

Summary
Activation-induced cytidine deaminase (AID) is required for immunoglobulin (Ig) gene class switch recombination (CSR), somatic hypermutation (SHM) and somatic hyperconversion. In general high levels of AID expression are found in mature B cells responding to antigens. However, AID expression and SHM have also been detected in developing B cells from transgenic mice that have a limited Ig repertoire. Here we demonstrate that AID expression and active CSR/SHM occur in developing B cells from wild-type mice. Further, our results suggest that somatic variants arising from developing B cells in the bone marrow further diversify in the spleen of unimmunized mice. AID expression in developing B cells is T-cell independent but involves engagement of B cell and Toll-like receptors. Early AID expression can increase the pre-immune repertoire of developing B cells, may provide an innate population of IgG- and IgA-expressing cells, and could be involved in receptor-editing of self-reactive immature B cells.
INTRODUCTION
One of most critical initial steps in lymphocyte development is the creation of a lymphocyte receptor repertoire. This occurs by the process of V(D)J recombination as mediated by the recombination-activating genes, Rag1 and Rag2 (Oettinger et al., 1990; Schatz et al., 1989). In the B cell lineage, V(D)J recombination leads to diversification of immunoglobulin (Ig) receptors by a random rearrangement of Ig gene segments during B cell development. Further diversification is achieved by somatic hypermutation (SHM) (Neuberger and Milstein, 1995), which introduces point mutations to rearranged Ig genes, or by somatic hyperconversion (SHC), which replaces portions of the Ig variable (V) region sequence with pseudo V region sequences (Reynaud et al., 1989). Additional modification also occurs by class switch recombination (CSR), a process that changes the Cμ constant region to another constant region gene (Cγ, Cα, or Cε) (Honjo et al., 1981) to define the Fc-mediated effector functions.
SHM, CSR and SHC require activation-induced cytidine deaminase (AID). AID is expressed upon activation of B lymphocytes and it has been widely accepted that CSR and SHM occur during germinal center (GC) reactions after antigen activation of mature B cells with T cell help (Kelsoe, 1995; Muramatsu et al., 1999). However, some studies have indicated that SHM may occur outside of the GCs (Kato et al., 1998; Wang et al., 2000; William et al., 2002), that SHM can occur without T-cell help (Monson et al., 2001; Toellner et al., 2002; Weller et al., 2001), and that SHM can occur even in the absence of an immune response (Reynaud et al., 1995; Weller et al., 2004). The exact mechanisms involved in T-independent SHM processes that occur outside of the GCs are not known.
The paradigm that SHM is limited to mature B cells has also been recently challenged by a report showing that, in mutant mice having a limited Ig repertoire (κ−/− and QM mice), SHM of λ light chain can occur in immature bone marrow (BM) B cells (Mao et al., 2004). This λ light chain SHM was T-independent and correlated with AID expression in the immature B cells. However, despite detection of AID expression in immature B cells both from the κ−/− and QM mutant mice and from wild-type normal C57BL/6 mice, no λ light chain SHM was found from the wild-type C57BL/6 mice. The basis for this difference is not known; one proposed hypothesis is that SHM events in immature B cells occur at lower levels in λ than in κ (Mao et al., 2004).
The expression of AID in developing B cells in both mutant and normal mice (Mao et al., 2004) raises questions regarding the mechanism of AID induction at these early stages of B cell development. In mature B cells, AID activation involves antigenic stimulation via B cell receptor (BCR), cytokines, and the interaction between B and T cells via CD40:CD40 ligand (Armitage et al., 1992). Stimulation of human and mouse mature B cells via Toll-like receptors (TLRs) also can induce activation and CSR (He et al., 2004; Lin et al., 2004). Perhaps BCR or TLR signaling might be involved in AID induction in developing B cells, although it is not clear what ligands might mediate this signaling. TLRs are evolutionarily conserved germline-encoded receptors that recognize pathogen-associated molecular patterns (PAMPs) and initiate innate immune responses. TLR signaling requires a set of adaptor molecules including myeloid differentiation factor 88 (MyD88), MAL, TRIF, and TRAM to induce appropriate gene expression. MyD88 is the most broadly used adapter, essential for signaling via TLR1/TLR2, TLR2/TLR6, TLR5, TLR7, and TLR9, though TLR3 does not depend upon MyD88 and TLR4 is only partially dependent upon it (Kawai et al., 1999; Yamamoto et al., 2002; Hoebe et al., 2003; Yamamoto et al., 2003; Yamamoto et al., 2003).
Bone marrow is an extremely heterogeneous microenvironment where a variety of biological events, such as cell proliferation, differentiation and death take place and single-stranded DNA (ssDNA), double-stranded DNA (dsDNA), RNA or Smith/ribonucleoprotein (Sm/RNP)-are released from apoptotic cells: molecules that are sensed via TLRs 3, 7, and 9. In mature B cells, such endogenous self-antigens can synergistically engage BCR/TLR and activate autoreactive B cells (Lau et al., 2005; Leadbetter et al., 2002). It is possible that such self-antigens released from dying cells in the BM also synergistically engage TLRs and self-reactive BCR to induce activation of developing B cells. Indeed, skewing of immunoglobulin subtypes was observed in the serum of mice lacking MyD88 and TRIF, in which no TLR signaling occurs (Gavin et al., 2006). However, the role of nucleic acid sensing was not specifically analyzed in this study.
To directly address the role of AID during B cell development we have now analyzed the expression of AID in the BM of normal mice. The results presented here, with demonstration of active CSR and SHM of Vκ4 strongly indicate that CSR and SHM are ongoing processes during B cell development of normal mice and that these processes are T cell-independent. The somatic variants generated in developing B cells appear to migrate to and further diversify in the spleen during the maturation process. In addition we present evidence that BCR and TLR engagement are involved in the activation of developing B cells to express AID. Our results therefore suggest that TLRs can play a crucial role in shaping the development of the adaptive immune system.
RESULTS
AID Is Expressed In Developing B Cells of Normal Mice.
To determine if CSR and SHM might be features of normal BM B cell development, AID expression was measured by PCR and quantitative real-time PCR (qPCR) following reverse transcription (RT) in sorted developing B cells from BM. As a negative control we used sorted developing B cells from AID-deficient (Aicda−/−) mice. Because Aicda−/− mice are on a mixed C57BL/6 and CBA background (Muramatsu et al., 2000) Aicda−/− mice were generated by intercrossing Aicda+/− mice. This allowed us to use Aicda+/+ littermates with the same mixed genetic background as the control Aicda−/− mice. In addition, we examined sorted developing B cells from the BM of C57BL/6, BALB/c, and CBA mice.
Bone marrow cells from three 6-week old mice of each strain were purified by fluorescence activated cell sorting (FACS) into live (PI excluding) pro/pre-B (hereafter pre-B) cells (AA4.1+B220+κ−λ−), and immature B cells (AA4.1+B220+κ+λ+) (Figure S1A). AID expression was also analyzed in sorted B cells isolated from Peyer's patches (PP). PPs are a good source of mature B cells undergoing active CSR and SHM as a result of activation by commensal antigens (Ehrenstein and Neuberger, 1999; Gonzalez-Fernandez and Milstein, 1993). PP B cells were separated into two populations by FACS; B220+IgMhi cells which represent mature naïve B cells that have not undergone CSR, and B220+IgMlo cells that are presumably undergoing CSR to IgG or IgA (Figure S1B). Finally, we measured AID expression in FACS sorted mature B cells from the spleen, which were defined as AA4.1−B220+κ+λ+ (Figure S1C). The FACS staining profiles of each cell population were almost identical in all the tested mouse strains (data not shown).
