

Abstract
Transcription factor paralogs may share a common role in staged or overlapping expression in specific tissues, as in the Hox family. In other cases, family members have distinct roles in a range of embryologic, differentiation or response pathways (as in the Tbx and Pax families). For the interferon regulatory factor (IRF) family of transcription factors, mice deficient in Irf1, Irf2, Irf3, Irf4, Irf5, Irf7, Irf8 or Irf9 have defects in the immune response but show no embryologic abnormalities1–7. Mice deficient for Irf6 have not been reported, but in humans, mutations in IRF6 cause two mendelian orofacial clefting syndromes8–10, and genetic variation in IRF6 confers risk for isolated cleft lip and palate11–15. Here we report that mice deficient for Irf6 have abnormal skin, limb and craniofacial development. Histological and gene expression analyses indicate that the primary defect is in keratinocyte differentiation and proliferation. This study describes a new role for an IRF family member in epidermal development.
To define the role of Irf6 in vivo, we generated mice deficient for Irf6. We identified an embryonic stem cell line carrying a gene trap allele for Irf6 in the Omnibank database (OST398253)16. Sequence analysis showed that the gene trap vector inserted into intron 1 of Irf6, 36 bp from the splice donor site (Fig. 1a). We detected the Irf6 gene trap allele (Irf6gt1) by PCR (Fig. 1b) and DNA blot analysis (data not shown). We mated mice heterozygous for the gene trap allele (Irf6gt1/+), and 18% (34 of 192) of the embryos showed abnormal external morphology (Fig. 1c,d). Genotyping demonstrated that all abnormal mice were homozygous for the gene trap allele (Irf6gt1/gt1), and their frequency was not significantly different from mendelian ratios (P = 0.06). Expression analysis showed Irf6 protein in skin and oral epithelium from E17.5 wild-type embryos, as previously shown17,18 but reduced expression in skin from heterozygous embryos and no detectable expression in skin from Irf6gt1/gt1 embryos (Fig. 1e), demonstrating that the gene trap is a null allele for Irf6.
Figure 1.
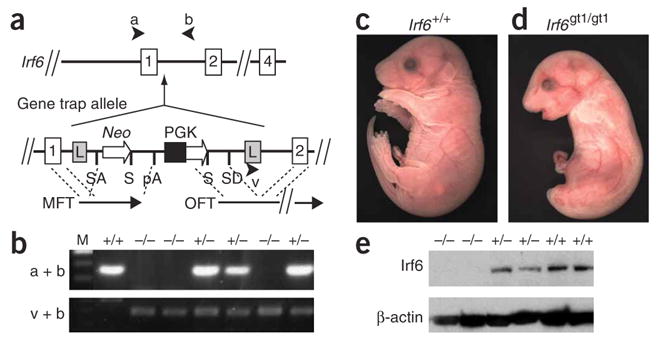
Genotypic and phenotypic analysis of mice deficient for Irf6. (a) Irf6 gene trap allele (Irf6gt1). The gene trap vector (VICTR48) inserted 36 bp from the splice donor site of intron 1 and contains flanking long terminal repeats (L), a splice acceptor (SA) and donor (SD) sites, the ORF for neomycin (Neo), stop codons (S), polyadenylation site (pA), the phosphoglycerate kinase (PGK) promoter, marker fusion transcript (MFT) and OmniBank sequence tag (OST) fusion transcript (OFT). (b) Genotypic analysis by PCR of genomic DNA derived from E17.5 embryos using primers a and b to detect wild-type (+) and using primers v and b to detect mutant (−) alleles. (c) Gross appearance of wild-type E17.5 embryos. (d) E17.5 embryo that is homozygous for gene trap allele (Irf6gt1/gt1). (e) Protein blot analysis of protein extracts obtained from E17.5 skin from mice with the indicated genotype. Blots were probed with antibodies directed against the indicated protein.
Initial analysis did not identify any obvious differences between wild-type and heterozygous E17.5 embryos. However, homozygous Irf6gt1/gt1 (null) embryos had taut, shiny skin, lacked external ears and had snouts and jaws shorter and more rounded than their wild-type littermates (Fig. 1c,d). The null embryos also had short forelimbs that lacked visible digits and a single caudal projection that lacked visible hindlimbs and tail.
Skeletal preparations showed that all four limbs and a tail had formed in the null embryos (Fig. 2a). However, the skeletal analysis showed abnormal vertebrae, tail (Fig. 2b), sternum, xiphoid process (Fig. 2c), digits (Fig. 2d) and skull, and there was a cleft of the secondary palate in the null embryos (Fig. 2e). The skeletal abnormalities are unexpected, as Irf6 expression was not detected in developing bone (data not shown). However, similar skeletal abnormalities were observed in mice deficient for Chuk (also known as Ikka)19. Chuk, too, is not expressed in developing bone, and epidermal Chuk expression is sufficient to rescue the skeletal phenotype20. As in the case of Chuk-deficient mice, we speculate that the skeletal abnormalities in the Irf6-null mice are secondary to defects in epidermal differentiation (see below).
