

Abstract
Saccharomyces cerevisiae vacuole inheritance requires two low molecular weight activities, LMA1 and LMA2. LMA1 is a heterodimer of thioredoxin and protease B inhibitor 2 (I2B). Here we show that the second low molecular weight activity (LMA2) is monomeric I2B. Though LMA2/I2B was initially identified as a protease B inhibitor, this protease inhibitor activity is not related to its ability to promote vacuole fusion: (i) Low Mr protease B inhibitors cannot substitute for LMA1 or LMA 2, (ii) LMA1 and LMA2 promote the fusion of vacuoles from a strain that has no protease B, (iii) low concentrations of LMA2 that fully inhibit protease B do not promote vacuole fusion, and (iv) LMA1, in which I2B is complexed with thioredoxin, is far more active than LMA2/I2B in promoting vacuole fusion and far less active in inhibiting protease B. These studies establish a new function for I2B.
Organelle inheritance is tightly regulated during the cell cycle (1, 2). In Saccharomyces cerevisiae (2), low copy-number organelles such as the nucleus (3, 4) and mitochondria (5, 6) are segregated into the bud. We have been studying the inheritance of the yeast vacuole. In early S-phase, the maternal vacuole projects a striking array of tubular-vesicular material, termed the segregation structure, into the bud (7). This vacuolar exchange continues until the onset of M-phase. The vacuolar vesicles in the bud then fuse to form the daughter cell organelle.
Several vac (vacuole inheritance) mutants are defective in this process (8–12). In a biochemical approach, we have developed an assay to study the process of vacuole inheritance in vitro (13–15). Isolated vacuoles, when incubated with ATP and cytosol, form tubular extensions that resemble the segregation structures seen in vivo (13). These structures are physiologically relevant, as they are not produced from vacuoles prepared from vac mutant strains.
Vacuole fusion, the terminal step of the inheritance mechanism, is also observed in vitro, requires VAC gene products, and can be monitored in a quantitative assay. This reaction, which requires ATP and cytosol, relies on the mixing of vacuole contents after fusion (14, 15). For this assay, vacuoles are purified from two strains. The first lacks the PHO8 gene, which encodes vacuolar pro-alkaline phosphatase, but contains the normal vacuolar proteases. The second lacks proteinase A, the vacuolar maturation enzyme, and therefore contains enzymatically inactive pro-alkaline phosphatase. Upon membrane fusion and mixing of the vacuole contents, the prophosphatase is proteolytically activated. Alkaline phosphatase activity, determined by colorimetric assay, is a direct measure of membrane fusion.
Fusion of vacuoles requires the small GTPase protein Ypt7p (16) and the membrane-trafficking proteins Sec17p (α-SNAP) and Sec18p (NSF) (17, 18). All three factors act early in the fusion pathway, where ATP-driven release of Sec17p by Sec18p primes the vacuoles for Ypt7p-dependent docking (18, 19). After docking, these factors are no longer required for membrane fusion.
Yeast cytosol contains several proteins that support vacuole fusion (20). These include a high molecular weight activity and two low molecular weight activities (LMA1 and LMA2). LMA1 is a heterodimer of thioredoxin 1 and protease B inhibitor 2 (I2B) and stabilizes vacuoles that have been “primed” by Sec 17p release (21). We now report that LMA2 is actually I2B, free of thioredoxin, and show that LMA2, though less active than LMA1, does not support vacuole fusion simply by inhibiting protease B.
MATERIALS AND METHODS
Yeast Strains.
S. cerevisiae strains were: K91-1A (MATa ura3 pho8::pAL134 pho13::pPH13 lys1) from Y. Kaneko (Institute of Fermentation, Osaka); BJ 3505 (MATa pep4::HIS6 prb1-Δ1.6R HIS3 lys2–208 trp1-Δ101 ura3–52 gal2 can) and DKY 6281 (MATa leu2-3 leau2-112 ura3-52 his3-Δ200 trp1-Δ901 lys2–801 suc2-Δ9 pho8::TRP1) from D. Klionsky (University of California, Davis); YS18 (MATα ura3-5 his3-11 leu2-3,-112 canR); YPS19 (MATα Δpbi2::URA3) from D. Wolf (University of Stuttgart, Stuttgart, Germany); EMY21-1C (MATa TRX1 TRX2 Δlys2::HIS3 ura3-1 Δade3-100 ade2 trp1-1 leu2-3 112 his3-11 can1-100), and EMY21-8D (MATa Δtrx1::TRP1 Δtrx2::LEU2) from E. Muller (University of Washington, Seattle). Yeast were grown in YPD medium.
Materials.
Oxalyticase was from Enzogenetics (Corvallis, OR). ATP, GTP, DEAE-Dextran, Ficoll 400, Sephacryl S-300HR, SP-Sepharose Fast Flow, PBE 94, and Polybuffer 96 were from Pharmacia. Creatine phosphokinase, creatine phosphate, Pipes, chymostatin, guanosine 5′[γ-thio]triphosphate (GTP[γS]) and Pefabloc SC were from Boehringer Mannheim. N-(3-triethylammoniumpropyl)-4-(p-diethylaminophenyl-hexatrienyl) pyridinium dibromide (FM4-64) was from Molecular Probes. Coomassie brilliant blue R, polyvinylidene difluoride membranes, and Protein Assay kit (using BSA as a standard) were from Bio-Rad. Mastoparan was from Peninsula Laboratories and microcystin LR from LC Services (Woburn, MA). Azocoll was from Calbiochem. LMA1 was purified as described (20). His-tagged Sec18p and other reagents were prepared as described (16, 17).
