
The mutations identified in these patients are thought to affect the function and localization of SHANK3 at PSD and dendritic spines. These results are consistent with the alterations of dendritic spines in individuals with learning disabilities15. In mice, Shank-3 promotes the maturation and the enlargement of dendritic spine heads and is even able to induce spine formation in aspiny neurons11. In ASD, an abnormality of synapse formation and maintenance was first suggested by the identification of mutations in X-linked NLGN3 and NLGN49, and next confirmed by functional studies of the causative mutations. Therefore, we hypothesize that the protein complex including neuroligins and SHANK participates in the assembly of specialized postsynaptic structures required for the development of language and social communication.
Supplementary Material
Acknowledgments
We thank the affecetd individuals and their families for participating in this study and all the collaborators of the Paris Autism Research International Sibpair (PARIS) Study: Sweden: Department of Child and Adolescent Psychiatry, Göteborg University, Göteborg: Christopher Gillberg, Maria Råstam, Carina Gillberg, Gudrun Nygren, Henrik Anckarsäter, Ola Ståhlberg; France: Department of Psychiatry, Groupe Hospitalier Albert Chenevier et Henri Mondor, Créteil: Marion Leboyer; INSERM U513, Université Paris XII, Créteil: Catalina Betancur; Service de Psychopathologie de l’Enfant et l’Adolescent, Hôpital Robert Debré, Paris: Catherine Colineaux, Deborah Cohen, Nadia Chabane, Marie-Christine Mouren-Siméoni; INSERM U679, Hôpital Pitié-Salpêtrière, Paris: Alexis Brice; Norway: Centre for Child and Adolescent Psychiatry, University of Oslo, Oslo: Eili Sponheim; Department of Pediatrics, Rikshospitalet, University of Oslo, Oslo: Ola H. Skjeldal; USA: Department of Pediatrics, Georgetown University School of Medicine, Washington D.C.: Mary Coleman; Children’s National Medical Center, George Washington University School of Medicine, Washington, D.C.: Philip L. Pearl; New York State Institute for Basic Research in Developmental Disabilities, Staten Island, New York: Ira L. Cohen, John Tsiouris; Italy: Divisione di Neuropsichiatria Infantile, Azienda Ospedaliera Senese, Siena: Michele Zappella; Austria: Department of General Psychiatry, University Hospital, Vienna: Harald Aschauer; Belgium: Centre de Génétique Humaine, Institut de Pathologic et de Génétique, Gerpinnes, Loverval: Lionel Van Maldergem
We also thank the DNA and cell bank of INSERM U679 (IFR des Neurosciences, Hôpital Pitié-Salpêtrière), the Centre d’Investigations Cliniques of the Hôpital Robert Debré, C. Bouchier and S. Duthoy for the use of sequencing facilities at the Génopole Pasteur, and A. Hchikat, L. Margarit, and G. Rouffet for technical assistance. This work was supported by the Pasteur Institute, INSERM, Assistance Publique-Hôpitaux de Paris, Fondation France Telecom, Cure Autism Now, Fondation de France, Fondation Biomédicale de la Mairie de Paris, Fondation pour la Recherche Médicale, EUSynapse European Commission FP6, AUTISM MOLGEN European Commission FP6, Fondation NRJ, the Swedish Science Council and the Deutsche Forschungsgemeinschaft DFG, SFB 497.
References
- 1.Folstein SE, Rosen-Sheidley B. Nat Rev Genet. 2001;2:943–955. doi: 10.1038/35103559. [DOI] [PubMed] [Google Scholar]
- 2.Persico AM, Bourgeron T. Trends Neurosci. 2006;29:349–358. doi: 10.1016/j.tins.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Vorstman JA, et al. Mol Psychiatry. 2006;11(1):18–28. doi: 10.1038/sj.mp.4001781. [DOI] [PubMed] [Google Scholar]
- 4.Manning MA, et al. Pediatrics. 2004;114:451–7. doi: 10.1542/peds.114.2.451. [DOI] [PubMed] [Google Scholar]
- 5.Bonaglia MC, et al. J Med Genet. 2006;43:822–8. doi: 10.1136/jmg.2005.038604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naisbitt S, et al. Neuron. 1999;23:569–82. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- 7.Boeckers TM, Bockmann J, Kreutz MR, Gundelfinger ED. J Neurochem. 2002;81:903–10. doi: 10.1046/j.1471-4159.2002.00931.x. [DOI] [PubMed] [Google Scholar]
- 8.Meyer G, Varoqueaux F, Neeb A, Oschlies M, Brose N. Neuropharmacology. 2004;47:724–33. doi: 10.1016/j.neuropharm.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 9.Jamain S, et al. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonaglia MC, et al. Am J Hum Genet. 2001;69:261–8. doi: 10.1086/321293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roussignol G, et al. J Neurosci. 2005;25:3560–70. doi: 10.1523/JNEUROSCI.4354-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baron MK, et al. Science. 2006;311:531–5. doi: 10.1126/science.1118995. [DOI] [PubMed] [Google Scholar]
- 13.Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. Nature. 2001;413:519–23. doi: 10.1038/35097076. [DOI] [PubMed] [Google Scholar]
- 14.Somerville MJ, et al. N Engl J Med. 2005;353:1694–701. doi: 10.1056/NEJMoa051962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carlisle HJ, Kennedy MB. Trends Neurosci. 2005;28:182–7. doi: 10.1016/j.tins.2005.01.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.