Figure 2.
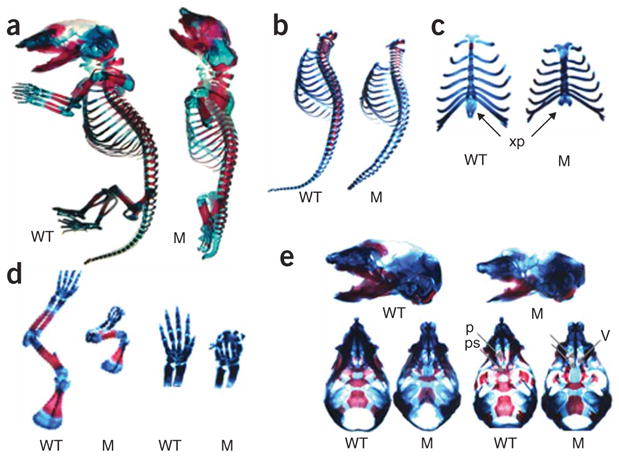
Skeletal defects in mice deficient for Irf6. (a) Skeletal structures of E17.5 wild-type (WT) and newborn mutant (M) embryos were stained for bone (alizarin red S) and cartilage (alcian blue). (b) The spine showed equal numbers of vertebrae in wild-type and null embryos. However, vertebrae in the mutant appeared smaller and showed delayed ossification. The mutant tail was substantially shorter. (c) There were equal numbers of ribs in the wild-type and null embryos, but the sternum appeared shorter with delayed ossification. The xiphoid process (xp) was bifid in the null embryo, demonstrating a failure of complete fusion of the thoracic cage. (d) The long bones were slightly shorter in null versus wild-type littermates, but both null and wild-type embryos showed normal surrounding muscle when skin was dissected. Proximal bones of the upper and lower limb were present in the null embryos, but distal structures, notably the digits, were severely abnormal. Forepaws were magnified to display synostosis of digits and absence of distal phalanges. (e) Skulls from wild-type and null embryos in lateral (top), superior (bottom left) and inferior (bottom right) views. Mandibles were removed for inferior views. The mandible in the null embryo was smaller with a narrower angle than in the wild-type, and the snout was also shorter in the null embryo. The palate shelves (p) in the wild-type embryo fuse along the midline, but the null embryo has a cleft palate. The palate shelves of null embryo are posteriorly and laterally displaced, allowing for direct viewing of nasal structures, including the vomer (v). The presphenoid (ps) is absent in the mutant. Similar results were obtained from three skeletal preparations.
Histological analysis of skin showed wrinkles in epidermis from embryonic day (E) 17.5 wild-type embryos (Fig. 3a) but not in epidermis from null embryos (Fig. 3b). In wild-type embryos, the four stratified layers of the epidermis (basal, spinous, granular and cornified) were all present and visible. In the epidermis from null embryos, the basal layer was present along with a greatly expanded spinous layer, but the outer two layers appeared absent. Electron microscopy analysis of wild-type skin confirmed the presence of the electron-dense keratohyalin granules and the cornified outer layer (Fig. 3c). The epidermis from null embryos lacked these landmarks (Fig. 3d). The cornified layer serves as a barrier whose function can be measured qualitatively with toluidine blue, a dye that does not penetrate mature skin. We observed that toluidine blue was excluded from the epidermis of E17.5 wild-type embryos but not from the epidermis of null embryos (Fig. 3e,f).
Figure 3.