Figure 1A shows the expression of AID and RAG-2, measured by PCR, in the indicated populations of BM and PP B-lineage cells isolated from the indicated strains of mice. As expected, RAG-2 expression is only detected in pre-B and immature B cells, validating our cell purification procedure. Strikingly, AID expression was detected in all B-lineage populations obtained from AID-sufficient mice, including pre-B and immature B cells of the BM. AID expression was detected by RT-PCR reactions in the sorted BM-derived immature B cell population of normal C57BL/6 mice as previously reported (Mao et al., 2004). In addition, expression of AID was detected in pre-B cells. Expression of AID by pre-B and immature B cells was clearly observed not only in C57BL/6 mice but also in age-matched (6 weeks old) BALB/c, Aicda+/+ littermate control (Figure 1A) and CBA mice (Figure 1B). No AID expression was seen in any cell population isolated from Aicda−/− mice.
Figure 1. Expression of AID in Pre-B and Immature B Lymphocytes from Inbred Strains of Mice.

(A) AID and RAG-2 expression were analyzed by RT-PCR in the sorted pre-B and immature B lymphocytes from BM and IgMhi and IgMlo mature B cells from PP of C57BL/6, Aicda−/−, Aicda+/+, CBA and BALB/c mice. The relative AID expression (B) in pre-B and immature B cells from different mouse strains and (C) of B cells in different lymphoid tissues from wild-type (Aicda+/+) littermate controls were quantified by quantitative real-time PCR (qPCR) from the sorted cells as described in Experimental Procedures. Consistent results were obtained by more than three times of the experiments. N.D.: not detected.
In order to quantitatively compare the levels of AID expression in different cell populations we turned to quantitative real-time PCR (qPCR) analysis (Figure 1B and 1C). C57BL/6 mice consistently expressed lower levels of AID than Aicda+/+ littermate controls, CBA or BALB/c mice. CBA and BALB/c mice showed about 1.5 to 3 times higher AID expression than C57BL/6 in pre-B and immature B cells. Aicda+/+ mice displayed levels of AID expression in immature B cells intermediate between C57BL/6 and CBA (Figure 1B). Also, the levels of AID expression in pre-B cells were consistently about 10% or less than immature B cells across different strains when normalized by β-actin (Figure 1B). Previously, it has been reported that about 16% of C57BL/6 immature B cells express AID by single-cell analysis (Mao et al., 2004). This previous finding, combined with our current quantitative analysis, indicates that AID-expressing pre-B cells represent about 1% of the population. Pre-B and immature B cells exhibited lower levels of AID expression compared to PP-derived B cells (Figure 1C). Notably, naïve splenic mature B cells had lower relative AID expression than BM immature B cells.
AID Expression in The BM Induces Ongoing CSR in Developing B Cells.
In order to ascertain that AID expressed in pre-B and immature B cells is active and functional, the presence of circle transcripts (CTs) and post-switch transcripts (PSTs) was assessed in these cells. CSR is accompanied by deletion of circular DNA (CD) from the immunoglobulin heavy chain (Igh) locus. Each CD contains the I-promoter, which is still active in looped-out CD and directs production of I-Cμ transcripts termed “circle transcripts (CTs)” (Kinoshita et al., 2001). CTs are produced in an AID-dependent manner and disappear from B cells very rapidly. Thus, the best demonstration of active ongoing CSR is the presence of CTs in the cells being analyzed. In addition, after CSR takes place, the germline μ promoter becomes associated with a particular CH gene that represents the isotype, stays active, and generates Iμ-CH transcripts termed “post-switch transcripts (PSTs)” (Muramatsu et al., 2000). PCR amplification of CT cDNA was followed by Southern blot analysis in order to enhance the sensitivity of detection. It is noteworthy that CT-α exhibit two transcripts due to alternative splicing donor sites in the Iα exon (Kinoshita et al., 2001).
CTs for IgA (CT-α) and IgG2b (CT-γ2b) were clearly detected in cDNA obtained from sorted pre-B and immature B cells from wild-type AID control (Aicda+/+), whereas no CTs were detected in cDNA of sorted pre-B or immature B cells from Aicda−/− mice in vivo (Figure 2A). Iμ-Cμ PCR products were used as loading controls and were detected by Southern blot analysis. The distribution of the isotypes expressed was not equal across different strains of mice. For instance CT-α and CT-γ2b were also detected from C57BL/6 pre-B and immature B cells (Figure 2B), whereas CT-γ2b and CT-γ3 were found in both pre-B and immature B cells of BALB/c mice (Figure 2C).
Figure 2. Ongoing CSR in Developing B Cells In Vivo.

CTs and PSTs for γ2b, γ3, and α were detected by Southern blot analyses of PCR-amplified cDNA from sorted pre-B and immature B cells in naïve (A) Aicda+/+, (B) C57BL/6, and (C) BALB/c, but not detected in naive (A) Aicda−/− mice. Iμ-Cμ is shown as cDNA loading control. Positive control samples were prepared as described in the Supplemental Data. Assays marked with asterisks (*) have identical positive controls because these were analyzed in a single blot. Sample loadings in (B) and (C) were not carefully quantitated, therefore variations in signal intensities may not be meaningful. (D) Surface expression of IgA was measured by FACS analyses. For this assay 7−11 week old Aicda−/− or Aicda+/− mice were used. FACS contour plots are shown for representative Aicda−/− or Aicda+/− mice stained either with anti-IgA or with an isotype-matched control antibody. (E) Percentages of AA4.1+B220+IgA+ or AA4.1+B220+Iso.Cotrl.+ populations determined from FACS analyses as in (D) are depicted as vertical scatter plots. Each dot represents the percentage of the population from an individual mouse. The table indicates the mean population percentages determined from the scatter plots for the indicated mouse strains (± s.d). Five Aicda−/− and ten Aicda+/− mice were tested. The P value was obtained by one-tailed Student's t-test. (F) The BM AA4.1+B220+IgA+ population was sorted and pooled from three Aicda+/− or two Aicda−/− mice. cDNA was synthesized from purified RNA, and AID, PST-α, α-transcript, CT-α, and GAPDH were amplified by PCR. Five-fold sample dilutions are shown for Aicda+/− and Aicda−/− to allow semi-quantitative comparisons of signals within each PCR assay. Comparison of signals between PCR assays is not meaningful due to differences in reaction efficiencies. The two CT-α panels are from the same blot; one panel is overexposed (O.E.) to increase sensitivity.
To confirm that CSR led to proper recombination, we amplified PSTs for each isotype using specific primers (Muramatsu et al., 2000). PSTs were found corresponding to the expected isotypes that had been indicated by CTs, confirming that some fractions of the sorted cells are undergoing active CSR (Figure 2A, 2B, and 2C).
To further determine whether CSR in developing B cells led to appropriate cell surface protein expression, we stained BM cells from Aicda+/− and Aicda−/− mice and examined the surface expression of IgA using FACS analysis. Viable (PI−) BM single cell suspensions were stained with antibodies specific for B220, AA4.1, and IgA. Figure 2D (lower) shows that a fraction of BM B cells from Aicda+/− mice express surface IgA, as predicted from the presence of CT-α and PST-α in sorted BM B cells. A much smaller fraction of BM B cells from Aicda−/− mice stained with the anti-IgA antibody (Figure 2D, lower). IgA staining in Aicda−/− mice is presumably due to some non-specific binding by the anti-IgA reagent. To confirm that the IgA staining of Aicda+/− B cells is specific, samples were also stained with an isotype control antibody (Figure 2D, upper). In contrast to the result with anti-IgA, there was a very low frequency of staining of both Aicda+/− and Aicda−/− B cells with the isotype control antibody. Figure 2E summarizes IgA surface staining of BM B cells from a number of mice. The difference in the fraction of IgA+ cells in Aicda+/− compared to Aicda−/− mice was reproducible (3.91% versus 1.28%, respectively) and statistically significant (P< 0.01).