Yeast Cytosol.
For purification of I2B, K91-1A cells were grown in 150L of YPD to OD600 = 3.0 and collected in a Sharples centrifuge. Cells were suspended in 6 l of water at 23°C, sedimented (5,000 × g, 5 min, 4°C), resuspended in 3 l of cytosol buffer (20 mM Pipes⋅KOH, pH 6/150 mM KOAc/250 mM sorbitol/5 mM MgCl2), sedimented, resuspended in 250–300 ml of cytosol buffer and frozen by dropwise addition to liquid nitrogen. Cells were lysed in liquid nitrogen by homogenization in a 5-l Waring blender. One quarter of the frozen cells were processed at a time for 5 min each, with occasional addition of liquid nitrogen. The lysed cell powder was thawed in a 4 l Erlenmeyer flask under hot tap water. After partial thawing, 200 mM phenylmethylsulfonyl fluoride in isopropanol was added to a final concentration of 0.5 mM. After centrifugation at 4°C as above, the supernatants were pooled. Mg-ATP and 1,000× protease inhibitor cocktail (1,000× PIC; 200 mM Pefabloc SC/5 mg/ml leupeptin/500 mM 1,10-phenanthroline/5 mg/ml pepstatin A) were added to 0.1 mM and 2× final concentrations. The lysate was centrifuged (45Ti, 4°C, 20 min) and supernatants were frozen as droplets in liquid nitrogen.
Cytosol preparations from other yeast stains were either by a smaller version of this procedure or by agitation with glass beads (13). For heated cytosol supernantant (HCS), cytosol was heated to 80°C in a boiling water, kept at 80°C for 4 min, then cooled in an ice water bath. The precipitate was removed by centrifugation (17,000 × g, 30 min, 4°C).
Colorimetric Vacuole Fusion Assay.
Fusion assays were described previously (20, 22), using salt-washed, frozen vacuoles and 17 ng of his-tagged recombinant Sec18p. One unit of activity is the hydrolysis of 1 μmol p-nitrophenol phosphate/min⋅μg of BJ 3505 vacuolar protein.
Inhibitors were used at the following concentrations: 1.5 mM GTP[γS], 22 μM mastoparan, 64 μg/ml Gdi1p, 30 μg/ml affinity purified anti-Sec17p, 30 μg/ml affinity purified anti-Sec18p, 75 μg/ml affinity purified anti-Ypt7p, 1.2 mM neomycin, and 15 μM microcystin-LR. When apyrase was used (66 unit/ml), the ATP regeneration system was omitted from the assay. Assays with anti-Sec17p and anti-Sec18p antibodies used fresh vacuoles that had not been salt washed. A mixture of equal parts of BJ 3505 and DKY 6281 vacuoles was incubated at 30°C for 10 min with 0.73 μg of IgG/μg of vacuoles. After 3 min at 0°C, vacuoles were resedimented (8,000 × g, 80 sec) and resuspended at 0.3 mg/ml in 20 mM Pipes⋅KOH (pH 6.8), 200 mM sorbitol. Fusion reactions (30 μl) contained 20 μl of vacuoles.
Morphological Vacuole Fusion Assay.
Fresh BJ 3505 vacuoles were diluted to 0.4 mg/ml in 20 mM Pipes⋅KOH (pH 6.8), 200 mM sorbitol, 50 mM KOAc, and 100 mM KCl and incubated at 30°C for 10 min. After centrifugation (8,000 × g, 80 sec), vacuoles were resuspended at 1.2 mg/ml in 20 mM Pipes⋅KOH (pH 6.8) and 200 mM sorbitol. Ten microliters of these vacuoles were used in a 30-μl standard fusion reaction, then stained with 1 μl of 20 μM FM4-64 (32) and visualized by fluorescence microscopy with TMAXp3200 film (Kodak) and a Nikon F3 camera.
Purification of I2B from Yeast Cytosol.
K91–1A yeast cytosol (400 ml) was passed through a 6.5 × 17 cm DEAE-cellulose column in buffer A [20 mM Hepes⋅KOH, pH 6.8/5 mM MgC12/0.1 mM MnCl2/10% (wt/vol) glycerol] at 6.0 ml/min, 4°C. Unbound material was applied at 6.0 ml/min to a 3.8 × 10 cm column of SP-Sepharose Fast Flow in buffer A at 4°C. The column was eluted with 250 ml of buffer A with 200 mM KCl and a 575 ml linear gradient of 200 mM-550 mM KCl in buffer A at 1.8 ml/min.