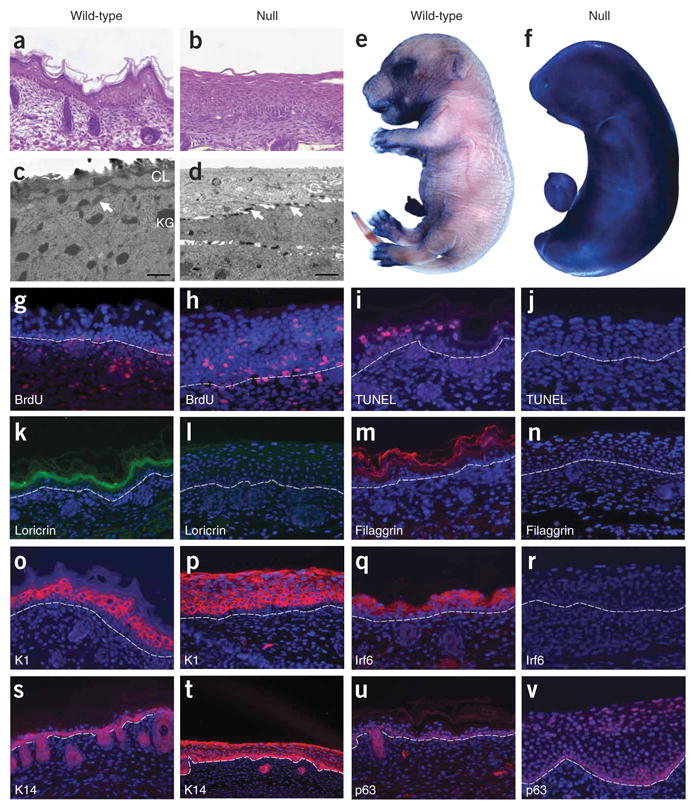
Histologic and molecular analyses of Irf6-null E17.5 embryos. (a–d) Hematoxylin and eosin–stained sections (a,b) and electron micrographs (c,d) of back skin. Note the presence of keratohyalin granules (KG) in the granular layer and a cornified layer (CL) of wild-type epidermis (c), which are absent in null epidermis (d). Arrows indicate desmosomes. Dye exclusion assays of whole embryos show a functional barrier in wild-type (e) but not in null (f) embryos. Cellular proliferation and programmed cell death were evaluated by BrdU (g,h) and TUNEL (i,j) staining, respectively. Immunofluorescent staining (red, except loricrin is green) for cornified and granular layers (k–m) and for spinous (o–r) and basal (s–v) layers. Proteins against which antibodies were directed are indicated. Nuclei were counterstained with DAPI (blue), and the basement membrane is indicated by the dashed line.
To determine whether the expanded spinous layer resulted from increased cell proliferation and/or decreased programmed cell death, we performed BrdU and TdT-mediated dUTP nick end labeling (TUNEL) analyses, respectively. The epidermis from E17.5 wild-type embryos incorporated BrdU in the basal layer but not in the suprabasal layers (Fig. 3g). However, the epidermis obtained from null embryos incorporated BrdU in the basal and spinous layers (Fig. 3h). We also observed TUNEL staining in the epidermis from wild-type embryos but not from null embryos (Fig. 3i,j). Thus, proliferation in the spinous layer and a failure of keratinocytes to terminally differentiate contribute to an increase in the thickness of the spinous layer in mice deficient for Irf6.
We performed microarray and immunostaining analyses to characterize the molecular changes in skin from Irf6 null embryos. We observed significant changes in the level of expression (≥1.8-fold with P ≤ 0.05) for 573 genes, including 42 of 80 protein markers for epidermal differentiation (Table 1 and Supplementary Table 1 online). The expression levels of 21 of 28 markers, including loricrin (Fig. 3k,l) and filaggrin (Fig. 3m,n), decreased significantly in the granular and cornified layers. In the expanded spinous layer, we observed no change or an increase in gene expression for all markers (n = 25), including keratin 1 (K1) (Fig. 3o,p). However, Irf6 was greatly decreased owing to the gene trap mutation (Fig. 3q,r). Notably, we also observed changes in expression for markers of the basal layer (Table 1). Keratin 14 (K14) and p63 were expressed ectopically in the spinous layer of the epidermis from null embryos (Fig. 3s–v). Thus, mice that are deficient for Irf6 do not silence K14 and p63 in the spinous layer. In total, based on the abnormal epidermal histology, barrier function, cell proliferation, cell death and gene expression, we conclude that Irf6 is necessary for regulating proliferation and terminal differentiation of keratinocytes.
Table 1.
Change in gene expression for markers representative of epidermal differentiation in skin from wild-type versus Irf6-null E17.5 embryos
| Gene | Accession number | Layer | Change in expression (as a multiple of WT control) | Chromosome |
|---|---|---|---|---|
| Small proline rich–like family | AK003705 | G,C | −310.8 | 3F1 |
| Loricrin | NM_008508 | G,C | −7.3 | 3F1 |
| Filaggrin | J03458 | G,C | −2.6 | 3F2 |
| Desmocollin 1 | NM_013504 | S,G | NS | 18 |
| Small proline-rich 2a | NM_011468 | S,G | 16.9 | 3F1 |
| Keratin 2-6b | NM_010669 | S(i) | 156.1 | 15F2 |
| Irf6 | NM_016851 | S | −20.4 | 1H6 |
| Stratifin | NM_018754 | S | 3.3 | 4D3 |
| Desmocollin 2 | NM_013505 | S | 8.1 | 18 |
| Keratin 2-1 | NM_008473 | S | NS | 15 |
| Keratin 1–10 | AK014360 | S | NS | 11 |
| S100a8 | NM_013650 | B,S(i) | 61.8 | 3F1 |
| S100a9 | NM_009114 | B,S(i) | 85.1 | 3F1 |
| IkB kinase 1 | NM_007700 | B,S | NS | 19 |
| Rac1 | BC003828 | B | NS | 5 |
| p63 | AF075436 | B | 1.7 | 16 |
| Keratin 1–14 | BC011074 | B | 2.4 | 11 |
| Desmocollin 3 | BB006344 | B | 2.9 | 18 |
Epidermal layers in which each gene is expressed in wild-type embryo are basal (B), spinous (S), granular (G) and cornified (C). Expression in a layer may be inducible (i), increase (positive), decrease (negative) or not change significantly (NS). WT, wild-type.