To confirm that the cells identified as IgA+ by FACS are the actively switching population, AA4.1+B220+IgA+ BM cells were sorted and assayed for the presence of AID expression, PST-α, CT-α, and α-transcript. All these markers of CSR were detected in cells from Aicda+/− but not Aicda−/− mice (Figure 2F). We also confirmed by DNA sequence analyses that all α-transcripts exhibited functional V-D-J-Cα recombinations (Figure S2).
Our data clearly demonstrate that ongoing CSR takes place at the RNA and the protein level during B cell development.
Active SHM in BM Immature B Cells and Further Diversification in the Spleen of Normal Inbred Mice
The physiological function of AID expression and SHM during B cell development may be to contribute to Ig diversification, by analogy with their role in Ig diversification in sheep and cattle (Reynaud et al., 1995). If this is the case somatic variants should be found also in the spleen of naive mice. We therefore looked at the frequency of SHM in κ light chains derived from the Igκ-V4 gene (Vκ4) family of sorted immature BM B cells, splenic immature B cells (also called, transitional B cells), and splenic naïve mature follicular B cells. According to published reports (Brekke and Garrard, 2004; Thiebe et al., 1999) all of the mouse Igκ-V germline genes have been sequenced and they consist of 140 germline genes; 93 of which are functional with the remaining 47 being pseudogenes. These genes are distributed into 18 families of Igκ-V germline genes and the Vκ4 family is the largest with 27 highly homologous functional genes and 6 pseudogenes. The database for Igκ-V genes is a composite of sequences from the C57BL/6, BALB/c, A/J, 129, C3H and DBA strains of mice. Comparison of Igκ-V genes from various inbred strains has led to the conclusion that there are few or no polymorphisms between most Igκ-V germline gene alleles from different inbred strains (Thiebe et al., 1999).
To avoid detecting SHM in contaminating RNA from plasma cells, plasmablasts, or pre-plasma memory cells, we used PCR to amplify rearranged genomic DNA from sorted populations. The PCR strategy is diagrammed in Figure 3A. The reverse primer was designed to anneal on the 3' intronic region of the Jκ5 exon and therefore the primers used will amplify rearranged Vκ4-Jκ5 segments of genomic DNA specifically. We reasoned that this strategy might maximize our chances of detecting SHM as autoreactive light chain genes that used Jκ5, and are therefore unable to undergo further light chain rearrangements, might use SHM as an alternative receptor editing mechanism. The frequency of clones expressing a particular germline Vκ4 in these groups of mice were quite similar (data not shown).
Figure 3. Isolation of BM Immature B cells, Splenic Transitional B cells, or Follicular B Cells by Cell Sorting for Genomic DNA PCR.
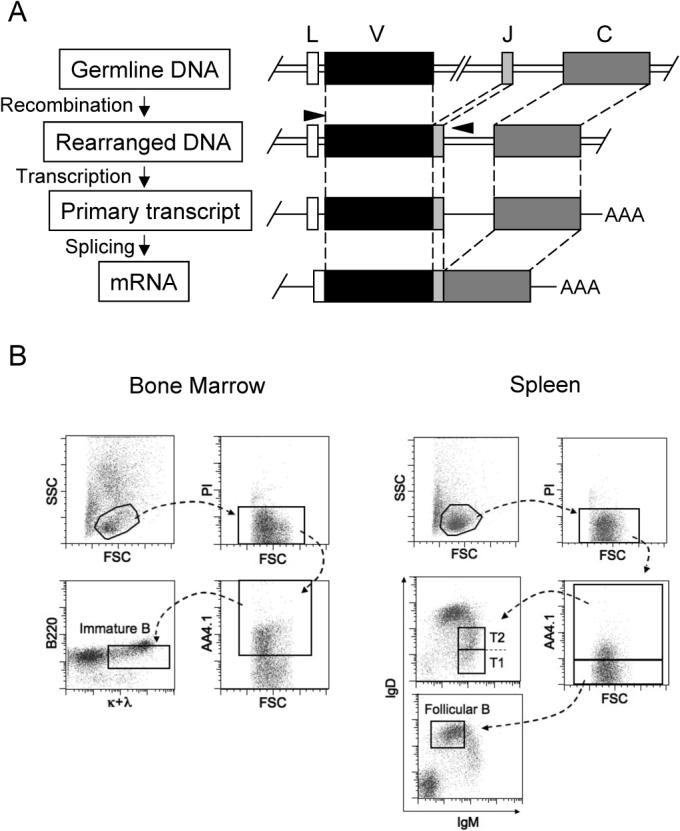
(A) Strategy to amplify rearranged Vκ4Jκ5 DNA using a specific primer set. Forward primer anneals selectively on the IgVκ4 gene leader sequence and the reverse primer anneals on 3' intronic region of the Jκ5 exon as shown as arrowheads, respectively. The primer sequences are described in Supplemental Methods. (B) BM immature B cells (AA4.1+B220+κ+λ+) were sorted as described in Figure S1A. Splenic transitional B cells (transitional type 1 and 2) and mature follicular B cells were sorted as AA4.1+IgMhiIgD+ and AA4.1−IgMloIgDhi, respectively.
Immature B cells (B220+AA4.1+κ+α+) were sorted from BM as described above. For the purification of splenic transitional or follicular B cells, lymphocytes were gated by side scatter (SSC) and forward scatter (FSC), and dead cells were excluded by propidium idodide (PI) staining. Viable lymphocytes were projected for AA4.1 surface expression to distinguish immature (AA4.1+) versus mature (AA4.1−) splenic B cells. Among immature splenic B cells, transitional type-I (T1: IgMhiIgDlo) and transitional type-2 (T2: IgMhiIgDhi) populations were combined and sorted as transitional B cells (AA4.1+IgMhiIgD+). On the other hand, among AA4.1− population that includes mature B cells and non-B cells, surface IgMloIgDhi cells were gated and sorted as naïve mature follicular B cells (AA4.1−IgMloIgDhi) (Figure 3B). Each compartment was sorted from single cell suspensions pooled from five C57BL/6 mice. DNA sequencing analysis of the isolated Vκ4 regions shows that the number of clones carrying mutation(s) and the mutation frequency in AID-deficient mice were at background levels: 4.4, 5.3, 5.6 nt changes per 104 bp from immature B, transitional B, and follicular B cells, respectively (Figure 4A and 4B). In comparison, we isolated significantly more clones of mutated Vκ4-Jκ5 genes from BM and splenic B cells of C57BL/6 mice (Figure 4A). The mutation frequencies were 19, 20.5, and 39.7 nt changes per 104 bp in clones amplified from immature B, transitional B, and follicular B cells respectively. The gradual increase in the number of clones that carry multiple mutations and the total frequency of nucleotide change suggests that mutations accumulate as B cells mature in the spleen (Figure 4A and 4B). Some examples of somatic variant sequences by ClustalW analysis are shown in Figure S3.
Figure 4. Active SHM in BM Immature B cells and Persistence of Somatic Variants in the Spleen of Naïve Mice.
