

Abstract
It is established that prolonged hypoxia leads to activation of KATP channels and action potential (AP) shortening, but the mechanisms behind the early phase of metabolic stress remain controversial. Under normal conditions IK1 channels are constitutively active while KATP channels are closed. Therefore, early changes in IK1 may underlie early AP shortening. This hypothesis was tested using transgenic mice with suppressed IK1 (AAA-TG). In isolated AAA-TG hearts AP shortening was delayed by ∼24 s compared to WT hearts. In WT ventricular myocytes, blocking oxidative phosphorylation with 1 mM cyanide (CN; 28°C) led to a 29% decrease in APD90 within ∼3-5 min. The effect of CN was reversed by application of 100 μM Ba2+, a selective blocker of IK1, but not by 10 μM glybenclamide, a selective blocker of KATP channels. Accordingly, voltage-clamp experiments revealed that both CN and true hypoxia lead to early activation of IK1. In AAA-TG myocytes, neither CN nor glybenclamide or Ba2+ had any effect on AP. Further experiments showed that buffering of intracellular Ca2+ with 20 mM BAPTA prevented IK1 activation by CN, although CN still caused a 54% increase in IK1 in a Ca2+-free bath solution. Importantly, both (i) 20 μM ruthenium red, a selective inhibitor of SR Ca2+-release, and (ii) depleting SR by application of 10 μM ryanodine+1 mM caffeine, abolished the activation of IK1 by CN. The above data strongly argue that in the mouse heart IK1, not KATP, channels are responsible for the early AP shortening during hypoxia.
Keywords: IK1, hypoxia, action potential, repolarization, myocytes
1. Introduction
It is firmly established that KATP channels [1] are critical sensory and regulatory elements in the response of the heart to various metabolic challenges, including ischemia and hypoxia. According to a widely accepted paradigm, KATP-mediated cardioprotection results from a reduced calcium load due to shortening of the action potential (AP) caused by KATP channel activation. Nevertheless, whereas the KATP-mediated cardioprotection is beneficial, the resulting AP shortening is generally proarrhythmic. Consistent with this, it has been found that activation of KATP channels by K+ channel openers is also proarrhythmic [2].
However, under normal conditions, cardiac IK1, not KATP channels, dominate the K+ conductance near the resting membrane potential. Therefore, any variations of IK1 during an initial period of metabolic inhibition would be dominant over that produced by KATP channels which activate after a significant delay. Consistently, several studies strongly support the view of IK1 being a critical player during the early phase of response to hypoxia. For example, Petrich et al (1991) [3] have shown that in the rabbit heart, hypoxia-induced early AP shortening and speeding up of the late phase of repolarization is sensitive to low concentrations of Ba2+, a specific blocker of IK1. The early increase in IK1, preceding the activation of KATP channels, was also observed in guinea-pig cardiomyocytes during metabolic inhibition with cyanide (CN), a blocker of oxidative phosphorylation [4]. An increase in IK1 is also associated with atrial fibrillation [5], where the metabolic status of cardiac myocytes is probably also affected. The latest data also strongly implicate activation of Kir2.1 channels, the major isoform underlying IK1 in cardiac myocytes, in response to hypoxia in rabbit arterial smooth muscle cells [6].
In this study, we take advantage of transgenic mice with suppressed IK1 (AAA-TG) in order to investigate the early effects of CN and true hypoxia on the activity of IK1 at the level of the whole heart and isolated cardiac myocytes. Our data convincingly show that in the mouse heart the early AP shortening during hypoxia is mediated by IK1, not KATP channels, thus pointing to activation of IK1 as the primary fast component of electrical response to metabolic stress.
2. Methods
2.1. Expression of Kir2.1 subunits in the mouse heart
Transgenic mice (TG) with suppressed IK1 in the heart were produced as described previously [7]. Briefly, dominant-negative Kir2.1-AAA channel subunits were fused with Green Fluorescent Protein (GFP) at the C terminus and expressed in the heart under control of the α-myosin heavy chain promoter to produce AAA-TG mice. For whole-heart experiments AAA-TG mice were identified by PCR analysis of genomic tail DNA using primers corresponding to the sequence of GFP. Specifically, in whole-heart experiments we used a transgenic line where ∼95% of cardiac myocytes express a dominant negative transgene, assuring a global suppression of IK1 throughout the heart (line 2 in ref [7]). In single-cell experiments, myocytes were isolated from both available AAA-TG lines, and cells expressing Kir2.1-AAA subunits were identified by GFP fluorescence [7].
2.2. Composition of solutions
Krebs–Henseleit bicarbonate (KHB) solution (mM)
NaHCO3 25, NaCl 118, KCl 4.7, MgSO4 1.2, NaH2PO4 1.2, CaCl2 2.5, EDTA 0.5, Glucose 10, with pH adjusted to 7.4 using HCl.
Tyrode solution (mM)
NaCl 137, KCl 5.4, MgCl2 0.5, NaH2PO4 0.16, NaHCO3 3, HEPES 5, Glucose 10, CaCl2 1, with pH adjusted to 7.4 with NaOH.
Extracellular solution for Ca2+ current (ICa) recordings (mM)
tetraethylammonium chloride 137, MgCl2 1, glucose 10, CaCl2 2, HEPES 10, with pH adjusted to 7.4 using NaOH.
Intracellular solution for ICa recordings (mM)
CsCl 111, tetraethylammonium chloride 20, glucose 10, EGTA 14, HEPES 10, MgATP 5, with pH adjusted to 7.2 using CsOH.
Extracellular solution for Na+/Ca2+ exchange current (INCX) recordings (mM)
NaCl 140, CsCl 4, CaCl2 2.5, MgCl2 1.2, glucose 10, HEPES 5, with pH adjusted to 7.4 using NaOH. Solution was supplemented with 10 μM nicardipine and 10 μM ouabain in order to inhibit ICa and Na+/K+ ATPase, respectively. The putative INCX was calculated as a difference between the current measured in the presence of extracellular Ca2+ and that measured in the presence of 5 mM Ni2+/ 0 Ca2+.
Intracellular solution for INCX recordings (mM)
CsCl 110; NaCl 20; HEPES 10; MgCl2 0.4; glucose 5; tetraethylammonium chloride 20; EGTA 5; MgATP 4; CaCl2 1, with pH adjusted to 7.2 using CsOH.
Stock solutions of glybenclamide (10 mM), pinacidil (100 mM) and nicardipine (10 mM) were prepared in dimethylsulfoxide (DMSO). A 10 mM stock solution of oubain was prepared in H2O. All stocks were kept frozen at −20 °C. A 2 M Sodium Cyanide (CN) stock solution in H2O was prepared fresh every four days and kept at +4 °C. pH of CN-containing solutions was adjusted to 7.35 with HCl just before the experiment.
KINT (mM)
KCl 130, EGTA 2, CaCl2 0.2, HEPES 10, K2ATP 5, with pH adjusted to 7.35 with KOH.