Rac1, a Rho guanosine triphosphatase, is expressed primarily in the basal layer of the epidermis and regulates proliferation and differentiation by maintaining the stem cell compartment21. To test for a possible relationship between Irf6 and Rac1, we compared the expression of Rac1 in skin from wild-type and null E17.5 embryos. From the microarray analysis, we did not observe significant quantitative changes in expression of Rac1 (Table 1). Also, we did not detect qualitative changes in expression of Rac1 using immunostaining (data not shown). These results suggest that Rac1 is epistatic to Irf6 in regulating keratinocyte differentiation or that they operate in independent pathways.
In the Irf6-null embryos, we observed epidermal adhesions at several sites, including the oral cavity (Fig. 4a,b), between the tail and hindlimbs (Fig. 4c,d) and in the esophagus (Fig. 4e,f). In contrast, we observed an oral adhesion in only 2 of 18 heterozygous embryos. The oral adhesions were unilateral and located superficial to the developing first or second molar (Fig. 4g). A similar phenotype was recently observed22 in mice heterozygous for the R84C variant in Irf6. The R84C variant is highly associated with popliteal pterygium syndrome8. In a paper published in this issue22, the authors observed oral adhesions in 89% (25 of 28) of heterozygous embryos, and all adhesions were bilateral. Moreover, they observed epidermal adhesions between the ventral surface of the tongue and the floor of the mouth. We did not observe adhesions in this location in embryos heterozygous for the gene trap allele. Both Irf6 mutant strains were on a similar mixed genetic background of C57Bl/6 and 129/SvJ, suggesting that the phenotypic changes were due to allele differences.
Figure 4.
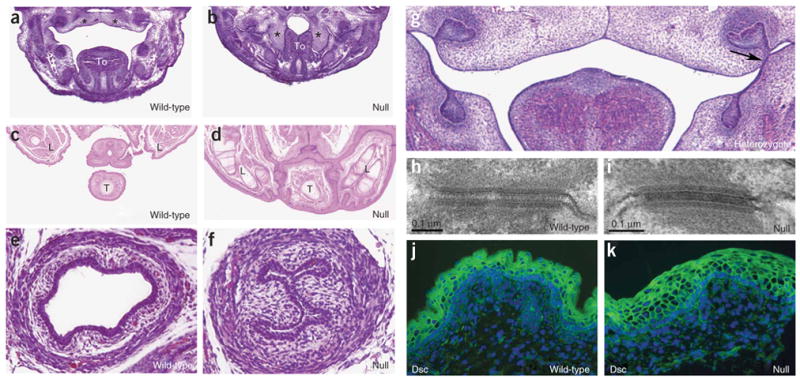
Epidermal adhesions and desmosomal structure in Irf6-null embryos. (a–f) Hematoxylin and eosin staining for sections of E14.5 frontal head (a,b), E17.5 rostral trunk (c,d) and E17.5 caudal trunk (e,f). Palate shelves elevate and begin to fuse in wild-type mice (a) but not in null embryos (b), where adhesions contribute to crowding of the oral cavity and prevent the palate shelves from elevating. Hindlimbs and tail are joined by epithelial adhesions (compare c with d). Esophagus is closed in null embryo (compare e with f). *: palate shelves; To: tongue; L: hindlimb; T: tail. Adhesion in the heterozygote was observed superficial to the first molar on the left side only (g, arrow). (h,i) Electron micrographs of desmosomes. Immunofluorescent staining (green) for desmocollin (Dsc) (j,k). We did not observe any difference in desmosomal structure and protein between wild-type and null embryos. Nuclei were counterstained with DAPI (blue).
To further characterize the epidermal adhesions observed in the null embryos, we performed electron microscopy and immunostaining analyses. We detected desmosomes, an important structure in the junctional complex between epithelial cells, in the epidermis of both wild-type and null E17.5 embryos (Fig. 4h,i). However, desmosomes in the epidermis of wild-type embryos were not observed in the most superficial layer, whereas desmosomes in the epidermis of null embryos were observed throughout the epidermis, including the most superficial regions (Fig. 3c,d). Microarray analysis (Table 1) showed that the expression of the desmosomal components either increased or did not change significantly. In addition, the subcellular localization of the desmocollin proteins did not seem to differ between wild-type and null epidermis (Fig. 4j,k). We speculate that the epidermal adhesions observed in the Irf6-null embryos result from the absence of the normal cornified layer. Without the cornified layer, cell interactions, including desmosomes, can occur between cells from adjoining tissues.