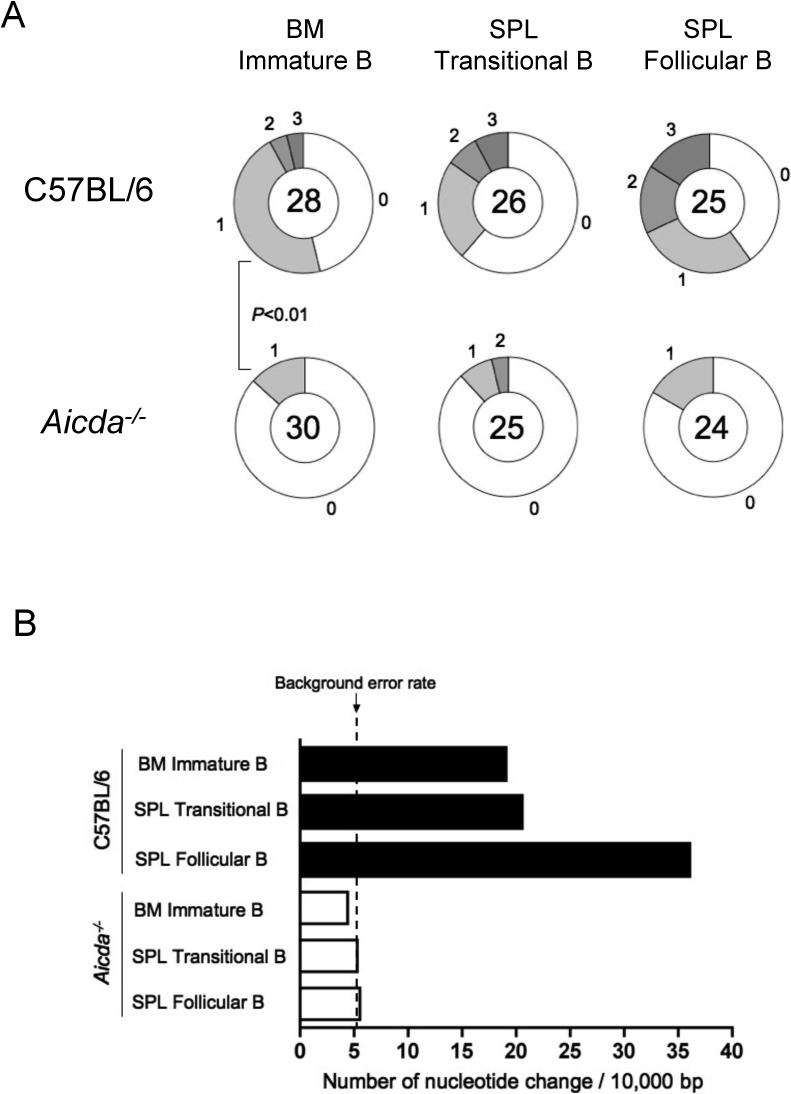
BM immature B cells (AA4.1+B220+κ+λ+), splenic transitional B cells (AA4.1+IgMhiIgD+), and follicular B cells (AA4.1−IgMloIgDhi) were sorted as shown in Figure 3. Rearranged Vκ4-Jκ5 light chain DNA was amplified with a specific primer set (Figure 3A), cloned and sequenced. (A) The sequencing results are depicted as pie charts as described above. Each P value was obtained by two-tailed Student's t-test. (B) Mutation frequencies were calculated by the number of nt change per 104 bp from C57BL/6 (filled bars) and from Aicda−/− (open bars) mice.
These results indicate that SHM can be found not only in BM immature B cells but also in naïve splenic B cells. Also, the mutations tend to be accumulated through the B cell maturation process in unimmunized mice, supporting the idea that AID-mediated SHM is a physiologically important diversification mechanism in the mouse pre-immune repertoire.
Somatic Hypermutation in Developing B Cells Is Not An Artifact by Contamination of Plasma Cells
B lymphocyte induced maturation protein-1 (Blimp-1) is a transcriptional repressor that is encoded by Prdm1 and a “master regulator” for the formation of plasma cells (Shapiro-Shelef et al., 2003). Mice lacking Prdm1 are early embryonic lethal and therefore, B cell-specific Blimp-1-deficient mice (Prdm1flox/floxCD19Cre/+) had been generated by crossing Prdm1flox/flox mice with transgenic mice that express Cre recombinase under the control of the CD19 promoter (CD19Cre/+). These mice had normal B cell development but defective in the formation of Ig-secreting plasma cells and pre-plasma memory B cells (Shapiro-Shelef et al., 2003).
To address whether somatic variants that we found from immature B cells are artifacts by contamination of BM-residing plasma cells in a more definitive way, we generated and examined B cell-specific Blimp-1-deficient mice (Prdm1flox/floxCD19Cre/+,hereafter Prdm1−/−) as previously described (Shapiro-Shelef et al., 2003). An intercross between F1 gave rise to Prdm1−/− mice along with wild-type littermate controls.
As previously described (Shapiro-Shelef et al., 2003), naïve Prdm1−/− mice had extremely low level of serum IgM compared to wild-type littermate controls (Figure 5A). Pre-B and immature B cells from Prdm1−/− and control mice were sorted as described above. The staining profiles from the two groups were indistinguishable (Figure S4A). AID expression between Prdm1−/− and wild-type developing B cells was determined by qPCR from sorted pre-B and immature B cells. We did not find any statistically significant difference between Prdm1−/− mice and wild-type littermate control in AID expression by this assay (Figure 5B). Frequency of SHM in Vκ4 was analyzed to ensure that SHM takes place in developing B cells in the absence of plasma cells or memory B cells. Indeed, comparable frequency of SHM was found between Blimp-1-deficient and wild-type B cells (Figure 5C).
Figure 5. Analysis of AID Expression and SHM in Blimp-1-Deficient Developing B Cells.
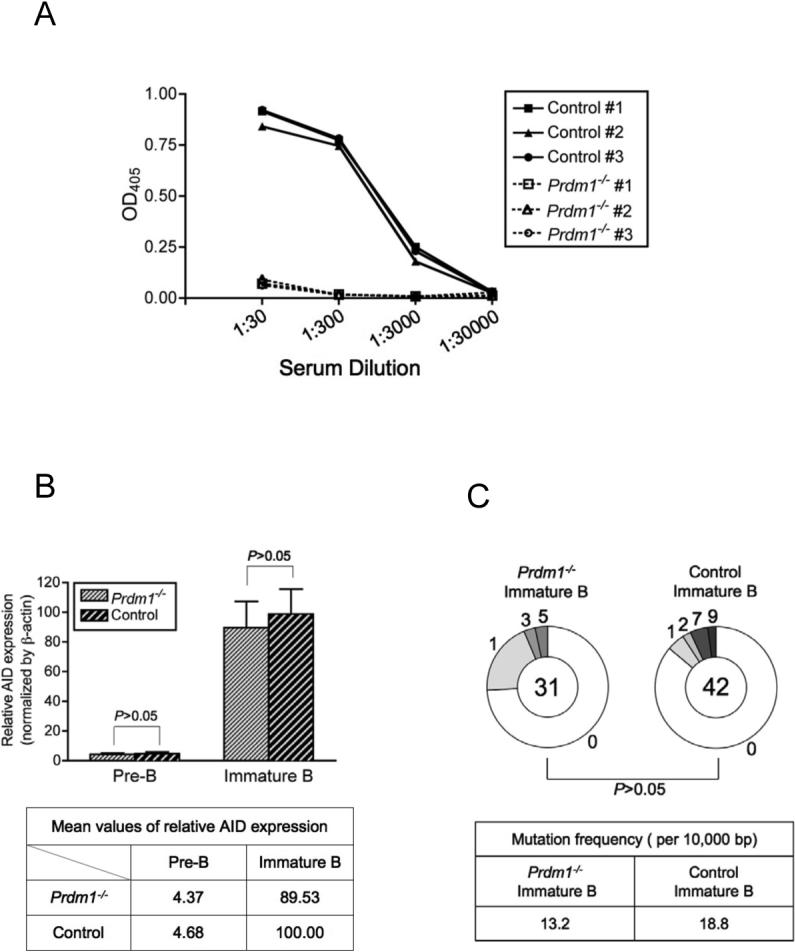
(A) Sera from 6 week-old naïve mice were tested for IgM by ELISA to screen Prdm1−/− and wild-type control mice. (B) Pre-B and immature B cells were sorted and relative AID expression was measured by qPCR as described in Experimental Procedures. One representative out of two similar results is shown. (C) SHM of the Vκ4 light chain was analyzed using cDNA generated from sorted immature B cells. The mutation frequency was also calculated as shown in the table. The P value was obtained by two-tailed Student's t-test.