Two peaks of activity emerged. The first peak (45 ml) was concentrated to 1.5 ml in Amicon Centriprep 10 concentrators (3,000 × g, 4°C), then incubated at 95°C for 4 min and centrifuged (17,000 × g, JA18, 4°C, 30 min). The supernatant was applied to a 1.5 × 16 cm Sephacryl S-300HR column in buffer B [25 mM ethanolamine-CH3COOH, pH 9.4, 10% (wt/vol) glycerol] at 4°C. Two peaks of activity emerged. The activity with the lower molecular weight (5 ml) was pooled and loaded at 0.5 ml/min onto a 1.0 × 6.4 cm PBE 94 column (equilibrated at 4°C in buffer B followed by 2 ml of buffer C, a 1:10 dilution of Polybuffer 96-CH3COOH, pH 6.0). The column was eluted with buffer C.
Protein Sequencing.
Fractions (5 ml) were concentrated to 200 μl by lyophilization, precipitated by methanol/chloroform (23), analyzed by high-Tris SDS/PAGE (20), and transfered to polyvinylidene difluoride membrane. The protein band was excised, moistened with 5 μl of methanol, submerged in water, divided into ≈1 mm2 pieces, and transfered to a microfuge tube with 30 μl of digestion buffer (24). Trypsin (Boehringer Mannheim sequencing grade, 0.1 μg) was added and the suspension incubated for 18 hr at 37°C. A further 20 μl of digestion buffer was added and the sample shaken for 30 min followed by centrifugation (23°C, 1,700 rpm, 5 min). The polyvinylidene difluoride pieces were transfered to a Hewlett–Packard injection vial and washed successively with 50 μl of digestion buffer and 50 μl of 0.1% (wt/vol) trifluoroacetic acid, followed by shaking and recentrifugation. The pooled washes were injected onto an HPLC column and the peptides were sequenced.
Production of I2B in Escherichia coli.
Oligonucleotides (GIBCO/BRL) with sequences flanking the coding region of the I2B gene (PBI2) (31) were: 3′-HindIII GCGTAAGCTTCAGGAATCAAT; PBI2, 5′-BspHI CAAGGTCCATCATGACAA. They were used to amplify PBI2 from yeast genomic DNA (Promega) using DNA polymerization mix (Pharmacia) and Taq DNA polymerase (GIBCO/BRL). The product was purified using the QIAquick PCR product purification kit (Qiagen, Chatsworth, CA).
PBI2-pBAD22 was produced by digestion of the I2B PCR product with BspHI and HindIII and digestion of pBAD22 (25) with NcoI and HindIII. DNA fragments were gel purified on a 1% agarose gel, ligated into the vector with T4 DNA ligase (26) and transformed into BL21 (DE3) (27).
Purification of I2B from E. coli.
I2B-pBAD22/BL21(DE3) was grown in TB (26) with 100 mg/l amp to OD600 = 0.3, and 200 ml of 20% (wt/vol) arabinose was added. Cells were grown to OD600 = 1.4, sedimented (5,000 × g, 10 min), resuspended in 40 ml of buffer D (50 mM KOAc, pH 5.5) with 1 mM phenylmethylsulfonyl fluoride, and sonicated. The lysate was centrifuged (37,000 × g, 30 min, 4°C) and the supernatant was warmed to 80° in a boiling water bath with continuous stirring followed by 4 min further incubation and centrifugation. The supernatant was loaded onto a 1.5 × 11.2 cm column of SP-Sepharose Fast Flow in buffer D at 4°C. The column was eluted with 40 ml of buffer D followed by a linear, 100 ml gradient of 0–500 mM KCl in buffer D. I2B emerged at ≈200 mM KCl.
Other Methods.
Gel filtration of yeast cytosols (20, 21) and vacuolar protease B assay (28) were as described. Frozen vacuoles (100 μl, 60 μg of protein) were incubated at 30°C for 3 min with swirling in a final volume of 300 μl with 100 mM Pipes⋅KOH, pH 6.8, 200 mM sorbitol, 0.15% (wt/vol) TX-100, and 5 mg of Azocoll. High-Tris SDS/PAGE was as described (20). Samples were concentrated with chloroform/methanol (23). Proteins were transfered to a polyvinylidene difluoride membrane using a PolyBlot apparatus (American Bionetics, Hayward, CA). I2B was detected with the enhanced chemiluminescence kit (Amersham) and enhanced chemiluminescence hyperfilm (Amersham) using a rabbit serum produced to the 12 C-terminal residues of I2B (ENVEEDKEVHTN) (21) and goat anti-rabbit conjugated to horseradish peroxidase (Bio-Rad).
RESULTS
Purification of I2B.