The morphological, skeletal and histological abnormalities in Irf6-null embryos are very similar to the phenotypes observed in the repeated epilation (Er) mouse23 and mice that are deficient for the IkB kinase 1 gene (Chuk, also known as Ikka)19. Recently, two groups demonstrated that the Er phenotype results from a frameshift mutation in stratifin (Sfn)24,25, a member of the 14-3-3 protein family. Two possible mechanisms for the common phenotype are that Irf6 is necessary for the expression of Chuk and Sfn in skin or that Irf6 protein interacts with Ikka and Sfn. The microarray analysis did not show any significant change in the expression level of Chuk in the skin from E17.5 null embryos and showed a 3.3-fold increase in the expression level of Sfn (Table 1). Protein blot analysis confirmed these results (Fig. 5a,b). Immunostaining showed that Sfn (Fig. 5c,d) and Ikka (Fig. 5e,f) were expressed in the spinous layer of wild-type embryos, with some Ikka expression in the basal layer. No change occurred in the epidermis of the null embryos. To test for a direct protein interaction between Irf6 and Ikka or Sfn, we performed coimmunoprecipitation. Antibodies specific for each of these proteins did not pull down either of the other two proteins, but the antibody for Ikka readily pulled down Ikkb, another member of the Ikk complex (data not shown). We conclude that Irf6 is not necessary for the expression of Chuk and Sfn in epidermis, that Irf6 does not form a detectable complex with Ikka and Sfn in epidermis and that other mechanisms are responsible for the common phenotype observed in mouse strains with mutations in each of these genes.
Figure 5.
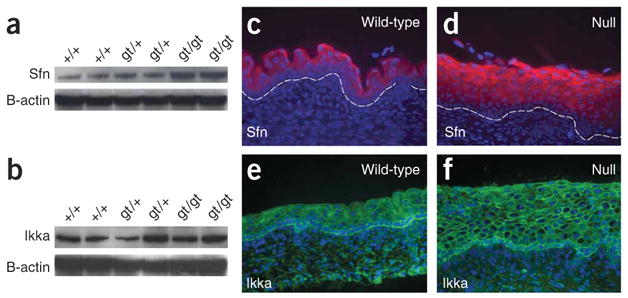
Expression of stratifin and Ikka in skin from Irf6-null E17.5 embryos. (a,b) Protein blot analyses show that stratifin expression (a) is greater in null embryos, whereas Ikka expression (b) does not seem to change. The respective blots were stripped and reprobed with β-actin as a control for loading. (c) Immunostaining shows that stratifin is expressed in the cytoplasm of spinous cells in skin from wild-type E17.5 embryos (c); it remains cytoplasmic but increases along with the expanded spinous layer in the skin from null embryos (d). (e,f) Immunostaining shows that Ikka is expressed predominantly in the basal layer in skin from both the wild-type (e) and Irf6-null (f) embryos. Ikka is localized to the cytoplasm and in the nucleus of some cells. This distribution does not appear to differ in skin from wild-type and Irf6-null embryos.
We report here that mice deficient for Irf6 have abnormal skin, limb and craniofacial morphogenesis. The major histological feature of the Irf6-null mice is the lack of a normal stratified epidermis. The null epidermis was thicker than in wild-type mice, and the suprabasal keratinocytes failed to stop proliferating and failed to terminally differentiate. We conclude that a significant role for Irf6 is to regulate proliferation and differentiation of keratinocytes.
In humans, mutations in IRF6 cause two related orofacial clefting disorders, Van der Woude (VWS) and popliteal ptyergium syndromes (PPS)8. Both VWS and PPS show an autosomal dominant pattern of inheritance, whereas mice heterozygous for the Irf6 null allele display a low-penetrance oral adhesion phenotype rather than a highly penetrant orofacial cleft phenotype. Although mice homozygous for the null Irf6 allele have a cleft palate, the cleft seems to be caused by a defect in elevation, which could occur as a primary defect or could be secondary to crowding of the craniofacial structures owing to the constrictive action of the skin or oral adhesions.
In addition to the oral facial clefts, individuals with PPS may present with a combination of eyelid adhesions (ankyloblepharon), oral adhesions, syndactyly of digits and pterygia11. Previously, we proposed that the additional abnormalities seen in individuals with PPS are due to a dominant-negative effect of the R84C variant8. The phenotypes of the Irf6 mutant mice described here and in the accompanying report22 are consistent with this model. First, the skin phenotypes in the mice homozygous for both Irf6 alleles resemble the pterygia seen in individuals with PPS, with the much more severe phenotype in mice representing the null condition. Second, the phenotype observed in mouse embryos heterozygous for the R84C variant has a higher penetrance (89% versus 11%) and is more severe (multiple versus single sites) than the phenotype observed in mouse embryos heterozygous for the null allele. We recognize that this genotype-phenotype relationship for Irf6 mutations is not absolute in either humans or mice. Thus, other factors contribute to the IRF6 spectrum of phenotypes.