2.3. Isolated heart perfusion and MAP recordings
Mice were anesthetized with Avertin as previously reported [7]. Hearts were rapidly excised, rinsed in Tyrode solution and immediately perfused with the same solution for 1–2 min at room temperature. Hearts were then mounted on a modified perfusion apparatus (Kent Scientific, USA) for retrograde aortic perfusion with oxygenated KHB solution (95% O2+5% CO2) at a constant pressure of 75–85 cm H2O (37 °C). The PO2 in the solution was measured using an automated blood gas analyzer (IL 1640, Instrumentation Laboratory, Warrington) and was above 580 mmHg in all experiments. The AV node was ablated by applying a gentle pinch using forceps, and hearts were then paced at a basic cycle length of 150 ms with pacing electrodes placed at the base of the left ventricle. Hearts were allowed to stabilize for 10-20 min. Hypoxia was induced by switching to a KHB solution equilibrated with nitrogen (95% N2+5% CO2).
Monophasic action potentials (MAPs) from beating hearts were recorded using an 0.25 mm (diameter) silver wire insulated with Teflon sleeves by gently placing it on the left ventricle [8]. The signals were recorded using a DP-304 differential amplifier (Warner Instruments, USA), Digidata 1322A and Clampex 8.2 software (Molecular Devices, USA). Signals were filtered at 0.1-1 kHz, sampled at 3 kHz, and analyzed off-line using Clampfit 8.2 (Molecular Devices, USA) and Microsoft Excel, 2003.
Quantification of MAP shortening was carried out at the level of 75% (MAPD75) rather than 90%, as in single cell experiments. One of the reasons is that in this type of experiment the magnitude of the observed changes and the reliability of measurements are most optimal with MAPD75. Although, the magnitude of MAPD changes is obviously increased at 90%, and even further at 95%, the well-known ‘motion artifact’ [9], characteristic for MAP recordings, makes MAPD measurements at these levels less reliable. Additionally, due to a highly ‘triangular’ shape of the mouse AP, and the fact that IK1 contributes primarily to the late (shallow) phase of the AP, MAPD changes at lower levels would be diminished and eventually lost, for example, at MAPD50.
2.4. Preparation of isolated cardiomyocytes and patch-clamp recordings
Cardiomyocytes were isolated from the left ventricular wall of the heart excised from 2–5 month old mice using collagenase treatment as described previously [7]. AP and whole-cell currents from single isolated cardiomyocytes were recorded in current-clamp or voltage-clamp configuration, respectively, using Axopatch 200B amplifier (Molecular Devices, USA). Signals were digitized with Digidata 1322A and Clampex 8.2 software, and analyzed off-line using Clampfit 8.2 (Molecular Devices, USA) and Microsoft Excel, 2003.
Series resistance compensation was routinely set above 80% to produce an access resistance less than 2 MΩ. Composition of solutions for patch-clamp recordings can be found above. All patch-clamp recordings were obtained at 28 °C.
For recordings of APs and IK1 myocytes were bathed in Tyrode solution modified as indicated in the text, and KINT served as a pipette solution.
IK1 was measured as a Ba2+-sensitive current in response to a 4 sec long voltage ramp from −100 mV to + 10 mV (reduced voltage range is presented in figures). Holding potential was set to −75 mV.
ICa was recorded in the following way: Membrane potential was first stepped from a −50 mV holding potential to −75 mV for 100 ms (to remove Ca2+ channel inactivation), then returned to the holding potential for 20 ms (to inactivate Na+ channels), and then stepped to a series of membrane potentials between −50 and +50 mV for 250 ms. Currents were recorded in the presence of 2 mM extracellular Ca2+, and the current-voltage relationship for ICa was then calculated as a difference between the peak current and the current remaining at the end of the voltage step.
INCX was measured as a Ni-sensitive current at the end of 300 ms long voltage steps from a −40 mV holding potential to potentials between −80 mV and +80 mV.
The density of KATP channels and their sensitivity to ATP were measured using inside-out patches at a +50 mV membrane potential. KINT served as both the pipette and bath solution. In patches, membrane capacitance cannot be accurately measured, therefore the KATP channel density is calculated per patch (not per pF). In contrast to whole-cell measurements, experiments using inside-out patches (i) are fast, allowing to test a great number of myocytes, (ii) permit measurements of ATP sensitivity, and (iii) permit recordings of IKATP which are not compromised by voltage-clamp errors caused by an enormous conductance of fully activated channels.
2.5. Application of hypoxic solution in patch-clamp experiments
In order to minimize the contamination of the perfusing hypoxic solution with atmospheric oxygen in patch-clamp experiments several precautions have been taken.
First, we used a glass syringe as the main container for Tyrode solution equilibrated with 100% nitrogen gas. Solution was bubbled continuously for >20 minutes at room temperature and pH adjusted to ∼7.3. Solution was then quickly loaded into the glass syringe just before the experiment, and a 1/4″ of mineral oil poured over the top of the solution to prevent any contact with atmospheric oxygen. Second, since the flow rate of solution in the patch-clamp experiment is rather slow, and the distance between the main solution container and perfusion chamber is quite long (∼2′), even a connection with a teflon tube may potentially be insufficient to prevent contamination with atmospheric oxygen. Thus, we used a second larger outer teflon tube ventilated continuously at a high flow rate with 100% nitrogen to further disrupt contact with air. Solution then entered an in-line heater mounted on the stage of the microscope near the perfusion chamber. The interior of the heater is made of stainless steel and thus the possibility of contamination with air is excluded. Third, in order to avoid contact of the solution with the air right at the chamber, myocytes, voltage-clamped in the whole-cell configuration on the floor of the chamber, were lifted up and moved into the center of the laminar stream of O2-free solution.
The total length of teflon tube in direct contact with the air is ∼1-1.5″, including the last ½″ segment. Experimental results show that the level of O2 depletion is sufficient to cause metabolic inhibition comparable to that produced by 1 mM CN.
2.6. Data analysis
The data are represented as a mean±S.E. (Standard Error). Statistical significance was estimated using a two-tailed t-test with equal variances. *, ** and *** indicate p values <0.05, <0.01 and <0.005, respectively.
3. Results
3.1. Early MAP shortening in response to hypoxia is abolished in TG hearts lacking IK1
The effects of hypoxia, mimicked by application of an O2-free solution, were first studied in Langendorff perfused hearts from WT and AAA-TG mice using MAP recordings (Fig. 1). In WT hearts, the response to hypoxia displays two distinct components. Early, presumably IK1-dependent, MAP shortening occurred nearly concurrently with the replacement of O2 by N2 in the perfusate (Fig. 1A; 2). This was then followed by a more rapid, presumably KATP-dependent phase (Fig. 1A; 3), which led to short ‘neuron-like’ MAPs (Fig. 1A; 4). This biphasic MAP shortening was observed in all four WT hearts tested. In contrast, only monophasic MAP shortening was observed in AAA-TG hearts (n=4). The early phase was absent, and only the second markedly rapid MAP shortening (Fig. 1A; 3) occurred at the same time as the rapid phase of MAP shortening observed in WT hearts.
Fig. 1. Early MAP shorting induced by hypoxia is abolished in AAA-TG mice.

A. A representative time course of MAP duration during hypoxia. In WT hearts, removal of O2 from the perfusate (application of N2;
 ) leads to an early, presumably IK1-dependent, abbreviation of MAP (2;
) leads to an early, presumably IK1-dependent, abbreviation of MAP (2;
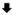 ) followed by a further rapid MAP shortening (3;
), presumably due to the activation of KATP channels. The time for early MAP shortening was defined as the time when MAPD75 falls by ∼10% of the Ctrl value measured just before application of N2 (1;
). The early phase of MAP shortening is absent in AAA-TG hearts, and only the rapid phase is observed, which starts at the same time as the KATP-dependent phase of MAP shortening in WT hearts (3;
). B. Representative MAPs from WT (gray) and AAA-TG (black) hearts recorded at the times as indicated in A. MAP amplitudes were normalized to each other to highlight the differences in shape. C, D. MAPD75 measured at the times as indicated in A. At the time when the early MAP shortening has just begun in AAA-TG hearts (3), in WT hearts MAPD75 has been already reduced by 28%. Statistical significance is indicated compared to MAPD75 before application of N2 (1). D. The time delay (Dt) between the application of N2 and the time when MAPD75 is decreased by 10% in WT and AAA-TG hearts. n=4 for both WT AAA-TG hearts.