To identify factors that support in vitro vacuole fusion, yeast cytosol was passed through a DEAE-cellulose column and the unbound material was loaded onto a SP-Sepharose Fast Flow column and eluted with a salt gradient. The ability of each fraction to catalyze vacuole fusion was assayed with a limiting amount of Sec18p (ref. 21 and Fig. 3B). Two peaks of activity emerged that were displaced to either side of the bulk of the protein (data not shown). The peak activity at the lower salt concentration was subjected to heat-treatment that, surprisingly, stimulated the fusion activity ≈3-fold (data not shown). This was probably due to the removal of inhibitors of the fusion pathway. After gel filtration through Sepharose S-300HR, two peaks of activity emerged. The active material of lower molecular weight was purified to homogeneity by chromatofocusing on a PBE 94 column. The activity emerged as a single peak with an apparent pI of 7.4 (Fig. 1B). A single protein of ≈7 kDa comigrated with the activity (Fig. 1A). This band was digested with trypsin and peptide fragments were purified by reverse-phase HPLC. One peptide had the sequence YTIKVPDVLHxNK that is identical to amino acids 41–53 of I2B, the yeast protease B inhibitor 2 (29).
Figure 3.
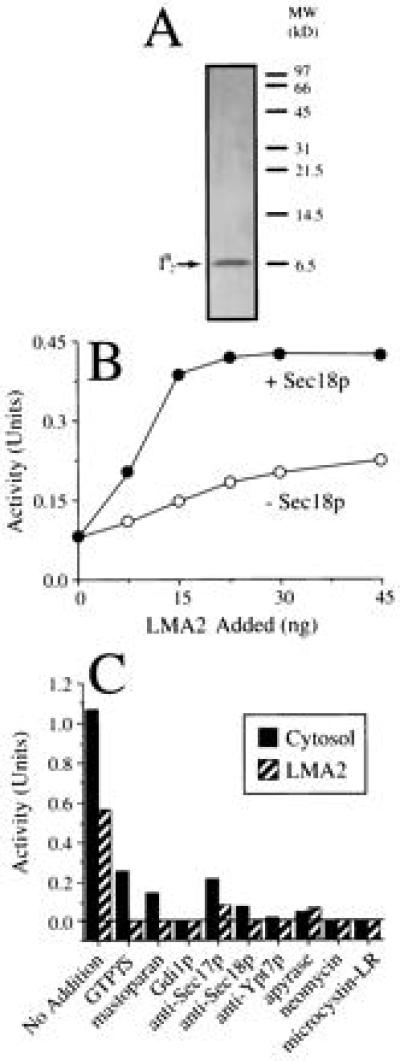
Recombinant I2B drives vacuole fusion along the Sec18p-dependent pathway. (A) LMA2 (I2B) was purified from a recombinant E. coli strain and analyzed by high-Tris SDS/PAGE and silver-staining. (B) Vacuole fusion reactions contained LMA2 in the presence or absence of Sec18p (see Materials and Methods). (C) Vacuole fusion was assayed in the presence of Sec18p and either 80 μg of yeast cytosol or 75 ng of LMA2. Assays were performed in the absence (no addition) or presence of various inhibitors of vacuole fusion (see Materials and Methods). The ATP-only background was 0.05 units.
Figure 1.
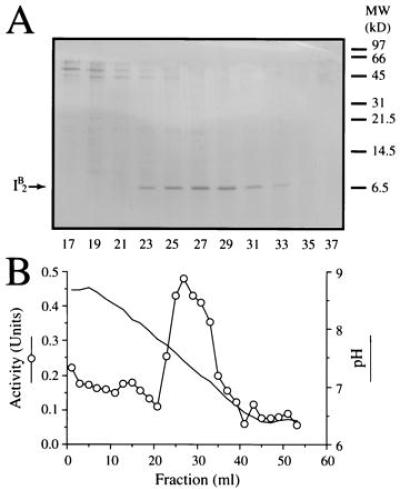
I2B promotes vacuole fusion. (A) Yeast cytosol was subjected to successive steps of DEAE-cellulose, SP-Sepharose Fast Flow, Sephacryl S-300HR chromatography, and chromatofocusing. Alternate fractions (200 μl) of the latter were analyzed by high-Tris SDS/PAGE followed by staining with Coomassie brilliant blue R. Positions of molecular weight markers are shown on the right and of I2B on the left. (B) The pH of alternate fractions and their ability to drive vacuole fusion in the presence of limiting amounts of Sec18p was determined.
IB2 is the LMA2 Activity.
I2B was originally identified as a cytosolic protein that inhibits yeast vacuolar proteinase B (30) and was later found as a component of the LMA1 heterodimer that can catalyze yeast vacuole fusion (20). Gel filtration has shown that yeast cytosol contains two low molecular weight activities termed LMA1 and LMA2 (21). Cytosol derived from a strain in which both thioredoxin genes have been deleted, however, only contains the LMA2 activity. Immunoblotting with anti-I2B antibodies of Sephacryl S-100HR fractions after gel filtration of wild-type cytosol confirms that I2B elutes with the LMA1 activity but also shows that the LMA2 fraction contains a disproportionate amount of I2B relative to its functional activity (Fig. 2A, fraction 52). Moreover, a similar analysis of cytosol from cells that contain no thioredoxin shows that I2B elutes exactly with LMA2 (Fig. 2B). Taken together, these data indicate that I2B and LMA2 are the same protein.
Figure 2.