The earliest phenotype observed in the skin of Irf6-null mice was ectopic K14 expression in the spinous layer, suggesting a role for Irf6 in repressing K14 transcription. A previous study showed that two other transcription factors expressed in the spinous layer, Skin-1a and Tst-1, contribute to the repression of K14 (ref. 26). Also, Skin-1a bound directly to CREB-binding protein (CBP), suggesting that repression of the K14 promoter may be due to interference of the CBP/p300 coactivator complex. Because IRF1 (ref. 27), IRF3 (ref. 28) and IRF7 (ref. 29) interact with CBP and/or p300, it is possible that IRF6 interacts with the CBP/p300 coactivator complex to help repress K14 expression in the spinous layer and to regulate other events in keratinocyte differentiation such as cessation of cell proliferation30. These new roles for a member of the IRF family expand the repertoire of IRF functions from stress response into development but suggest a bridge that spans these apparently disparate functions.
METHODS
Generation of mice deficient for Irf6
We obtained the embryonic stem (ES) cell line OST398253 that contains the gene trap allele for Irf6 from Lexicon Genetics. The ES cells were microinjected into C57BL/6 blastocysts, and we obtained five male chimeras that were mated with C57BL/6 mice. Offspring with agouti coat color were genotyped for germline transmission of the gene trap allele. Maintenance and handling of mice were approved by the Animal Care Unit at the University of Iowa.
Genotyping of mutant mice
We used a PCR-based assay to genotype the gene trap cell line and subsequent mutant mice. Genomic DNA was isolated as recommended (Model 850 Alpha, Autogen) from mouse tail clips or embryonic tissues. PCR primers a and b flank the insertion site and detect the wild-type allele by amplifying a 461-bp product. Primer v, derived from the long terminal repeat of the vector, and primer b detect the gene trap allele by amplifying a 283-bp product. Primer sequences are listed in Supplementary Table 2 online.
Protein blot analysis
Protein was purified as recommended (PARIS, Ambion) and quantified with a bicinchoninic acid (BCA) assay (Sigma). Total protein was separated in a 10% denaturing polyacrylamide gel (Invitrogen). We used 10, 20 and 50 μg of protein from skin per lane to detect Irf6, Sfn and Ikka, respectively. Transfer to a nylon membrane was performed as recommended (Invitrogen). We generated rabbit polyclonal antibodies against Irf6 (ref. 17) and purchased antibodies for Sfn and Ikka (Santa Cruz Biotech). The primary antibodies were detected as recommended using the ECL Plus kit (Amersham Biosciences). The blots were stripped and reprobed with a monoclonal antibody to β-actin. Blots were exposed to film for ≤1 min to detect β-actin, Irf6 and stratifin and were exposed for 15 min to detect Ikka.
Morphological analysis of mice
Null embryos were obtained from Irf6+/gt1 matings. The presence of a copulation plug was defined as E0.5. Embryos were removed from euthanized mothers, analyzed and genotyped as above. Examination of skeletal morphology was done at E17.5. The skin was removed from wild-type and null embryos; it was then fixed in 40% formaldehyde/glacial acetic acid/70% ethanol mix (1:1:8 ratio), rinsed and then stained with alcian blue 8GX and alizarin red S (Sigma) to stain cartilage and calcified bone, respectively.
Histological analysis of mouse tissue
Embryos were fixed overnight in freshly prepared 4% paraformaldehyde and paraffin processed using standard methods. Sections (5 μm) were deparaffinized and stained with hematoxylin and eosin (SurgiPath) to visualize structures.
In vivo barrier assay
The in vivo epidermal barrier assay was performed on E17.5 embryos using a dye exclusion assay. Embryos were fixed in 100% methanol for 5 min, rinsed briefly with PBS, stained in 0.1% toluidine blue for 1 min and washed several times with PBS.
Immunohistological analysis of mouse skin
Immunostaining was performed by incubating deparaffinized or unfixed frozen sections with antibodies at 4 °C overnight. Polyclonal antibodies were detected with Alexa 568–conjugated (Invitrogen) or FITC-conjugated (Jackson Immuno Research Laboratories) goat anti-rabbit secondary antibody as recommended. Monoclonal antibodies were detected either with the MOM kit (Vector Laboratories) followed by Strep-Alexa 568 (Invitrogen) or directly with a fluorescein isothiocyanate (FITC)-conjugated anti-loricrin (Covance). Slides were rinsed and mounted with Vectashield with 4,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories) for visualization of nuclei. Antibodies were purchased for K1 (Novocastra), K14 (Novocastra), filaggrin (Novocastra), Irf6 (ref. 17), Sfn (Santa Cruz Biotech), Ikka (Santa Cruz Biotech), Rac1 (Chemicon) and desmocollin (Abcam).