) followed by a further rapid MAP shortening (3;
), presumably due to the activation of KATP channels. The time for early MAP shortening was defined as the time when MAPD75 falls by ∼10% of the Ctrl value measured just before application of N2 (1;
). The early phase of MAP shortening is absent in AAA-TG hearts, and only the rapid phase is observed, which starts at the same time as the KATP-dependent phase of MAP shortening in WT hearts (3;
). B. Representative MAPs from WT (gray) and AAA-TG (black) hearts recorded at the times as indicated in A. MAP amplitudes were normalized to each other to highlight the differences in shape. C, D. MAPD75 measured at the times as indicated in A. At the time when the early MAP shortening has just begun in AAA-TG hearts (3), in WT hearts MAPD75 has been already reduced by 28%. Statistical significance is indicated compared to MAPD75 before application of N2 (1). D. The time delay (Dt) between the application of N2 and the time when MAPD75 is decreased by 10% in WT and AAA-TG hearts. n=4 for both WT AAA-TG hearts.
Fig. 2. CN-induced early AP shortening is abolished in isolated myocytes with suppressed IK1.

Examples of APs in WT (A) and AAA-TG (B) myocytes. A. In WT cells, 10 μM glybenclamide (Gb) does not significantly affect AP. Application of 1 mM CN in the presence of Gb leads to AP shortening within ∼3 min. Further addition of 100 μM Ba2+ completely reverses the effect of CN. (Insert) AP shortening caused by 100 μM pinacidil is not blocked by 100 μM Ba2+. B. Application of CN, Gb or Ba2+ exerts no effect on the AP in AAA-TG myocytes within the same period of time (∼3-5 min). C. Summarized data show that in WT myocytes 10 μM Gb has virtually no effect on APD90 while 1 mM CN reduces APD90 by nearly 30% within 3 min (n=10). Further application of 100 μM Ba2+ increases APD90 by ∼17% above the control level (n=10). In AAA-TG myocytes with suppressed IK1, CN (3 min application), Gb or Ba2+ have no affect on APD90 (n=7). For each of the three conditions, a relative change in APD90 values is presented; therefore a dashed line at 100% represents control data.
Fig. 3. Time course of the CN-induced current at −60 mV in WT myocytes.

An example of the time course of the whole-cell current amplitude measured at a −60 mV membrane potential during application of 1 mM CN at 28 °C. (Insert) −60 mV roughly corresponds to the peak of outward IK1. (1) Currents are stable for a prolonged period of time until the application of CN. (2) CN leads to a quick increase in current amplitude which remains quasi-stable until (3) a sharp increase after >15 min due to activation of KATP channels.
Fig. 4. Application of CN leads to early upregulation of IK1 in WT myocytes.

A and B. Representative Ba2+-sensitive (A) and Ba2+-insensitive (B) currents recorded before and after a 3 min application of 1 mM CN at 28 °C. In A, the dotted trace represents a difference, or CN-activated, current. Currents were recorded in response to a 4 s voltage ramp from −100 mV to 10 mV (reduced voltage range is shown) in the absence and presence of 100 μM Ba2+. C. Current densities at −60 mV. Ba2+-sensitive currents are increased by ∼50%, and Ba2+-insensitive currents are not affected by CN treatment (n=11, p<0.05 vs Ctrl, respectively).
As we have shown previously, suppression of IK1 leads to a significant prolongation of AP in AAA-TG hearts [7]. Here, under control conditions, MAPD75 in AAA-TG hearts was larger by 44% when compared to WT hearts: 49.4±4.1 ms and 34.3±2.4 ms, respectively (p<0.005, n=4, (Fig. 1 C&D). Upon application of hypoxic solution, in WT hearts a 10% decrease in MAPD75 was observed 30.0±5.4 s after application of N2, while in AAA-TG hearts the MAPD75 decrease was significantly delayed to 53.8±7.5 s (p<0.05, n=4, Fig. 1E). This ∼24 s delay led to a situation such that at the time when the MAP shortening had just begun in AAA-TG hearts, MAPD75 has been already decreased by 28% in WT hearts (Fig. 1; comp. C and D).
Once the quick late phase of MAP shortening began, the rates of rapid decrease were not different between WT and AAA-TG hearts. After 150 s of hypoxia, MAPD75 reached small steady state values: 11.3±0.5 ms and 17.5±4.2 ms in WT and AAA-TG hearts, respectively (n=4 each; not statistically different). The above results strongly support the hypothesis that IK1 plays a significant role in the early stages of electrical response to metabolic stress.
3.2. Suppression of IK1 abolishes CN-induced early AP shortening in isolated myocytes
To further address the above hypothesis, the effects of metabolic stress on AP were studied in isolated myocytes from WT and AAA-TG hearts. Oxidative phosphorylation was blocked with 1mM CN in the presence of 10 mM glucose, and APs recorded using the current-clamp technique (T=28°C). For recordings of AP smaller pipettes were used allowing for higher quality gigaseals and minimal depolarization of membrane potential in AAA-TG myocytes essentially lacking stabilizing IK1 [7]. Specifically, the resting potentials were −72.7±0.4 (n=8) and −71.6±0.5 (n=7) in WT and AAA-TG myocytes, respectively (not significantly different; p>0.1). As shown in Fig. 2A, application of 10 μM glybenclamide, a potent blocker of KATP channels, did not significantly affect AP in WT myocytes, suggesting that KATP channels do not contribute to AP repolarization, and that the metabolic status of the myocytes is not affected by the isolation procedure. Addition of 1 mM CN caused significant AP shortening within 3-5 min, primarily in the range of membrane potentials negative to −40 mV (Fig. 2A), which corresponds to a slow phase of repolarization where the contribution of IK1 is most significant. Specifically, APD90 was decreased by 29.1±3.5% of the initial value (p<0.005, n=10, Fig. 2C). Further addition of 100 μM Ba2+, a specific blocker of IK1, not only completely reversed the effect of CN (Fig. 2A), but also increased APD90 by 16.9±8.3% compared to control (p<0.05, n=10, Fig. 2C). As expected, prolonged application of CN (>15-30 min) ultimately led to a sudden dramatic shortening of the AP followed by a complete loss of excitability due to activation of KATP channels (not shown).
To exclude the possibility of KATP channel contribution to early AP shortening, we took advantage of AAA-TG mice with suppressed IK1. In AAA-TG myocytes, CN had no effect on the AP within 5 min of treatment (n=7, Fig. 2B,C), and neither glybenclamide nor Ba2+ affected the AP within the same period of time (5 min). As with WT myocytes, in AAA-TG cells, AP shortening caused by the opening of KATP channels ultimately occurred after 15-30 min of treatment with CN (not shown).