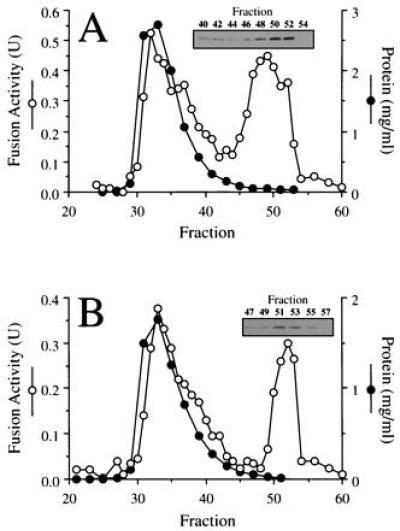
I2B is LMA2. Previously characterized (20) fractions from gel filtration of wild-type (A) or thioredoxin double-deletion cytosol (B) that had been assayed for protein and vacuole fusion activity (20) were assayed for I2B by immunoblot.
LMA2 Supports Normal Vacuole Fusion.
To confirm the identity of LMA2 (I2B), it was purified (Fig. 3A) from an E. coli strain in which expression of the PBI2 protein (31) is under the arabinose promoter. Purified, recombinant LMA2 was incubated with vacuoles and ATP in the presence of a limiting amount of recombinant Sec18p or Sec18p buffer (Fig. 3B). LMA2 was far less active in the absence of Sec18p, which alone yielded no signal. However, LMA2 and Sec18p acted synergistically (Fig. 3B). The LMA2-based reaction is sensitive to each of the characterized fusion inhibitors (Fig. 3C) that also inhibit the cytosol-driven vacuole fusion reaction (refs. 14, 16, and 18 and Fig. 3C).
Since I2B (LMA2) and thioredoxin are subunits of LMA1 (20, 21), we investigated the relative activities of LMA1 and LMA2. I2B (LMA2) and LMA1 concentrations were determined by quantitative immunoblotting with anti-I2B antibody (data not shown), and vacuole fusion assays were performed with either LMA1 (containing 75 ng of I2B) or 75 ng of purified I2B itself. LMA2 did not support fusion as rapidly as an equivalent amount of LMA1 (Fig. 4A). Although the extent of fusion catalyzed by LMA1 or LMA2 at low I2B concentrations was similar (Fig. 4B), the yield of the LMA1-driven reaction continued to increase with the protein concentration while that of the LMA2-driven reaction reached a plateau.
Figure 4.
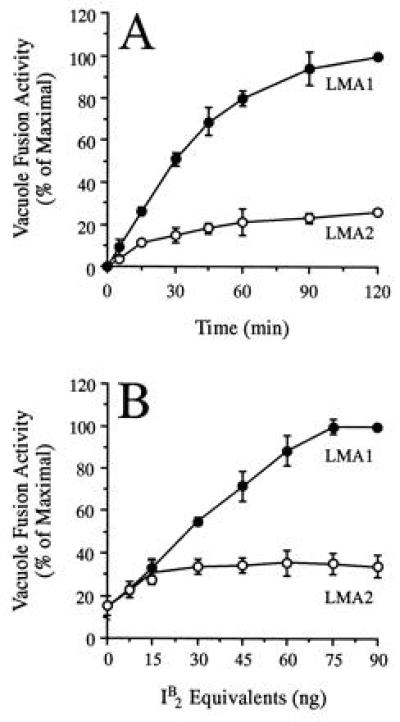
LMA2 is less efficient at promoting vacuole fusion than LMA1. (A) Aliquots were removed from the equivalent of ten regular fusion assays containing equal amounts of either LMA1 or LMA2 and placed on ice. Each aliquot was assayed for alkaline phosphatase activity. (B) Vacuoles were incubated with Sec18p and LMA1 or LMA2 containing equivalent amounts of I2B. The data are the average of three experiments ± SD and are shown as a percentage of the maximal fusion signal (A, 0.98 units; B, 1.05 units).
Since I2B can inhibit vacuolar protease B, it might catalyze vacuole fusion by functioning as a protease inhibitor. To test this, vacuoles were prepared from BJ3505 cells that contain neither protease A nor protease B and were incubated with Sec18p or with Sec18p and LMA2. At the start of the reaction, an aliquot of each mixture was placed on ice to prevent vacuole fusion. Upon completion of the reaction, aliquots were stained with the vacuole specific fluorophore FM4-64 (32). Little vacuole fusion was observed in the presence of Sec18p (Fig. 5B) compared with the ice control (Fig. 5A). However, after incubation with both Sec18p and LMA2, large vacuoles were seen, indicating that fusion had occurred (Fig. 5C).
Figure 5.