The results from Blimp-1-deficient mice definitively indicate that our observations of AID expression and SHM in BM developing B cells are not artifacts of plasma cell contamination.
AID Expression, CSR and SHM In Developing B Cells Are T-Independent.
To determine whether expression of AID and CSR in pre-B and immature B cells were dependent on T cell help, cDNAs from sorted developing B cells from athymic nude (Foxn1nu/Foxn1nu) mice were analyzed for AID and CT expression. These nude mice have a C57BL/6 genetic background.
Nude mice showed normal pre-B and immature B cell surface marker profiles compared to the wild-type littermate controls (Figure S4B). RAG-2 expression is shown for pre-B and immature B cells from both nude mice and the controls (Figure 6A). Using both PCR (Figure 6A) and qPCR assays (Figure 6B), AID expression levels in pre-B and immature B cells from nude mice were similar to those found in wild-type littermate controls. In addition, CTs and PSTs for IgA and IgG2b were detected from pre-B and immature B cells of both nude and the control mice, clearly indicating that ongoing CSR takes place in developing B cells of both athymic nude and wild-type control mice (Figure 6C). We also compared the frequency of Vκ4 variants in immature B cells from nude and wild-type littermate control mice. We found very similar frequency of Vκ4 variants: 12.4 versus 13.2 nucleotide changes per 104 bp from nude and wild-type immature B cells, respectively (Figure 6D). These results strongly support the idea that AID expression, ongoing CSR and active SHM are T cell independent.
Figure 6. AID Expression and Ongoing CSR in Athymic Nude Developing B Cells.
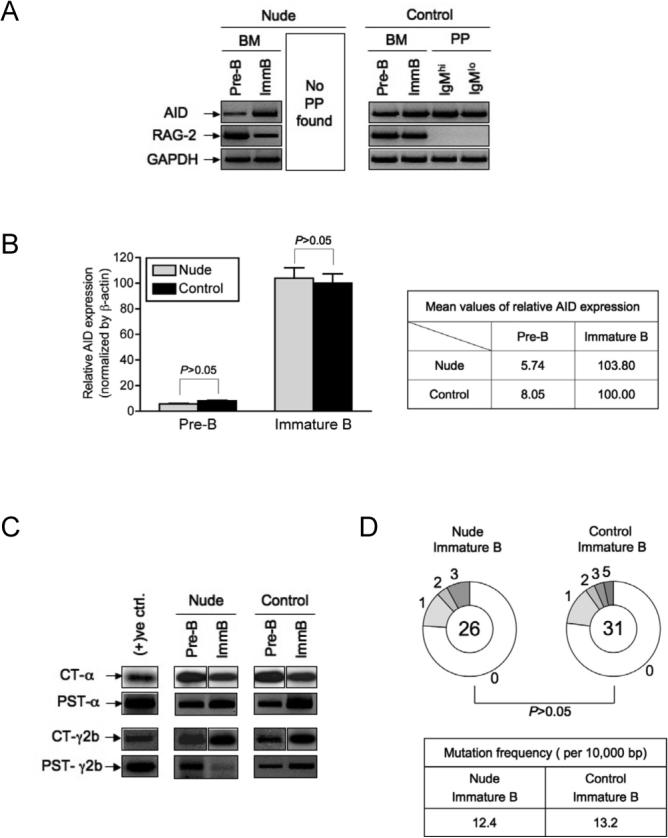
(A) AID and RAG-2 expression were examined by RT-PCR. (B) Relative AID expression from sorted pre-B and immature B cells of nude and wild-type control mice was measured by qPCR as described in Experimental Procedures. One out of three similar results is displayed. (C) CTs and PSTs for α and γ2b were detected from developing B cells of nude and wild-type littermate controls. Composite results from separate blots are presented for CTs. Loading was not sufficiently normalized to allow quantitative comparisons between samples. (D) SHM of the Vκ4 light chain was analyzed as described in Figure 5C.
Signals via BCR and TLR Are Critical for AID Expression in Developing B Cells in vivo.
To explore a possible role of TLR or BCR signaling in AID expression during B cell development in vivo, we examined relative AID expression level of pre-B and immature B cells in MyD88- and Bruton's tyrosine kinase (Btk)-deficient mice compared to their wild-type littermate controls. Btk is a molecule that is important for signaling mediated by BCR cross-linking (Rawlings et al., 1993; Thomas et al., 1993). We also tested “triple D” (3D) mice, which are homozygous for a recessive mutation of Unc93b1, generated by ENU (N-ethyl-N-nitrosurea) mutagenesis performed on a pure C57BL/6J background (Tabeta et al., 2006). 3D mice are defective in nucleic acid sensing through TLR3, TLR7 and TLR9 and also impaired in MHC class I and class II presentation of exogenous antigens.
Btk−/− mice showed a marked reduction in AID expression compared to the wild-type C57BL/6 background (Figure 7A). Notably, AID expression in immature B cells had a profound difference (about 90% decrease in relative AID expression in immature B cells compared to those of C57BL/6), whereas there was negligible difference found in pre-B cells. It suggests that in immature B cell stages, BCR cross-linking is the major signal to induce AID expression. Myd88−/− (Figure 7B) and Unc93b13d/3d (Figure 7C) mice also showed a significant decrease of AID expression in pre-B and immature B cells compared to their wild-type littermate controls. Interestingly, both Myd88−/− and Unc93b13d/3d mice displayed a significant decrease in AID expression not only in immature B cells but also in pre-B cells. For example, about 75% or 70% decrease in AID expression (normalized by β–actin expression) was detected in pre-B cells while about 70% or 60% decrease was found in immature B cells from Myd88−/− or Unc93b13d/3d, respectively, compared to their wild-type controls. This finding suggests that in pre-B cells where light chain rearrangement of BCR is not complete, TLR-mediated signals, and especially signals initiated by nucleic acids, may play a dominant role in AID expression. However, it is noteworthy that we did not see a complete shut-down of AID expression in Btk−/−, Myd88−/−, nor Unc93b13d/3d, indicating that stimuli other than BCR cross-linking or TLR signaling can contribute to induce AID expression in developing B cells.
Figure 7. Decreased AID Expression in Developing B Cells of Btk−/−, Myd88−/− and Unc93b13d/3d Mice.

The relative levels of AID expression in pre-B and immature B cells from (A) Btk−/−, (B) Myd88−/−, and (C) Unc93b13d/3d (3D) mice were measured by qPCR from the sorted cells. C57BL/6 (A and C) of the same age and (B) the wild-type littermates (Myd88+/+) were used as controls. Both (A) and (B) are one representative out of three experiments and (C) was done once. Each P value was obtained by two-tailed Student's t-test.
DISCUSSION
B cell antigen receptor (BCR) diversification in early mouse BM B cells is known to involve combinatorial gene rearrangements mediated by V(D)J recombination mechanisms. We have now found that V(D)J rearrangement is not the only mechanism for BCR diversification in early mouse B cells. Our results show that the AID is expressed in BM pre-B and immature B cells of normal wild-type mice. AID expression in these cells leads to active CSR and SHM as part of the normal, T-cell independent, developmental program. AID levels in developing B cells differ between various mouse strains; these differences suggest genetic factors that affect the level and timing of AID expression in various B-cell stages. BCR and TLR are involved in the expression of AID in early B cells, suggesting that signaling through these receptors is important for the induction of AID expression.