3.3. The early K+ current underlying CN-induced AP shortening is IK1
All together, the above data show that the 29% reduction in APD90 induced by CN (Fig. 2A,C) is due to the activation of IK1. To directly test this hypothesis we have characterized the properties of the early CN-activated current in the range of hyperpolarized potentials where the AP is primarily affected, i.e. negative to −40 mV (Fig. 2).
Fig. 3 shows a typical time course of the development of the whole-cell current at −60 mV (a membrane potential close to the peak of outward IK1) during CN treatment. Application of 1 mM CN led to a fast current increase, reaching a quasi-steady state level within 3-5 min at 28 °C (Fig. 3; phase 2). After that time, currents remained stable for several minutes until a sudden manifold increase in conductance due to activation of KATP channels occurred (Fig. 3; phase 3). While in all experiments the early effects of CN (phase 2) occurred within 3-5 minutes, the time delay before the activation of KATP channels varied significantly, ranging from 15 min to more than 30 min (was not quantified). In the initial control experiments in the absence of CN, currents remained stable for an extended period of time (>30 min).
It should be noted that the time course of response to metabolic inhibition was faster in experiments with isolated hearts (Fig. 1A) than that observed in isolated cells (Fig. 3). We think that the differences in (a) temperature − 37 °C vs 28 °C, and (b) pacing rates − 150 ms BCL vs none, in isolated hearts and single cells, respectively, contributed most to the observed discrepancy.
The relative stability of phase 2 provided us with the opportunity to characterize the early CN-activated current in more detail (Fig. 4). Currents were recorded in response to a slow voltage ramp in the absence and presence of 100 μM Ba2+. Fig. 4A shows that application of 1 mM CN for ∼3 min leads to an increase in Ba2+-sensitive current in both the inward and outward direction, negative and positive to the reversal potential, respectively. At −60 mV the amplitude of outward current was increased from 0.74±0.07 pA/pF to 1.11±0.08 pA/pF (p<0.05, n=11, Fig. 4C), a 50% increase. In contrast, no significant changes were observed in the Ba2+-insensitive component of the CN-activated current (Fig. 4B&C). Importantly, the Ba2+-sensitive difference current displayed strong inward rectification, undoubtedly confirming it to be IK1.
Application of CN had no effect on currents in transgenic AAA-TG myocytes (Fig. 4C). We have shown previously that in AAA-TG myocytes (identified using the intense green fluorescence of Kir2.1-AAA-GFP transgene) IK1 is suppressed >95% and, therefore, small remaining currents consist primarily of minor leak and other minor ion channel or pump currents (not characterized) [7]. Nevertheless, we have further confirmed essential suppression of IK1 in the current study. Currents measured in AAA-TG myocytes at −60 mV before and after application of 100 μM Ba2+ were indistinguishable: −0.59±0.04 pA/pF and −0.56±0.04 pA/pF, respectively (n=6).
Similar to experiments with AP, KATP channels were finally activated after >15 min of CN perfusion in both WT and AAA-TG myocytes (n>10, for each type), leading to a linearized current-voltage relationship with high slope conductance (not shown).
3.4. IK1 is activated during true hypoxia
To exclude the possibility that the early activation of IK1 is simply due to peculiar effects of CN, we have carried out experiments using true hypoxic conditions. Specifically, whole-cell currents were measured in WT myocytes during application of nominally O2-free (N2 saturated) solution in the presence of 10 mM glucose (see Methods). Similar to the effect of CN, hypoxia also activated a strongly inwardly rectifying current (Fig. 5), although the time for the current amplitude to reach a quasi steady state level was somewhat delayed compared to CN treatment (4-6 min vs 3-5 min, respectively). Specifically, at −60 mV the Ba2+-sensitive current (IK1) was increased from 0.56±0.06 pA/pF to 0.89±0.12 pA/pF, a nearly 60% increase (p<0.05, n=4, Fig. 5), and no significant change was observed in Ba2+-insensitive currents (not shown). Again, as expected, prolonged (15-30 min) perfusion with O2-free solution ultimately led to activation of KATP channels (not shown). Thus the data strongly support the hypothesis that early AP shortening originally observed in isolated hearts (Fig. 1) is indeed due to an increase in IK1.
Fig. 5. True hypoxia leads to early activation of IK1.

A. Representative Ba2+-sensitive current traces recorded in a WT myocyte before and after a 4 min application of nominally O2-free solution (N2) at 28 °C. The current activated by hypoxia displays strong inward rectification, confirming it is IK1. B. At −60 mV the Ba2+-sensitive current is increased by 60% when compared to control (n=4, p<0.05).
3.5. KATP channels are not involved in early AP shortening
In WT myocytes CN-induced early AP shortening might still be caused by activation of KATP channels, which can potentially be affected at the concentration of Ba2+ used to block IK1 in the above experiments. The data in Fig. 2A (insert) exclude this possibility since 100 μM Ba2+ does not reverse AP shortening caused by 100 μM pinacidil, a potent KATP channel opener (p<0.05, n=6, Fig. 2A; insert).
Although experiments with AAA-TG myocytes provide the most compelling evidence that IK1, not KATP channels, underlie early AP shortening, the conclusions are based on the assumption that KATP channels are not affected by transgenic manipulation of IK1. For example, it may be argued that overexpression of mutant Kir2.1-AAA subunits in AAA-TG hearts may significantly downregulate native KATP channels by some unknown mechanism, thus explaining a lack of the effect of CN in AAA-TG myocytes. The data in Fig. 6 argue against this possibility. First, we compared the density of KATP channels using inside-out patches excised from WT and AAA-TG myocytes. Although a 25% decrease in KATP channel density was indeed observed in AAA-TG myocytes compared to WT cells (p<0.01; n=91 and n=57, respectively), the effect was clearly too small to account for the inefficiency of CN to cause early AP shortening in these cells. Additionally, the sensitivity of KATP channels to ATP (K1/2) was not affected in AAA-TG myocytes (Fig. 6): 37.8±1.7 μM (n=91) and 38±2.4 μM (n=57) in WT and AAA-TG myocytes, respectively. Therefore, the data strongly suggest that remodeling of KATP channels does not underlie the differential response of WT and AAA-TG myocytes to CN application.
Fig. 6. KATP channels in WT and AAA-TG myocytes.

K+ currents were measured at +50 mV in high symmetrical K+ using excised membrane patches, and KATP current amplitudes calculated as a difference between currents in 0 and 1 mM ATP. A. In AAA-TG myocytes, KATP current density (per patch) is reduced by ∼25% compared to that in WT cells (p<0.01). B. A representative example of an ATP dose-response relationship in WT cells obtained under the same conditions as above. C. Normalized IKATP (IREL) from individual patches were fit with a Hill equation (insert) to estimate the slope (h) and concentration of half maximum block (K1/2). The data at each concentration show only a minor variation, thus standard error (SE) bars are too small compared to symbols. Neither the Hill coefficient (h) nor the sensitivity to ATP (K1/2) are affected in any significant way in AAA-TG myocytes, therefore, for visualization purpose, the averaged data were fit again with the Hill equation and the fit is plotted as indicated (continuous line). n=91 and n=57 for WT and AAA-TG cells, respectively, in all experiments.
3.6 ICa and INCX are not changed in AAA-TG myocytes
ICa and INCX also contribute to AP repolarization. In order to check whether the remodeling of these currents is responsible for a lack of AP shortening in AAA-TG myocytes, we compared the current densities of ICa and INCX in WT and AAA-TG myocytes.