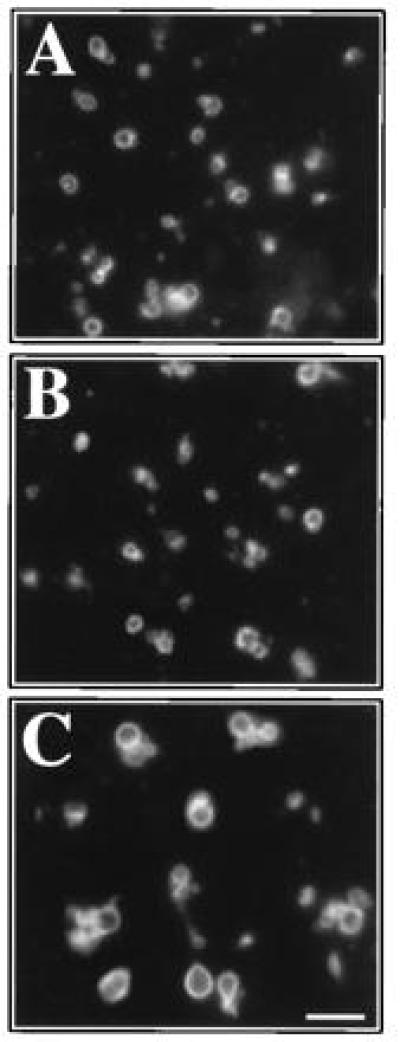
LMA2 supports the fusion of vacuoles lacking protease B. Fresh, salt-washed vacuoles were incubated for 2 hr at 0°C (A) or 27°C (B and C) in the presence of Sec18p (B) or Sec18p and LMA2 (A and C). Vacuoles were stained with FM4-64 and visualized by fluorescence microscopy. (Bar = 4 μm.)
To confirm that the activity of LMA2 as a protease inhibitor is unrelated to its ability to support vacuole fusion, reactions were performed in the presence of chymostatin, a serine protease inhibitor. Chymostatin does not inhibit protease A (33), an aspartate protease, but is a strong inhibitor of protease B, a serine protease. Chymostatin should therefore not prevent the maturation of pro-alkaline phosphatase (that largely depends on protease A) after vacuole fusion. Proteinase B activity is 93% inhibited by 7pM chymostatin (Fig. 6A) in an assay (33) that is highly specific for this protease (33) and that contains the same vacuole concentration as a fusion reaction. At the same concentration, chymostatin does not stimulate vacuole fusion (Fig. 6A). In contrast, LMA2 stimulates vacuole fusion at concentrations that completely inhibits protease B. A 20-fold lower concentration of LMA2 inhibits protease B by 97% but does not support vacuole fusion (Fig. 6A). Thus inhibition of protease B activity does not lead to vacuole fusion, and the activity of LMA2 in supporting vacuole fusion does not correlate with its inhibition of protease B.
Figure 6.
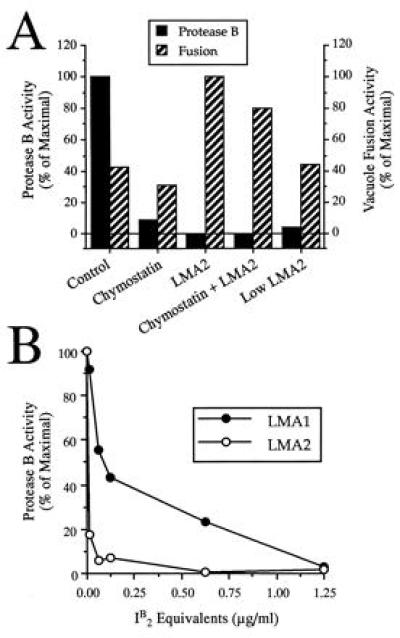
Inhibition of protease B does not promote vacuole fusion. (A) Vacuoles were assayed for protease B activity or fusion in the absence of added reagents or in the presence of 7pM chymostatin, 2.5 μg/ml LMA2, a mixture of both or 125 ng/ml of LMA2 alone (Low LMA2). The data were expressed as a percentage of maximal protease B activity or fusion activity (0.42 unit). (B) Vacuoles were assayed for protease B activity in the presence of LMA1 or LMA2 that contained equivalent amounts of I2B. Data are presented as a percent of protease activity in the absence of added protein and are the average of two experiments.
To test whether the binding of thioredoxin to I2B alters its ability to inhibit protease B, the protease B activity was assayed with LMA1 or with LMA2 that contained equivalent amounts of I2B (see above). While LMA1 can inhibit protease B, it did so far less effectively than LMA2 (Fig. 6B). The fact that LMA1 is more effective than LMA2 in promoting vacuole fusion (Fig. 4), but is less efficient in inhibiting protease B (Fig. 6B) shows that LMA2 does not support vacuole fusion by inhibiting protease B.
Yeast Cytosol Contains at Least One Other Heat-Stable Fusion Factor.
Yeast cytosol contains heat stable factors that can stimulate vacuole fusion (17) and LMA2 was purified from cytosol using a procedure that involved a heating step. To determine whether LMA2 alone is responsible for the activity of HCS, cytosols were prepared from a mutant yeast strain (YPS19) in which the I2B gene had been deleted (31) and from its parental strain (YS18). YS18 and YPS19 HCS had similar fusion activities (Fig. 7A), showing that I2B is not the only heat-stable cytosolic factor.
Figure 7.