It is important to note that several observations indicate that small numbers of BM-residing plasma or memory B cells cannot account for our findings of AID-expression in early B cells. First, plasma cells are substantially larger than developing B cells and are gated out of our early B cell populations by size. Second, our developing B cell populations are AA4.1+B220+ but BM plasma cells do not express surface AA4.1 or B220 (Kallies et al., 2004; McKearn et al., 1984). Third, Blimp-1 is a master transcription factor that is required for formation and maintenance of long-lived plasma cells and pre-plasma memory B cells in BM (Shapiro-Shelef et al., 2003; Shapiro-Shelef et al., 2005). Yet, high levels of Blimp-1 expression are restricted to rare cells in lymphoid tissues (from 0.1 to 0.5%) and suppress the expression of both B220 and AID (Kallies et al., 2004), indicating that plasma or memory B cells are unlikely to be present in our AA4.1+B220+AID+ populations. Fourth, comparable levels of AID expression and ongoing CSR are found in early B cells from nude and wild-type littermate controls mice even though nude mice lack GC reactions, plasma cells, or memory B cell formation. Finally, B cell-specific Blimp-1-deficient mice that lack plasma cells show no significant differences in AID expression in both pre-B and immature B cells when compared to wild-type littermates (Figure 5). Taken together these considerations provide compelling evidence against any significant contamination of mature B cells in our BM-derived pre-B and immature B cell populations.
A recent study using mutant mice carrying reporter gene constructs designed to detect AID-expressing cells by flow cytometric analysis shows no AID reporter expression in bone marrow cells in vivo even though detectable AID expression could be found by real-time PCR analysis (Crouch et al., 2007). However, we find clear AID expression and AID function in developing bone marrow B cells, suggesting that the reporter construct analyses may be insufficiently sensitive to detect lower levels of AID expression even when these are capable of both CSR and SHM. Kelsoe and his colleagues also recently reported that AID is expressed in mouse developing B cells in vivo, and that splenic immature (or transitional) B cells can switch isotypes and differentiate to plasma cells upon in vitro TLR stimulation (Ueda et al., 2007), consistent with our findings that developing B cells can be activated by self or external TLR ligands and can express functional AID .
Our results indicating that active CSR can occur in early developing mouse B cells alters the prevailing paradigm of CSR limited to mature B cells stimulated in the periphery and usually in GCs. Our findings could also account for the unexpected CSR that has been found in mutant mice (μMT mice) that cannot produce membrane-bound μ-chains. The μMT mutant mice have a pro-B cell developmental block resulting in a lack of peripheral B cells with surface IgM (Kitamura et al., 1991). Despite this defect in B cell development, μMT mice have been found to have normal levels of serum IgA (in a C57BL/6 background) (Macpherson et al., 2001) or serum IgG and IgA (in a BALB/c background) (Orinska et al., 2002; Hansan et al., 2002). The ability of pre-B cells to undergo CSR, and thereby produce membrane-bound IgG or IgA to circumvent the pro-B block, would provide a straightforward explanation for IgG and IgA expression in the μMT mice. The differential Ig isotype expression in C57BL/6 and BALB/c μMT mice could reflect genetic differences affecting CSR in these mouse strains; this is reminiscent of the genetic background effects we find for CSR in pre-B and immature B cells in normal mice.
For both normal and μMT mice, culturing B220+ BM cells in vitro with IL-7 has been reported to lead to AID expression, IgG germline transcription, and the detection of PSTs (Seagal et al., 2003). These results are consistent with our findings of AID expression and CSR even in unstimulated BM early B cells. However, because μMT/lpr mice showed an increase in the production of IgG-producing B cells in these in vitro cultures, it was suggested that IgG-positive B cells developing in the BM in vivo are deleted by a Fas/FasL-mediated pathway (Seagal et al., 2003). However, μMT mice exhibit nearly normal levels of serum IgG and IgA, indicating that, in vivo, IgG- and IgA-expressing BM B cells are not necessarily deleted even in the presence of an intact Fas/FasL pathway. Rather, CSR in the early stages of in vivo B cell development might provide a protective mechanism for young, pre-immune animals by generating IgG- or IgA-producing cells. As suggested previously, CSR at the level of pre-B cells could also represent an alternative backup pathway for B cell development (Macpherson et al., 2001).
Abelson murine leukemia virus (Ab-MuLV) infection of mouse BM cells can lead to establishment of transformed tumor cell lines that have a pre-B cell phenotype (Burrows et al., 1981; DePinho et al., 1984; Sugiyama et al., 1986). Some of these pre-B cell-lines exhibit CSR of γ2b or γ3 heavy chain constant region genes, discordant with the notion that CSR is limited to mature B cells. These findings have generally been ascribed to inappropriate CSR regulation in these transformed cell lines. Our demonstration of CSR in normal pre-B cells could explain the active CSR found in Ab-transformed pre-B cell-lines, but a recent report showing that Ab-MuLV infection of BM pre-B cells can greatly enhance AID expression provides an alternate mechanism to account for CSR in these cell-lines (Gourzi et al., 2006). Interestingly, Ab-MuLV induction of AID in infected pre-B cells inhibits tumorigenicity and it has been suggested that AID-induced DNA damage, leading to decreased proliferation and increased NK cell targeting, might be involved in this inhibition (Gourzi et al., 2006). It would appear that such effects of AID expression on proliferation and NK cell targeting are limited to virus-infected cells because (1) normal mature B cells proliferate and expand greatly after stimulations that induce AID expression and CSR (Muramatsu et al., 2000), and (2) as our results indicate, AID-expressing BM pre-B cells in normal mice appear to differentiate normally into immature B cells that express AID at higher levels. Thus, AID expression appears to play at least two roles in early B cells; induction in virally-infected pre-B cells leading to inhibition of the tumorigenicity of these infected cells, and induction in normal early B cells to provide receptor diversification during B cell development.
AID expression in immature B cells also mediates SHM. A previous report had shown AID expression, but no SHM of λ light chain, in immature B cells from wild-type C57BL/6 mice (Mao et al., 2004). We now find that AID expression can, in normal mice, lead to SHM of Vκ4 in the BM pre-B and immature B cells. We do not know why SHM was not detected previously in λ light chain of C57BL/6 mice but this could reflect a lower SHM frequency in λ relative to Vκ4 or might be due to a process that selectively amplifies early B cells carrying mutations in Vκ4 (see below).
What roles might SHM be playing in early B cells? Obviously this mechanism could provide additional pre-immune repertoire diversity in mature B cells to supplement the diversity from V(D)J recombination. AID expression in early B cells could also be involved in receptor editing of self-reactive BCR using SHM as a mechanism. Previous studies have indicated that B cells expressing rearranged Vκ4 genes are frequently negatively selected, perhaps because they are autoreactive (Kalled and Brodeur, 1990). We propose that the Vκ4 hypermutation that we have detected in immature B cells might be linked to receptor editing during the maturation of these cells. TLR dependency of AID expression during B cell development could be related to this hypothesis. In mature B cells, synergistic engagement of TLR/BCR can activate quiescent or autoreactive B cells in vitro (Leadbetter et al., 2002) and we suggest that autoreactive developing B cells can be activated by the same synergistic engagement of TLR/BCR. Thus, expression of AID during B cell development could both provide Ig diversity and serve as an additional mechanism of receptor editing in autoreactive B cells.
When SHC, which is known to also require AID, was first found in immature B cells in the chicken bursa (Reynaud et al., 1987), it was a notable deviation from B cell diversification by V(D)J recombination in the bone marrow of mice and humans. However, our results, showing AID-mediated Ig diversification in the mouse bone marrow, suggest that AID expression and function are also a part of the normal early B cell developmental program in mice, as in chickens, sheep, and other species (Cooper, 2002). Our findings lessen the appearance of evolutionary differences in B cell development between these species.