As shown in Fig. 7A, neither the peak amplitude of ICa nor its voltage dependence are affected in any significant way in AAA-TG myocytes, thus ruling out the contribution of ICa to early AP shortening.
Fig. 7. ICa and INCX in WT and AAA-TG myocytes.

A. (1) Representative Ca2+ currents in WT myocytes in response to a voltage protocol shown in (2). (3) Current-voltage relationships for peak ICa in WT and AAA-TG myocytes. There is no significant difference between the two groups (n=11, n=10, respectively). B. (1) Representative background currents in WT myocytes in response to voltage steps to potentials between −80 mV and +80 mV. (2) and (3) Current-voltage relationships for total, Ni2+-insensitive and Ni2+-sensitive background currents in WT and AAA-TG myocytes measured at the end of voltage steps. There is no significant difference between any corresponding currents in the two groups (n=14, n=9, respectively). See Methods for solution composition.
Similarly, the data in Fig. 7B show that both the Ni2+-sensitive current (putative NCX), and the total current recorded in the presence of extracellular Ca2+ are also not significantly different between WT (n=14) and AAA-TG (n=9) myocytes. Therefore, the contribution of NCX to early AP shortening can also be excluded.
3.7 SR Ca2+ release is involved in the upregulation of IK1 by hypoxia
It has been shown that a significant increase in intracellular Ca2+ (Ca2+i) during hypoxia may occur rather early, well before myocyte shortening [1]. Therefore, we sought to determine whether the early activation of IK1 is linked to changes in Ca2+i.
Since the Ba2+-insensitive current is not affected by CN (Fig. 4), changes in the total current represent changes in IK1. Therefore, to quantify the effect of CN we measured only the total outward current density at −60 mV.
As shown in Fig. 8, in the presence of 2 mM EGTA and 200 μM Ca2+ (pCa∼8.0) in the pipette solution, the total outward currents were increased from 0.33±0.05 pA/pF to 0.54±0.08 pA/pF (p<0.05, n=11). Elevating Ca2+ pCa∼5.0 did not significantly influence the effect of CN on total currents: 0.23±0.06 pA/pF and 0.60±0.10 pA/pF (p<0.05, n=4) before and after CN application, respectively. In contrast, buffering of Ca2+i with 20 mM BAPTA (nominally zero Ca2+; estimated pCai>12) prevented CN-induced activation of IK1 (n=6), strongly suggesting that Ca2+i signaling is involved in the activation of IK1 by hypoxia.
Fig. 8. SR Ca2+ release is involved in the activation of IK1 during hypoxia.

A. Strong intracellular Ca2+ buffering abolishes activation of IK1 by CN. Current densities at −60 mV were measured before and after application of 1 mM CN (∼3 min) using different concentrations of Ca2+ and types of Ca2+ buffer in the pipette solution (n=4, 11 and 6 from left to right). pCai values were calculated using the following concentrations of Ca2+-binding components and Ca2+. pCa2+i ∼5 − 5 mM ATP, 2 mM EGTA 190 μM Ca2+; pCa2+i ∼8 − 5 mM ATP, 170 μM Ca2+; pCa2+i >12 − 5 mM ATP, 20 mM BAPTA, 0.1 μM Ca2+; pH 7.3, T=25 °C. Calculations were performed using WinMaxc programs, http://www.stanford.edu/∼cpatton/maxc.html. B. Removal of extracellular Ca2+o does not affect the activation of IK1 by CN (n=6). In contrast, depletion of Ca2+ stores by extracellular application of 10 mM caffeine (Caff) plus 10 μM ryanodine (Rya) or the inhibition of Ca2+ release by 10 μM ruthenium red (RR; included into pipette solution) abolishes IK1 activation (n=7 and 5, respectively).
We then investigated what the potential source of Ca2+i could be. In the absence of Ca2+ in the bath solution (containing 0.5 mM EGTA), CN still caused an increase in the total currents from 0.27±0.04 pA/pF to 0.58±0.05 pA/pF (Fig. 8B, p<0.05, n=6), further excluding ICa in the early activation of IK1. Surprisingly, the effect of CN was abolished after the myocytes were briefly treated (2-3 min) with 10 mM caffeine and 10 μM ryanodine to deplete Ca2+i stores (Fig. 8B, n=7). Similarly, inclusion of 10 μM ruthenium red, an inhibitor of Ca2+ release, in the pipette solution also blocked the increase of IK1 by CN (Fig. 8B, n=5), strongly arguing that SR Ca2+ release is involved in the activation of IK1 by hypoxia.
4. Discussion
This study uncovers the mechanism of early AP shortening during metabolic stress. Specifically, the data clearly show that in the mouse heart early AP shortening induced by CN poisoning or true hypoxia is mediated by a significant increase in IK1, preceding the activation of KATP channels.
4.1. Can IK1 compete with KATP?
Since the discovery of an ATP-dependent K+ channel by Noma (1983) [1], a wealth of experimental data has been collected which undoubtedly implicate these channels as critical players in the response of cardiac muscle to metabolic challenges [10]. The power of KATP channel activation originates from a high density of these channels in the sarcolemmal membrane, such that the opening of less than 1% of them is sufficient to significantly abbreviate the AP [11], while their full activation leads to complete loss of excitability.
Importantly, 1% of KATP activation mentioned above corresponds to only a few nS of conductance [11], while the conductance of IK1, a major constitutively active K+ conductance, is estimated to be about 10-15 nS (at the resting membrane potential; mouse myocytes [12]). Thus, potential changes in IK1 during metabolic stress may lead to effects comparable to what is considered significant in the KATP arena, and would even dominate over KATP channels during the early phase of stress.
4.2. Previous evidence for IK1 activation by hypoxia
There are several studies which point to the involvement of IK1 in the electrical response of the heart to metabolic stress, but the biggest challenge in all of them was to differentiate between the effects produced by KATP and other ion channels. For example, Ruiz-Petrich (1991) [3] showed that in the rabbit heart the acceleration of final AP repolarization during hypoxia is Ba2+ sensitive, implicating activation of IK1. A number of follow-up studies also suggest that channels other than KATP may contribute to early AP shortening [13-15]. Specific conclusions, however, were largely based on the efficiency of KATP specific agents, such as glybenclamide, to block AP shortening caused by metabolic inhibition. The latter should be taken with caution, including this study, since it has been long established that the efficacy of glybenclamide, and other relevant drugs, may be strongly compromised by prolonged metabolic inhibition [16].
Nevertheless, direct evidence for IK1 activation by hypoxia are available. For example, Muramatsu et al (1990) [4] showed using whole-cell current measurements in guinea-pig myocytes that CN poisoning leads to early activation of a K+ current, long preceding KATP channel activation, which displays strong inward rectification similar to IK1. Also, Park et al (2005) [6] recently showed that in rabbit arterial smooth muscle cells, 2 min of hypoxia activated Kir2.1 channels, which are believed to be the major isoform underlying IK1 in mouse cardiac myocytes.
4.3. Activation of IK1 underlies early AP shortening during hypoxia
In this study we took advantage of TG mice with suppressed IK1 to overcome a number of difficulties related to the limited efficiency of a pharmacological approach. The original support for early activation of IK1 comes from experiments with isolated hearts. A delayed shortening of the MAP in AAA-TG hearts lacking IK1 during hypoxia is strongly indicative of early activation of IK1 in WT hearts (Fig. 1), but leaves a lot of room for other explanations.