Transmission electron microscopy of mouse skin
Skin from wild-type and null animals was fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer. Samples were washed and postfixed in 1% osmium and then embedded in Epon 12. Thin sections (70 nm) were counterstained with uranyl acetate and lead citrate before visualization.
BrdU assay
Pregnant mothers were injected with BrdU (Sigma) (100 μg per g pregnant dam weight) 2 h before killing. BrdU immunostaining was performed as above with a monoclonal antibody to BrdU (Dako).
TUNEL assay
The TUNEL assay (Trevigen) was performed as recommended. Slides were counterstained with DAPI to identify TUNEL-positive cells.
Microarray analysis
Total RNA was isolated from E17.5 skin. First, we observed that the average mass of skin was significantly higher for null mice (70 mg ± 20 mg (s.d.) for wild-type skin (n = 3) versus 150 mg ± 30 mg (s.d.) for null skin (n = 3); P = 0.02) and was consistent with the histological findings that skin obtained from null mice was thicker than skin obtained from wild-type littermates. Total RNA was isolated as recommended (RNeasy, Qiagen), except that tissue was treated with proteinase K (20 mg/ml) for 20 min at 55 °C. RNA was assessed for quality using the Model 2100 Bioanalyzer (Agilent Technologies). We processed 5 μg of total RNA for use on the microarray by using the Affymetrix GeneChip one-cycle target labeling kit (Affymetrix) according to the manufacturer’s recommended protocols. The resultant biotinylated complementary RNA (cRNA) was fragmented and then hybridized to the GeneChip Mouse Genome 430 2.0 Array (Affymetrix). The arrays were washed, stained and scanned using the Affymetrix Model 450 Fluidics Station and Affymetrix Model 3000 Scanner by the University of Iowa DNA Core Facility using the manufacturer’s recommended protocols. Expression values were generated using the Microarray Suite (MAS) v5.0 software (Affymetrix). CEL files were read into dChip, normalized and modeled using the perfect match-mismatch difference model. Negative values were truncated to a default value of 10. The ’present’ and ’absent’ calls used were those from MAS5. Excluding the Affymetrix control probes on the chip, a total of 30,250 probes were ‘present’.
Statistical analysis
We defined genes that were expressed differentially as those showing at least a 1.8-fold difference between wild-type and mutant arrays. The comparison parameters were as follows: 90% confidence bound for change in expression as a multiple of the control, P ≤ 0.05 for testing whether wild-type expression was equal to mutant expression, and a paired t-test with P ≤ 0.05. To compare the observed number of genotypes from heterozygous matings with the expected mendelian ratios, we used χ2 analysis. To compare the mass of wild-type and mutant skin, we used Student’s t-test.
Accession codes
Gene Expression Omnibus (GEO): microarray data, GSE5800.
Supplementary Material
Acknowledgments
The authors wish to acknowledge technical assistance from E. Sweezer, R. Cao, T. Kinney, S. Bullard, H. Mishima and A. Lidral. We also thank K. Walters and J. Shao of the Central Microscopy Research Facility and K. Knudtson of the DNA Core Facility at the University of Iowa. We wish to thank R. Richardson and M. Dixon for sharing results before publication. This work was supported in part by US National Institutes of Health grants DE16215 (J.C.M., B.C.S., M.L., B.Y.), DE13513 (B.C.S.) and DE08559 (J.C.M.).