EXPERIMENTAL PROCEDURES
Mice
All experiments with mice were performed in accordance with the regulations and with the approval of Tufts/NEMC IACUC. C57BL/6, CBA, CD19Cre/+ (Cd19tm1(cre)Cgn), B6.Cg-Foxn1 (nude mice), and littermate controls of nude mice were purchased from the Jackson Laboratory. AID-deficient mice were obtained from Dr. J. Stavnezer (University of Massachusetts Medical School, Worcester, MA) with permission from Dr. T. Honjo (Kyoto University, Kyoto, Japan) and the colony is maintained in our facility. Myd88−/− mice and wild-type littermate controls were obtained from Drs. A. Marshak-Rothstein (Boston University School of Medicine, Boston, MA) and L. Hu (Tufts-New England Medical Center, Boston, MA). Unc93b13d/3d mice were provided by Dr. A. Marshak-Rothstein. Btk−/− and BALB/c mice were provided from Drs. H. Wortis and N. Rosenberg (both in Tufts University School of Medicine, Boston, MA), respectively. Permission to use the Btk−/− mice was kindly given by Dr. Rudi Hendricks (Erasmus University Medical Center, Rotterdan, The Netherlands). All mice were maintained in a pathogen-free mouse facility of Tufts University School of Medicine.
Flow Cytometry and Cell Sorting
Cells were stained for flow cytometry using standard procedures. Propidium iodide (PI) was added just prior to sorting on a MoFlo instrument (Dako Cytomation) and to FACS analysis was done using FACSCalibur (BD Biosciences). Detailed staining procedures are described in Supplemental Data.
Genomic DNA Purification, RNA Purification and Generation of First-Strand cDNA
Genomic DNA was prepared by AquaPure Genomic DNA Kit (Bio-Rad) or by DNeasy Tissue Kit (Qiagen) according to the manufacturer's instruction. Cells were lysed in TRIzol reagent (Invitrogen) to purify RNA. First-strand of the cDNA was synthesized in a 20 μl reaction with oligo(dT)20 and SuperScript III (Invitrogen) by following the manufacturer's instruction.
PCR
For AID, RAG-2, CT, PST, and GAPDH, 2 μl of the first-strand cDNA was amplified in a 20 μl reaction using Platinum Taq DNA Polymerase (Invitrogen). Oligonucleotide primers and conditions for PCR are described in Supplemental Data.
Cloning and Sequencing
Detailed information for oligonucleotide primers and conditions for amplification of rearranged Vκ4 is described in Supplemental Data. In brief, Platinum Taq DNA polymerase High Fidelity (Invitrogen) was used. Amplified Vκ4 products were cloned into pCRII-TOPO vector (Invitrogen). Plasmid DNAs were purified using QIAprep Spin Miniprep Kit (Qiagen) and the PCR products were sequenced at Tufts University Core Facility (Tufts University School of Medicine, Boston, MA) or at Genewiz Inc. (Plainfield, NJ).
Quantitative Real-Time PCR
All qPCR experiments were carried out and analyzed as following: first-strand cDNA synthesis was performed on four-fold serial dilutions of purified RNA. Triplicate for each cDNA reaction was used for amplification with a pre-designed mouse AID- or β-actin (endogenous control)-specific TaqMan primer/probe set (Applied Biosystems) in an ABI PRISM 5700 Sequence Detection System (Applied Biosystems). Relative quantification was determined after establishing standard curves for mouse AID and β-actin expression. qPCR data represent means that were obtained from triplicates of each four-fold dilution of first-strand cDNA reaction ± s.d.
Southern Blot Analysis
PCR products of CT were separated with 2% agarose gel electrophoresis, transferred to Zeta-Probe Blotting Membranes (Bio-Rad). Oligonucleotide probe for Cμ was end-labeled with [32P]γATP as previously described (Kinoshita et al., 2001). The labeled probe was purified by NucAway Spin Columns (Ambion) before hybridization.
ACKNOWLEDGMENTS
We would like to thank Drs. Elina Kari and Robert Berland for critical review of our manuscript. We also thank Drs. Janet Stavnezer and Ann Marshak-Rothstein for providing AID-deficient mice (Dr. Stavnezer) and MyD88-deficient and Unc93b13d/3d mice (Dr. Marshak-Rothstein) and for helpful discussion. We are grateful to Dr. Linden Hu for MyD88-deficient mice as well. We also thank Allen Parmelee, Stephen Kwok, Christopher Martin and Jenna Bergerson for valuable assistance. We thank the Eshe Fund for their generous support. This work was supported by National Institutes of Health grants AI45104 (T.I.-K.) and AI24465 (E.S).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
REFERENCES
- Armitage RJ, Fanslow WC, Strockbine L, Sato TA, Clifford KN, Macduff BM, Anderson DM, Gimpel SD, Davis-Smith T, Maliszewski CR, et al. Molecular and biological characterization of a murine ligand for CD40. Nature. 1992;357:80–82. doi: 10.1038/357080a0. [DOI] [PubMed] [Google Scholar]
- Brekke KM, Garrard WT. Assembly and analysis of the mouse immunoglobulin kappa gene sequence. Immunogenetics. 2004;56:490–505. doi: 10.1007/s00251-004-0659-0. [DOI] [PubMed] [Google Scholar]
- Burrows PD, Beck GB, Wabl MR. Expression of mu and gamma immunoglobulin heavy chains in different cells of a cloned mouse lymphoid line. Proc Natl Acad Sci U S A. 1981;78:564–568. doi: 10.1073/pnas.78.1.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MD. Exploring lymphocyte differentiation pathways. Immunol Rev. 2002;185:175–185. doi: 10.1034/j.1600-065x.2002.18515.x. [DOI] [PubMed] [Google Scholar]
- Crouch EE, Li Z, Takizawa M, Fichtner-Feigl S, Gourzi P, Montaño C, Feigenbaum L, Wilson P, Janz S, Papavasiliou FN, Casellas R. Regulation of AID expression in the immune response. J Exp Med. 2007 doi: 10.1084/jem.20061952. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePinho R, Kruger K, Andrews N, Lutzker S, Baltimore D, Alt FW. Molecular basis of heavy-chain class switching and switch region deletion in an Abelson virus-transformed cell line. Mol Cell Biol. 1984;4:2905–2910. doi: 10.1128/mcb.4.12.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenstein MR, Neuberger MS. Deficiency in Msh2 affects the efficiency and local sequence specificity of immunoglobulin class-switch recombination: parallels with somatic hypermutation. Embo J. 1999;18:3484–3490. doi: 10.1093/emboj/18.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AL, Hoebe K, Duong B, Ota T, Martin C, Beutler B, Nemazee D. Adjuvant-enhanced antibody responses in the absence of toll-like receptor signaling. Science. 2006;314:1936–1938. doi: 10.1126/science.1135299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Fernandez A, Milstein C. Analysis of somatic hypermutation in mouse Peyer's patches using immunoglobulin kappa light-chain transgenes. Proc Natl Acad Sci U S A. 1993;90:9862–9866. doi: 10.1073/pnas.90.21.9862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourzi P, Leonova T, Papavasiliou FN. A role for activation-induced cytidine deaminase in the host response against a transforming retrovirus. Immunity. 2006;24:779–786. doi: 10.1016/j.immuni.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Hasan M, Polic B, Bralic M, Jonjic S, Rajewsky K. Incomplete block of B cell development and immunoglobulin production in mice carrying the muMT mutation on the BALB/c background. Eur. J. Immunol. 2002;32:3463–3471. doi: 10.1002/1521-4141(200212)32:12<3463::AID-IMMU3463>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- He B, Qiao X, Cerutti A. CpG DNA induces IgG class switch DNA recombination by activating human B cells through an innate pathway that requires TLR9 and cooperates with IL-10. J Immunol. 2004;173:4479–4491. doi: 10.4049/jimmunol.173.7.4479. [DOI] [PubMed] [Google Scholar]
- Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- Honjo T, Nakai S, Nishida Y, Kataoka T, Yamawaki-Kataoka Y, Takahashi N, Obata M, Shimizu A, Yaoita Y, Nikaido T, Ishida N. Rearrangements of immunoglobulin genes during differentiation and evolution. Immunol Rev. 1981;59:33–67. doi: 10.1111/j.1600-065x.1981.tb00455.x. [DOI] [PubMed] [Google Scholar]
- Kalled SL, Brodeur PH. Preferential rearrangement of V kappa 4 gene segments in pre-B cell lines. J Exp Med. 1990;172:559–566. doi: 10.1084/jem.172.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, Nutt SL. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J Exp Med. 2004;200:967–977. doi: 10.1084/jem.20040973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato J, Motoyama N, Taniuchi I, Takeshita H, Toyoda M, Masuda K, Watanabe T. Affinity maturation in Lyn kinase-deficient mice with defective germinal center formation. J Immunol. 1998;160:4788–4795. [PubMed] [Google Scholar]
- Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-Deficient Mice to Endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- Kelsoe G. The germinal center reaction. Immunol Today. 1995;16:324–326. doi: 10.1016/0167-5699(95)80146-4. [DOI] [PubMed] [Google Scholar]
- Kinoshita K, Harigai M, Fagarasan S, Muramatsu M, Honjo T. A hallmark of active class switch recombination: transcripts directed by I promoters on looped-out circular DNAs. Proc Natl Acad Sci U S A. 2001;98:12620–12623. doi: 10.1073/pnas.221454398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- Macpherson AJ, Lamarre A, McCoy K, Harriman GR, Odermatt B, Dougan G, Hengartner H, Zinkernagel RM. IgA production without mu or delta chain expression in developing B cells. Nat Immunol. 2001;2:625–631. doi: 10.1038/89775. [DOI] [PubMed] [Google Scholar]
- Mao C, Jiang L, Melo-Jorge M, Puthenveetil M, Zhang X, Carroll MC, Imanishi-Kari T. T cell-independent somatic hypermutation in murine B cells with an immature phenotype. Immunity. 2004;20:133–144. doi: 10.1016/s1074-7613(04)00019-6. [DOI] [PubMed] [Google Scholar]
- McKearn JP, Baum C, Davie JM. Cell surface antigens expressed by subsets of pre-B cells and B cells. J Immunol. 1984;132:332–339. [PubMed] [Google Scholar]
- Monson NL, Foster SJ, Brezinschek HP, Brezinschek RI, Dorner T, Lipsky PE. The role of CD40-CD40 ligand (CD154) interactions in immunoglobulin light chain repertoire generation and somatic mutation. Clin Immunol. 2001;100:71–81. doi: 10.1006/clim.2001.5049. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- Neuberger MS, Milstein C. Somatic hypermutation. Curr Opin Immunol. 1995;7:248–254. doi: 10.1016/0952-7915(95)80010-7. [DOI] [PubMed] [Google Scholar]
- Oettinger MA, Schatz DG, Gorka C, Baltimore D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 1990;248:1517–1523. doi: 10.1126/science.2360047. [DOI] [PubMed] [Google Scholar]
- Orinska Z, Osiak A, Lohler J, Bulanova E, Budagian V, Horak I, Bulfone-Paus S. Novel B cell population producing functional IgG in the absence of membrane IgM expression. Eur J Immunol. 2002;32:3472–3480. doi: 10.1002/1521-4141(200212)32:12<3472::AID-IMMU3472>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, Mohr RN, Bazan JF, Howard M, Copeland NG, et al. Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science. 1993;261:358–361. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- Reynaud CA, Anquez V, Grimal H, Weill JC. A hyperconversion mechanism generates the chicken light chain preimmune repertoire. Cell. 1987;48:379–388. doi: 10.1016/0092-8674(87)90189-9. [DOI] [PubMed] [Google Scholar]
- Reynaud CA, Dahan A, Anquez V, Weill JC. Somatic hyperconversion diversifies the single Vh gene of the chicken with a high incidence in the D region. Cell. 1989;59:171–183. doi: 10.1016/0092-8674(89)90879-9. [DOI] [PubMed] [Google Scholar]
- Reynaud CA, Garcia C, Hein WR, Weill JC. Hypermutation generating the sheep immunoglobulin repertoire is an antigen-independent process. Cell. 1995;80:115–125. doi: 10.1016/0092-8674(95)90456-5. [DOI] [PubMed] [Google Scholar]
- Schatz DG, Oettinger MA, Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell. 1989;59:1035–1048. doi: 10.1016/0092-8674(89)90760-5. [DOI] [PubMed] [Google Scholar]
- Seagal J, Edry E, Keren Z, Leider N, Benny O, Machluf M, Melamed D. A fail-safe mechanism for negative selection of isotype-switched B cell precursors is regulated by the Fas/FasL pathway. J Exp Med. 2003;198:1609–1619. doi: 10.1084/jem.20030357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 2003;19:607–620. doi: 10.1016/s1074-7613(03)00267-x. [DOI] [PubMed] [Google Scholar]
- Shapiro-Shelef M, Lin KI, Savitsky D, Liao J, Calame K. Blimp-1 is required for maintenance of long-lived plasma cells in the bone marrow. J Exp Med. 2005;202:1471–1476. doi: 10.1084/jem.20051611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama H, Maeda T, Akira S, Kishimoto S. Class-switching from mu to gamma 3 or gamma 2b production at pre-B cell stage. J Immunol. 1986;136:3092–3097. [PubMed] [Google Scholar]
- Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7:156–164. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- Thiebe R, Schable KF, Bensch A, Brensing-Kuppers J, Heim V, Kirschbaum T, Mitlohner H, Ohnrich M, Pourrajabi S, Roschenthaler F, et al. The variable genes and gene families of the mouse immunoglobulin kappa locus. Eur J Immunol. 1999;29:2072–2081. doi: 10.1002/(SICI)1521-4141(199907)29:07<2072::AID-IMMU2072>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Sideras P, Smith CI, Vorechovsky I, Chapman V, Paul WE. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science. 1993;261:355–358. doi: 10.1126/science.8332900. [DOI] [PubMed] [Google Scholar]
- Toellner KM, Jenkinson WE, Taylor DR, Khan M, Sze DM, Sansom DM, Vinuesa CG, MacLennan IC. Low-level hypermutation in T cell-independent germinal centers compared with high mutation rates associated with T cell-dependent germinal centers. J Exp Med. 2002;195:383–389. doi: 10.1084/jem.20011112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Liao D, Yang K, Patel A, Kelsoe G. T-Independent Activation-Induced Cytidine Deaminase Expression, Class-Switch Recombination, and Antibody Production by Immature/Transitional 1 B Cells. J Immunol. 2007;178:3593–3601. doi: 10.4049/jimmunol.178.6.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huang G, Wang J, Molina H, Chaplin DD, Fu YX. Antigen persistence is required for somatic mutation and affinity maturation of immunoglobulin. Eur J Immunol. 2000;30:2226–2234. doi: 10.1002/1521-4141(2000)30:8<2226::AID-IMMU2226>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Weller S, Braun MC, Tan BK, Rosenwald A, Cordier C, Conley ME, Plebani A, Kumararatne DS, Bonnet D, Tournilhac O, et al. Human blood IgM “memory” B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood. 2004;104:3647–3654. doi: 10.1182/blood-2004-01-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller S, Faili A, Garcia C, Braun MC, Le Deist FF, de Saint Basile GG, Hermine O, Fischer A, Reynaud CA, Weill JC. CD40-CD40L independent Ig gene hypermutation suggests a second B cell diversification pathway in humans. Proc Natl Acad Sci U S A. 2001;98:1166–1170. doi: 10.1073/pnas.98.3.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 2002;297:2066–2070. doi: 10.1126/science.1073924. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, Hoshino K, Takeuchi O, Kobayashi M, Fujita T, Takeda K, Akira S. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002;420:324–329. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.