On the contrary, results of experiments with isolated cells are strongly conclusive.
The first result is that the early AP shortening induced by CN poisoning in WT myocytes is completely eliminated in AAA-TG myocytes (Fig. 2B).
Second, glybenclamide could not prevent early AP shortening, while Ba2+ was efficient in doing so. Therefore, the early activation of KATP channels can be largely ruled out. However, as mentioned above, the efficiency of glybenclamide has to be treated with caution in these particular experiments. Additionally, in this study we also show that KATP channel density and ATP sensitivity is essentially unaffected in AAA-TG hearts (Fig. 6).
Experiments with Ba2+ ions also deserve attention. It is known that KATP channels are significantly less sensitive to block by extracellular Ba2+. For example, the Kd (0 mV) value for Ba2+ block of the least sensitive Kir2.1 channels is 62 μM [17], while the Kd (0 mV) value for KATP channels is in the mM range (1.18 mM [18]; 3.7 mM [19]). Nevertheless, Ba2+ block of KATP channels possesses a stronger voltage dependence, therefore complicating the estimation of Ba2+ block at hyperpolarized potentials, where most of the AP shortening occurs. In order to experimentally exclude the possibility of KATP involvement and to avoid complex calculations of potential block, we show that pinacidil-induced AP shortening (Fig. 2A; Insert) is not affected by 100 μM Ba2+.
Importantly, in contrast to ischemia, the level of high energy phosphates may not be significantly affected during early stages of hypoxia. For example, Allen et al (1985) [20] showed that in the ferret heart neither ATP nor ADP levels are affected within a few minutes of hypoxia, again arguing against KATP channel activation.
The third, probably most compelling evidence, come from the direct measurements of an early activated K+ current in WT myocytes. The mere fact that the early-activated K+ current displays strong inward rectification undoubtedly identifies it as IK1. In addition, this early-activated K+ current is strongly Ba2+-sensitive, with no effect of CN being observed in the Ba2+-insensitive component, thus further eliminating the contribution of KATP channels to this phenomenon (Fig. 4).
Also important, the early activated IK1 is not the peculiar result of poisoning by CN, since the same outcome is observed in myocytes exposed to true hypoxic conditions (Fig. 5).
It should be noted that since the experiments with isolated myocytes were carried out in the whole-cell configuration (T=28 °C), allowing for partial intracellular dialysis of small molecules, the magnitudes of AP shortening and IK1 activation may be different from that occurring in intact hearts. Nevertheless, even intracellular dialysis with solutions containing moderate concentrations of common buffers such as 2 mM EGTA and 10 mM HEPES cannot prevent changes in the intracellular environment caused by CN or hypoxia, suggesting that the effects observed in isolated cells may be even larger in intact hearts.
Whether IK1, but not KATP, channels are responsible for early AP shortening in other species or experimental models remains highly controversial. In particular, it has been shown that in the guinea-pig heart, selective blockade of KATP channels by HMR 1098 eliminates APD shortening in a low flow ischemia model, a condition similar to hypoxia in the current experiments [21]. In contrast, it has been shown that in the rabbit heart, glybenclamide had no effect on AP shortening up to 25 min of hypoxia [22]. There is more controversy with the data obtained in rodents. One of the striking findings is that in rat cardiac myocytes HMR 1098 does not block KATP channels or reverse AP shortening caused by metabolic inhibition (CN+ iodoacetic acid, no glucose) [23]. Irrespective of the mechanism of low efficiency of HMR 1098 in the above study, it shows that the data cannot be easily extrapolated from one species to another, or even from one specific technique to a similar one.
4.4. Are other channels or pumps involved in early AP shortening?
Most of the changes in AP occurred at potentials negative to −40 mV where IK1 channels are most active. This strongly limits a number of other potential players, but does not completely rule them out and make IK1 exclusive. For example, stress-induced changes in time-dependent currents, such as a Ca2+ current, may potentially contribute to early AP shortening in a dynamic manner. The data in Fig. 2B, however, nearly eliminate this possibility since selective inhibition of IK1 in AAA-TG myocytes completely prevents early AP shortening.
Nevertheless, the validity of the AAA-TG model still relies on the assumption that other major contributors to AP repolarization are not affected by transgenic IK1 manipulation. Therefore, one needs to be sure that these other conductances are intact in AAA-TG myocytes.
The data in Fig. 7A clearly show that there is no remodeling of the Ca2+ current in AAA-TG myocytes, further confirming that Ca2+ channels are not involved in early AP shortening. Time-independent currents such as that produced, for example, by the Na+/Ca2+ exchanger, or other uncharacterized conductances, can be largely excluded based on the results in Fig. 4, which show activation of only the Ba2+-sensitive IK1. Additionally, the data in Fig. 7B show that neither the total time independent current nor the Ni2+-insensitive (Na+/Ca2+ exchanger) conductance are remodeled in the AAA-TG mouse, leaving IK1 as the exclusive player in early AP shortening during hypoxia. Importantly, the magnitude of early AP shortening and IK1 upregulation are in a quantitative agreement sufficient to explain most of the results.
4.5 SR Ca2+ release is involved in the activation of IK1 by hypoxia
This study deals primarily with the cellular mechanism of early AP shortening, and only limited data are available with regard to the potential molecular mechanisms.
For example, while the results in Fig. 8B exclude the role of extracellular Ca2+ in IK1 activation, we found that Ca2+i plays a significant role. In particular, a buffering of Ca2+i with 20 mM BAPTA or pharmacological inhibition of SR release completely abolished the effect of CN on IK1 (Fig. 8), suggesting an essential role of SR in the phenomenon.
Consistent with our data, Dipp et al (2001) [24] have shown that in pulmonary smooth muscle cells hypoxia leads to a release of Ca2+ from the SR without affecting the Ca2+ influx from extracellular solution.
It may seem paradoxical that an increase in Ca2+i leads to IK1 activation since it has been found that Ca2+i blocks Kir channels in a voltage dependent manner in ventricular myocytes [25]. However, the blocking concentrations of Ca2+i are too high to have any physiological significance, with Ca2+ block being even less potent than that of Mg2+ [25], and therefore, other mechanisms of Ca2+i-dependent regulation of IK1 will have to be considered in future experiments.
4.6. Other potential mechanisms of IK1 activation
It is established, for example, that the activity of Kir channels is strongly dependent on the level of membrane PIP2 [26] and on their phosphorylation state [27], parameters which can easily be affected early in hypoxia. Nevertheless, involvement of free intracellular PAs, the major inwardly rectifying factors, seems more realistic [28]. This hypothesis is based on the facts that (i) IK1 channels are only partially blocked by PAs, allowing for a significant up or down-regulation, and (ii) the concentration of free, active PAs is determined by the intracellular concentrations of their numerous binding partners [29]. For example, the PAs are part of an ATP-Mg-PA complex [30, 31], and inorganic phosphates are also known chelating agents for PAs. Therefore, changes in the concentration of free PAs, and thus changes in IK1, can be as quick and versatile as that of their binding components. Consistent with this, it has been shown that in the rabbit heart, hypoxia leads to a manifold increase in the concentration of free inorganic phosphates in less than 3 minutes [20], which inevitably should affect the balance of free PAs, and thus affect IK1 rectification, within the same fast time frame.