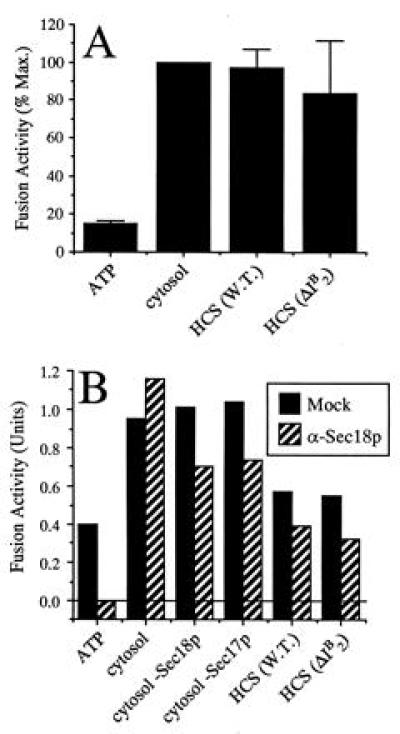
Yeast cytosol contains heat-stable activities other than LMA2. (A) Frozen, salt-washed vacuoles were incubated with ATP alone, 30 μg of I2B-wild-type cytosol (K91-1A), 1.2 μg of I2B-wild-type HCS, or 1.2 μg of I2B-deletion HCS. Data were normalized to the maximal (cytosol) signal (1.25 units) and the average of four experiments is shown ± SD. (B) Fresh vacuoles that had not been salt-washed were preincubated with preimmune or anti-Sec18p IgG. After removal of the antibody, vacuoles were incubated with ATP and 30 μg of cytosol, either with or without immunodepletion of Sec17p or Sec18p, or with 1.2 μg of HCS from wild-type or I2B-deletion cytosol. The average of two experiments is shown.
One such heat-stable activity is a “Sec18p-activating factor” (17). Preincubation of vacuoles with antibodies to Sec18p renders them inactive unless a subsequent fusion reaction is supplemented with active Sec18p, Sec18p-immunodepleted cytosol, or HCS (17). However, we have been unable to detect any Sec18p-activating factor activity associated with LMA2 (data not shown). HCS from yeast strains YS18 and YPS19 were equally able to overcome the Sec18 antibody block, confirming that the LMA2 and Sec18p-activating factor activities are distinct (Fig. 7B).
DISCUSSION
We recently identified LMA1, a low-molecular weight heterodimeric protein from S. cerevisiae that participates in vacuole inheritance both in vivo and in our in vitro assay (20). One component of this complex is thioredoxin while the other is I2B (21), demonstrating that I2B has a second, novel function. Here we report the independent purification of uncomplexed I2B as an activity that supports vacuole fusion in vitro. Deletion of both TRX1 and TRX2 eliminates LMA1 but not LMA2 activity (20). I2B from the thioredoxin double-mutant copurifies with the LMA2 activity and expression of the cloned gene in E. coli yields a protein of full activity, demonstrating that the LMA2 is I2B, free of thioredoxin.
Deletion of the I2B gene yields a vac phenotype in vivo (21). Furthermore, the LMA2-driven reaction is sensitive to all of the tested inhibitors of the cytosol-driven reaction. Since LMA2 can drive the fusion of vacuoles that lack protease B, it does not function in vacuole fusion by means of its protease B inhibitor activity. Furthermore, inhibition of vacuolar protease B using chymostatin does not stimulate vacuolar fusion, and low levels of LMA2 that almost completely inhibit protease B are insufficient to support the fusion reaction. It is also unlikely that LMA2 could be inactivating other proteases and so stimulate vacuole fusion for three reasons. First, I2B, as a noncompetitive inhibitor of protease B, must bind to the protease at a point other than its active site, and thus is unlikely to inhibit other proteases by binding to their catalytic clefts. Second, I2B has a very high (nM) affinity for protease B (1) and so is an unlikely candidate for a broad-spectrum protease inhibitor; it has almost no inhibitory activity to proteinase A or carboxypeptidase Y (1). Third, we have never observed stimulation of fusion by other protease inhibitors.
LMA2 is not the only heat-stable active factor in yeast cytosol that can support vacuole fusion. Heated cytosol supernatants from cytosol lacking I2B can still efficiently support vacuole fusion, indicating that at least one other heat-stable factor is involved. This activity may be the Sec18p-reactivating factor that may act by recruiting membrane-bound Sec18p into an active fusion complex (17). This activity is distinct from LMA2. In addition, the identity of the high molecular weight activity of yeast cytosol (20) has yet to be established.
Although LMA2 is capable of supporting vacuole fusion in the absence of thioredoxin, its activity is substantially lower than that of LMA1. We have as yet been unable to demonstrate any fusion activity for uncomplexed yeast thioredoxin in our system (Z.X. and P.S., unpublished observations).
Since the redox activity of thioredoxin is not required for LMA1 function (21) and LMA2 has no known catalytic activity, we postulate that these proteins function as a ligand for a membrane-bound receptor. We are currently attempting to identify this putative receptor, which should yield a significant insight into the mode of action of LMA1 and LMA2.
Acknowledgments
We thank Drs. Dieter Gallwitz, Yoshinobu Kaneko, Eric Muller, Peter Novick, James Rothman, Dietrich Scheglmann, Thomas Söllner, and Dieter Wolf for generously providing us with plasmids, strains, and antibodies; Dr. Mary Ann Gawinowicz for protein sequencing; Marilyn Rice Leonard for excellent technical assistance; and Drs. Mark Craighead, Graham Warren, and Andreas Mayer for advice and discussions. P.S. was supported by a Long-Term Fellowship from European Molecular Biology Organization. This work was supported by a grant from the National Institute of General Medical Sciences.