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- 1.Matsuyama T, et al. Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell. 1993;75:83–97. [PubMed] [Google Scholar]
- 2.Sato M, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 3.Mittrucker HW, et al. Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science. 1997;275:540–543. doi: 10.1126/science.275.5299.540. [DOI] [PubMed] [Google Scholar]
- 4.Takaoka A, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 5.Honda K, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 6.Holtschke T, et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–317. doi: 10.1016/s0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- 7.Kimura T, et al. Essential and non-redundant roles of p48 (ISGF3 gamma) and IRF-1 in both type I and type II interferon responses, as revealed by gene targeting studies. Genes Cells. 1996;1:115–124. doi: 10.1046/j.1365-2443.1996.08008.x. [DOI] [PubMed] [Google Scholar]
- 8.Kondo S, et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kayano S, et al. Novel IRF6 mutations in Japanese patients with Van der Woude syndrome: two missense mutations (R45Q and P396S) and a 17-kb deletion. J Hum Genet. 2003;48:622–628. doi: 10.1007/s10038-003-0089-0. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, et al. Novel mutations in the IRF6 gene for Van der Woude syndrome. Hum Genet. 2003;113:382–386. doi: 10.1007/s00439-003-0989-2. [DOI] [PubMed] [Google Scholar]
- 11.Zucchero TM, et al. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Engl J Med. 2004;351:769–780. doi: 10.1056/NEJMoa032909. [DOI] [PubMed] [Google Scholar]
- 12.Blanton SH, et al. Variation in IRF6 contributes to nonsyndromic cleft lip and palate. Am J Med Genet A. 2005;137:259–262. doi: 10.1002/ajmg.a.30887. [DOI] [PubMed] [Google Scholar]
- 13.Ghassibe M, et al. Interferon regulatory factor-6: a gene predisposing to isolated cleft lip with or without cleft palate in the Belgian population. Eur J Hum Genet. 2005;13:1239–1242. doi: 10.1038/sj.ejhg.5201486. [DOI] [PubMed] [Google Scholar]
- 14.Scapoli L, et al. Strong evidence of linkage disequilibrium between polymorphisms at the IRF6 locus and nonsyndromic cleft lip with or without cleft palate, in an Italian population. Am J Hum Genet. 2005;76:180–183. doi: 10.1086/427344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srichomthong C, Siriwan P, Shotelersuk V. Significant association between IRF6 820G->A and non-syndromic cleft lip with or without cleft palate in the Thai population. J Med Genet. 2005;42:e46. doi: 10.1136/jmg.2005.032235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zambrowicz BP, et al. Disruption and sequence identification of 2,000 genes in mouse embryonic stem cells. Nature. 1998;392:608–611. doi: 10.1038/33423. [DOI] [PubMed] [Google Scholar]
- 17.Bailey CM, et al. Mammary serine protease inhibitor (Maspin) binds directly to interferon regulatory factor 6: identification of a novel serpin partnership. J Biol Chem. 2005;280:34210–34217. doi: 10.1074/jbc.M503523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knight AS, Schutte BC, Jiang R, Dixon MJ. Developmental expression analysis of the mouse and chick orthologues of IRF6: the gene mutated in Van der Woude syndrome. Dev Dyn. 2006;235:1441–1447. doi: 10.1002/dvdy.20598. [DOI] [PubMed] [Google Scholar]
- 19.Hu Y, et al. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 20.Sil AK, Maeda S, Sano Y, Roop DR, Karin M. IkappaB kinase-alpha acts in the epidermis to control skeletal and craniofacial morphogenesis. Nature. 2004;428:660–664. doi: 10.1038/nature02421. [DOI] [PubMed] [Google Scholar]
- 21.Benitah SA, Frye M, Glogauer M, Watt FM. Stem cell depletion through epidermal deletion of Rac1. Science. 2005;309:933–935. doi: 10.1126/science.1113579. [DOI] [PubMed] [Google Scholar]
- 22.Richardson RJ, et al. IRF6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat Genet. doi: 10.1038/ng1894. advance online publication 15 October 2006. [DOI] [PubMed] [Google Scholar]
- 23.Fisher C, Jones A, Roop DR. Abnormal expression and processing of keratins in pupoid fetus (pf/pf) and repeated epilation (Er/Er) mutant mice. J Cell Biol. 1987;105:1807–1819. doi: 10.1083/jcb.105.4.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herron BJ, et al. A mutation in stratifin is responsible for the repeated epilation (Er) phenotype in mice. Nat Genet. 2005;37:1210–1212. doi: 10.1038/ng1652. [DOI] [PubMed] [Google Scholar]
- 25.Li Q, Lu Q, Estepa G, Verma IM. Identification of 14–3-3sigma mutation causing cutaneous abnormality in repeated-epilation mutant mouse. Proc Natl Acad Sci USA. 2005;102:15977–15982. doi: 10.1073/pnas.0508310102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugihara TM, Kudryavtseva EI, Kumar V, Horridge JJ, Andersen B. The POU domain factor Skin-1a represses the keratin 14 promoter independent of DNA binding. A possible role for interactions between Skn-1a and CREB-binding protein/p300. J Biol Chem. 2001;276:33036–33044. doi: 10.1074/jbc.M103000200. [DOI] [PubMed] [Google Scholar]
- 27.Dornan D, et al. Interferon regulatory factor 1 binding to p300 stimulates DNA-dependent acetylation of p53. Mol Cell Biol. 2004;24:10083–10098. doi: 10.1128/MCB.24.22.10083-10098.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin BY, et al. Crystal structure of IRF-3 in complex with CBP. Structure. 2005;13:1269–1277. doi: 10.1016/j.str.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 29.Yang H, Lin CH, Ma G, Baffi MO, Wathelet MG. Interferon regulatory factor-7 synergizes with other transcription factors through multiple interactions with p300/CBP coactivators. J Biol Chem. 2003;278:15495–15504. doi: 10.1074/jbc.M212940200. [DOI] [PubMed] [Google Scholar]
- 30.MacPartlin M, et al. p300 regulates p63 transcriptional activity. J Biol Chem. 2005;280:30604–30610. doi: 10.1074/jbc.M503352200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.