Intracellular pH seems to be another key parameter involved in upregulation of IK1 by CN [32] or hypoxia, although a pH-dependent mechanism is seemingly against the common view that (i) metabolic inhibition leads to intracellular acidification, and (ii) Kir2 channels are inhibited by low intracellular pH. This contradiction, however, is superficial, since ischemia and hypoxia, especially early hypoxia, are quite different processes. For example, the fact that CN application causes large reductions in intracellular pH is not a universal fact in all animals, and is certainly not a universal fact for different conditions. For example, Allen et al (1985) [33] showed that in the ferret heart, the intracellular pH is actually transiently increased, not decreased, after only 3 min of hypoxia without any change in the level of intracellular ATP/ADP. Eisner et al (1989) [34] observed a similar phenomenon in isolated rat myocytes by mimicking hypoxia with application of CN in the presence of glucose, as in our experiments.
Interestingly, alkalinization leads to deprotonation of PAs and relief of block at depolarized potentials [35], which would be consistent with a PA-dependent mechanism of IK1 activation. In addition, increase in pH is shown to increase binding of PA to ATP-Mg (and other nucleotides) complex [31], which would lead to a decrease in free PAs and thus increase in IK1.
4.7. Physiological significance
The significance of IK1 activation in arrhythmogenesis mirrors in many aspects that of KATP channels, although the results of this study place IK1 as the first responder to hypoxic stress. For example, it has been shown in a number of animal models that arrhythmogenic consequences of metabolic stress, a condition known to activate KATP channels, can be prevented by glybenclamide, a potent KATP channel blocker [36, 37]. Alternatively, activation of KATP channels by K+ channel openers has been shown to have a proarrhythmic effect [2].
In line with the above, upregulation of IK1 in TG mice also leads to multiple abnormalities in excitability [12], and provides a substrate for highly stable reentrant arrhythmias [38]. Consistent with this, it has been shown that in the guinea-pig heart, blockade of IK1 by Ba2+ ions can terminate induced ventricular fibrillation [39].
In humans, a number of gain-of-function mutations in the Kir2.1 gene were recently discovered to be the cause of Short QT Syndrome (SQT3) [40] and familial atrial fibrillation [41]. Thus, early in hypoxia, upregulation of IK1 may underlie a significant proarrhythmic potential.
Importantly, hypoxia and low-flow ischemia, but not complete ischemia, are the most common conditions experienced by the heart. In ∼90% of cases of sudden cardiac death, coronary arteries are narrowed but not completely blocked. Since even in the normally beating heart the extraction of oxygen is very high, such that 65-70% of it is removed from the blood as it passes through the heart, this may lead to frequent episodes of acute hypoxia/low-flow ischemia due to, for example, release of adrenaline during increased physical activity, which often acts as a trigger for sudden death. In fact, although ischemic conditions are clinically relevant, in most patients with an acute coronary occlusion, myocardial flow in the infarct region is not null but typically ∼15% of normal [42], a condition best mimicked by low-flow ischemia and hypoxia. Therefore, early hypoxia-induced activation of IK1 may potentially underlie a significant number of unexplained sudden cardiac deaths. If true, understanding the role of IK1 in excitability may be key to new ways of prevention and treatment of deadly arrhythmias, the most devastating cardiac condition in the world.
In conclusion, our studies show that in the mouse heart early AP shortening during hypoxia is mediated by activation of IK1.
Acknowledgments
This study was supported by a RO1 HL69052 grant from NHBLI. We would like to thank Brian Panama for critically reading this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–8. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- 2.Fischbach PS, White A, Barrett TD, Lucchesi BR. Risk of ventricular proarrhythmia with selective opening of the myocardial sarcolemmal versus mitochondrial ATP-gated potassium channel. J Pharmacol Exp Ther. 2004;309:554–9. doi: 10.1124/jpet.103.060780. [DOI] [PubMed] [Google Scholar]
- 3.Ruiz-Petrich E, de Lorenzi F, Chartier D. Role of the inward rectifier IK1 in the myocardial response to hypoxia. Cardiovasc Res. 1991;25:17–26. doi: 10.1093/cvr/25.1.17. [DOI] [PubMed] [Google Scholar]
- 4.Muramatsu H, Sato R, Okumura H. Early increase in K+ conductance during metabolic inhibition by cyanide in guinea pig ventricular myocytes. Nippon Ika Daigaku Zasshi. 1990;57:308–21. doi: 10.1272/jnms1923.57.308. [DOI] [PubMed] [Google Scholar]
- 5.Dobrev D, Wettwer E, Kortner A, Knaut M, Schuler S, Ravens U. Human inward rectifier potassium channels in chronic and postoperative atrial fibrillation. Cardiovasc Res. 2002;54:397–404. doi: 10.1016/s0008-6363(01)00555-7. [DOI] [PubMed] [Google Scholar]
- 6.Park WS, Han J, Kim N, Ko JH, Kim SJ, Earm YE. Activation of inward rectifier K+ channels by hypoxia in rabbit coronary arterial smooth muscle cells. Am J Physiol Heart Circ Physiol. 2005;289:H2461–7. doi: 10.1152/ajpheart.00331.2005. [DOI] [PubMed] [Google Scholar]
- 7.McLerie M, Lopatin AN. Dominant-negative suppression of I(K1) in the mouse heart leads to altered cardiac excitability. J Mol Cell Cardiol. 2003;35:367–78. doi: 10.1016/s0022-2828(03)00014-2. [DOI] [PubMed] [Google Scholar]
- 8.Knollmann BC, Katchman AN, Franz MR. Monophasic action potential recordings from intact mouse heart: validation, regional heterogeneity, and relation to refractoriness. J Cardiovasc Electrophysiol. 2001;12:1286–94. doi: 10.1046/j.1540-8167.2001.01286.x. [DOI] [PubMed] [Google Scholar]
- 9.Franz MR. Current status of monophasic action potential recording: theories, measurements and interpretations. Cardiovasc Res. 1999;41:25–40. doi: 10.1016/s0008-6363(98)00268-5. [DOI] [PubMed] [Google Scholar]
- 10.Nichols CG, Lederer WJ. Adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Am J Physiol. 1991;261:H1675–H1686. doi: 10.1152/ajpheart.1991.261.6.H1675. [DOI] [PubMed] [Google Scholar]
- 11.Nichols CG, Ripoll C, Lederer WJ. ATP-sensitive potassium channel modulation of the guinea pig ventricular action potential and contraction. Circ Res. 1991;68:280–7. doi: 10.1161/01.res.68.1.280. [DOI] [PubMed] [Google Scholar]
- 12.Li J, McLerie M, Lopatin AN. Transgenic Up-Regulation of IK1 in The Mouse Heart Leads to Multiple Abnormalities of Cardiac Excitability. Am J Physiol Heart Circ Physiol. 2004;287:H2790–802. doi: 10.1152/ajpheart.00114.2004. [DOI] [PubMed] [Google Scholar]