ABBREVIATIONS
- LMA
low molecular weight activity
- IB
inhibitor of vaculor protease B
- HCS
heated cytosol supernatant
References
- 1.Betz H, Hinze H, Holzer H. J Biol Chem. 1974;249:4515–4521. [PubMed] [Google Scholar]
- 2.Warren G, Wickner W. Cell. 1996;84:395–400. doi: 10.1016/s0092-8674(00)81284-2. [DOI] [PubMed] [Google Scholar]
- 3.Eshel D, Urrestarazu S, Vissers J, Jauniaux J, van Vliet-Reedijk J, Planta R, Gibbons I. Proc Natl Acad Sci USA. 1991;90:11172–11176. doi: 10.1073/pnas.90.23.11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muhua L, Karpova T, Cooper J. Cell. 1994;78:9194–9202. doi: 10.1016/0092-8674(94)90531-2. [DOI] [PubMed] [Google Scholar]
- 5.McConnell S, Yaffe M. J Cell Biol. 1992;118:385–395. doi: 10.1083/jcb.118.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simon V R, Swayne T C, Pon L A. J Cell Biol. 1995;130:345–354. doi: 10.1083/jcb.130.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weisman L S, Wickner W. Science. 1988;241:589–591. doi: 10.1126/science.3041591. [DOI] [PubMed] [Google Scholar]
- 8.Weisman L, Emr S, Wickner W. Proc Natl Acad Sci USA. 1990;87:1076–1080. doi: 10.1073/pnas.87.3.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weisman L S, Wickner W. J Biol Chem. 1992;267:618–623. [PubMed] [Google Scholar]
- 10.Shaw J M, Wickner W. EMBO J. 1991;10:1741–1748. doi: 10.1002/j.1460-2075.1991.tb07698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicholson T A, Weisman L S, Payne G S, Wickner W. J Cell Biol. 1995;130:835–845. doi: 10.1083/jcb.130.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y-X, Zhao H, Harding T M, Gomes de Mesquita D S, Woldringh C L, Klionsky D J, Munn A L, Weisman L. Mol Biol Cell. 1996;7:1375–1389. doi: 10.1091/mbc.7.9.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conradt B, Shaw J, Vida T, Emr S, Wickner W. J Cell Biol. 1992;119:1469–1479. doi: 10.1083/jcb.119.6.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conradt B, Haas A, Wickner W. J Cell Biol. 1994;126:99–110. doi: 10.1083/jcb.126.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haas A, Conradt B, Wickner W. J Cell Biol. 1994;126:87–97. doi: 10.1083/jcb.126.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haas A, Scheglmann D, Lazar T, Gallwitz D, Wickner W. EMBO J. 1995;14:5258–5270. doi: 10.1002/j.1460-2075.1995.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haas A, Wickner W. EMBO J. 1996;15:3296–3305. [PMC free article] [PubMed] [Google Scholar]
- 18.Mayer A, Wickner W, Haas A. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 19.Mayer A, Wickner W. J Cell Biol. 1997;136:307–317. doi: 10.1083/jcb.136.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Z, Wickner W. J Cell Biol. 1996;132:787–794. doi: 10.1083/jcb.132.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Z, Mayer A, Muller E, Wickner W. J Cell Biol. 1997;136:299–306. doi: 10.1083/jcb.136.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haas A. Methods Cell Sci. 1996;17:283–294. [Google Scholar]
- 23.Wessel D, Flugge U I. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 24.Fernandez J, DeMott M, Atherton D, Mische S M. Anal Biochem. 1992;201:255–264. doi: 10.1016/0003-2697(92)90336-6. [DOI] [PubMed] [Google Scholar]
- 25.Guzman L M, Belin D, Carson M J, Beckwith J. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 27.Douville K, Price A, Eichler J, Economou A, Wickner W. J Biol Chem. 1995;270:20106–20111. doi: 10.1074/jbc.270.34.20106. [DOI] [PubMed] [Google Scholar]
- 28.Saheki T, Holzer H. Eur J Biochem. 1974;42:621–626. doi: 10.1111/j.1432-1033.1974.tb03377.x. [DOI] [PubMed] [Google Scholar]
- 29.Maier K, Müller H, Tesch R, Trolp R, Witt I, Holzer H. J Biol Chem. 1979;254:12555–12561. [PubMed] [Google Scholar]
- 30.Maier K, Müller H, Holzer H. J Biol Chem. 1979;254:8491–8497. [PubMed] [Google Scholar]
- 31.Schu P, Suarez Rendueles P, Wolf D H. Eur J Biochem. 1991;197:1–7. doi: 10.1111/j.1432-1033.1991.tb15874.x. [DOI] [PubMed] [Google Scholar]
- 32.Vida T A, Emr S D. J Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones E W. Methods Enzymol. 1991;194:428–453. doi: 10.1016/0076-6879(91)94034-a. [DOI] [PubMed] [Google Scholar]