- 13.Ruiz Petrich E, Leblanc N, deLorenzi F, Allard Y, Schanne OF. Effects of K+ channel blockers on the action potential of hypoxic rabbit myocardium. Br J Pharmacol. 1992;106:924–30. doi: 10.1111/j.1476-5381.1992.tb14436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruiz Petrich E, Ponce Zumino A, Schanne OF. Early action potential shortening in hypoxic hearts: role of chloride current(s) mediated by catecholamine release. J Mol Cell Cardiol. 1996;28:279–90. doi: 10.1006/jmcc.1996.0026. [DOI] [PubMed] [Google Scholar]
- 15.Baiardi G, Zumino AP, Petrich ER. Effects of barium and 5-hydroxydecanoate on the electrophysiologic response to acute regional ischemia and reperfusion in rat hearts. Mol Cell Biochem. 2003;254:185–91. doi: 10.1023/a:1027384215339. [DOI] [PubMed] [Google Scholar]
- 16.Findlay I. Sulphonylurea drugs no longer inhibit ATP-sensitive K+ channels during metabolic stress in cardiac muscle. J Pharmacol Exp Ther. 1993;266:456–67. [PubMed] [Google Scholar]
- 17.Shieh RC, Chang JC, Arreola J. Interaction of Ba2+ with the pores of the cloned inward rectifier K+ channels Kir2.1 expressed in Xenopus oocytes. Biophys J. 1998;75:2313–2322. doi: 10.1016/S0006-3495(98)77675-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quayle JM, Standen NB, Stanfield PR. The voltage-dependent block of ATP-sensitive potassium channels of frog skeletal muscle by caesium and barium ions. J Physiol. 1988;405:677–97. doi: 10.1113/jphysiol.1988.sp017355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takano M, Ashcroft FM. The Ba2+ block of the ATP-sensitive K+ current of mouse pancreatic beta-cells. Pflugers Arch. 1996;431:625–31. doi: 10.1007/BF02191912. [DOI] [PubMed] [Google Scholar]
- 20.Allen DG, Morris PG, Orchard CH, Pirolo JS. A nuclear magnetic resonance study of metabolism in the ferret heart during hypoxia and inhibition of glycolysis. J Physiol. 1985;361:185–204. doi: 10.1113/jphysiol.1985.sp015640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gögelein H, Englert HC, Kotzan A, Hack R. HMR 1098: An Inhibitor of Cardiac ATP-Sensitive Potassium Channels. Cardiovascular Drug Reviews. 2000;18:157–174. [Google Scholar]
- 22.de Lorenzi F, Cai S, Schanne OF, Ruiz Petrich E. Partial contribution of the ATP-sensitive K+ current to the effects of mild metabolic depression in rabbit myocardium. Mol Cell Biochem. 1994;132:133–43. doi: 10.1007/BF00926922. [DOI] [PubMed] [Google Scholar]
- 23.Rainbow RD, Norman RI, Hudman D, Davies NW, Standen NB. Reduced effectiveness of HMR 1098 in blocking cardiac sarcolemmal K(ATP) channels during metabolic stress. J Mol Cell Cardiol. 2005;39:637–46. doi: 10.1016/j.yjmcc.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 24.Dipp M, Nye PC, Evans AM. Hypoxic release of calcium from the sarcoplasmic reticulum of pulmonary artery smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2001;281:L318–25. doi: 10.1152/ajplung.2001.281.2.L318. [DOI] [PubMed] [Google Scholar]
- 25.Matsuda H, Cruz Jdos S. Voltage-dependent block by internal Ca2+ ions of inwardly rectifying K+ channels in guinea-pig ventricular cells. J Physiol. 1993;470:295–311. doi: 10.1113/jphysiol.1993.sp019859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 1998;391:803–6. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 27.Romanenko V, Fang Y, Byfield F, Travis AJ, Vandenberg C, Rothblat GH, et al. Cholesterol Sensitivity and Lipid Raft Targeting of Kir 2.1 Channels. Biophys J. 2004;87(6):3850–61. doi: 10.1529/biophysj.104.043273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994;372:366–369. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe S, Kusama-Eguchi K, Kobayashi H, Igarashi K. Estimation of polyamine binding to macromolecules and ATP in bovine lymphocytes and rat liver. J Biol Chem. 1991;266:20803–9. [PubMed] [Google Scholar]
- 30.Meksuriyen D, Fukuchi-Shimogori T, Tomitori H, Kashiwagi K, Toida T, Imanari T, et al. Formation of a complex containing ATP, Mg2+, and spermin. Structural evidence and biological significance. J Biol Chem. 1998;273:30939–44. doi: 10.1074/jbc.273.47.30939. [DOI] [PubMed] [Google Scholar]
- 31.Nakai C, Glinsmann W. Interactions between polyamines and nucleotides. Biochemistry. 1977;16:5636–41. doi: 10.1021/bi00644a039. [DOI] [PubMed] [Google Scholar]
- 32.Muramatsu H, Sato R, Okumura H. Early increase in K+ conductance during metabolic inhibition by cyanide in guinea pig ventricular myocytes. Nippon Ika Daigaku Zasshi. 1990;57:308–21. doi: 10.1272/jnms1923.57.308. [DOI] [PubMed] [Google Scholar]
- 33.Ellis D, Noireaud J. Intracellular pH in sheep Purkinje fibres and ferret papillary muscles during hypoxia and recovery. J Physiol. 1987;383:125–41. doi: 10.1113/jphysiol.1987.sp016400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eisner DA, Nichols CG, O'Neill SC, Smith GL, Valdeolmillos M. The effects of metabolic inhibition on intracellular calcium and pH in isolated rat ventricular cells. J Physiol. 1989;411:393–418. doi: 10.1113/jphysiol.1989.sp017580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo D, Lu Z. Mechanism of IRK1 channel block by intracellular polyamines. J Gen Physiol. 2000;115:799–814. doi: 10.1085/jgp.115.6.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolleben CD, Sanguinetti MC, Siegl PK. Influence of ATP-sensitive potassium channel modulators on ischemia-induced fibrillation in isolated rat hearts. J Mol Cell Cardiol. 1989;21:783–8. doi: 10.1016/0022-2828(89)90717-7. [DOI] [PubMed] [Google Scholar]
- 37.Gwilt M, Henderson CG, Orme J, Rourke JD. Effects of drugs on ventricular fibrillation and ischaemic K+ loss in a model of ischaemia in perfused guinea-pig hearts in vitro. Eur J Pharmacol. 1992;220:231–6. doi: 10.1016/0014-2999(92)90752-p. [DOI] [PubMed] [Google Scholar]
- 38.Noujaim SF, Pandit SV, Berenfeld O, Vikstrom K, Cerrone M, Mironov S, et al. Up-regulation of the inward rectifier K+ current (IK1) in the mouse heart accelerates and stabilizes rotors. J Physiol. 2007;578:315–326. doi: 10.1113/jphysiol.2006.121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warren M, Guha PK, Berenfeld O, Zaitsev A, Anumonwo JM, Dhamoon AS, et al. Blockade of the inward rectifying potassium current terminates ventricular fibrillation in the guinea pig heart. J Cardiovasc Electrophysiol. 2003;14:621–31. doi: 10.1046/j.1540-8167.2003.03006.x. [DOI] [PubMed] [Google Scholar]
- 40.Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, et al. A Novel Form of Short QT Syndrome (SQT3) Is Caused by a Mutation in the KCNJ2 Gene. Circ Res. 2005;96:703–4. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- 41.Xia M, Jin Q, Bendahhou S, He Y, Larroque MM, Chen Y, et al. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun. 2005;332:1012–9. doi: 10.1016/j.bbrc.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 42.Sabia PJ, Powers ER, Ragosta M, Sarembock IJ, Burwell LR, Kaul S. An association between collateral blood flow and myocardial viability in patients with recent myocardial infarction. N Engl J Med. 1992;327:1825–31. doi: 10.1056/NEJM199212243272601. [DOI] [PubMed] [Google Scholar]