

Abstract
We have explored genome-wide expression of genes related to glycobiology in exon 1 transgenic Huntington’s disease (HD) mice using a custom designed GLYCOv2 chip and Affymetrix microarray analyses. We validated, using quantitative real-time PCR, abnormal expression levels of genes encoding glycosyltransferases in the striatum of R6/1 transgenic mice, as well as in postmortem caudate from human HD patients. Many of these genes show differential regional expression within the CNS, as indicated by in situ hybridization analysis, suggesting region-specific regulation of this system in the brain. We further show disrupted patterns of glycolipids (acidic and neutral lipids) and/or ganglioside levels in both the forebrain of the R6/1 transgenic mice and caudate samples from human HD subjects. These findings reveal novel disruptions in glycolipid/ganglioside metabolic pathways in the pathology of HD and suggest that the development of new targets to restore glycosphingolipid balance may act to ameliorate some symptoms in HD.
Keywords: gangliosides, Huntington’s disease, gene expression, neurodegeneration, caudate, R6/1 transgenic mouse, human
Introduction
Huntington’s disease (HD) is a progressive neurodegenerative disorder characterized by motor, psychiatric, and cognitive disturbances. HD is a dominant genetic disorder caused by an expansion of a CAG trinucleotide repeat region in exon 1 of the HD gene resulting in an abnormally long stretch of glutamine residues in the N-terminal region of the encoded huntingtin protein (Huntington’s Disease Collaborative Research Group 1993). This polyglutamine expansion has been proposed to result in a novel toxic gain of function of the mutant protein (MacDonald and Gusella, 1996), which displays aberrant interactions with normal protein partners in vivo, and accumulate in neurons to form intranuclear inclusions (Ross, 2002). However, loss of function of wild-type huntingtin has also been implicated in pathology (Cattaneo et al., 2005). Huntingtin, a 3144 amino acid protein, exhibits widespread expression in both brain and peripheral tissues (DiFiglia et al., 1995). A core pathological feature of HD is severe atrophy and neurodegeneration of the striatum, and to a lesser extent, the cerebral cortex (Vonsattel et al., 1985). How the mutated protein causes specific neurodegeneration within the striatum, despite its ubiquitous expression, is unknown.
Several mechanisms have been proposed to explain the pathological effects of huntingtin mutation, such as impairment of protein folding and degradation, imbalance in calcium homeostasis, disruption in axonal transport and synaptic transmission, and transcriptional dysregulation (Ross, 2002). Additionally, disturbances in the expression of genes related to cholesterol and fatty acid metabolism have been identified in striatal cells by microarray analysis (Sipione et al., 2002) and, more recently, dysfunction in the cholesterol biosynthetic pathway was described in mice and cell culture models of HD (Valenza et al., 2005). Additionally, huntingtin has been shown to interact with acidic phospholipids at the plasma membrane (Kegel et al., 2005). These findings have instigated a renewed interest in lipid abnormalities in HD, as studies nearly 40 years ago, reported disrupted lipid and glycosphingolipid levels in brains of HD subjects. Abnormal lipid composition was shown in the striata of HD patients as early as 1967 (Borri et al., 1967), as well as an increase in cholesterol and a decrease in the glycolipid content of the cerebral cortex (Hooghwinkel et al., 1968). Alterations in ganglioside concentrations in the caudate and putamen of HD patients have also been reported (Bernheimer H., 1979, Higatsberger et al., 1981). It should be noted, however, that these early studies were conducted prior to the identification of the HD gene and criteria for HD diagnosis in these studies was not clarified. Disruption in ganglioside metabolism is associated with several neurodegenerative disorders (Kolter and Sandhoff, 2005), however no further investigations of lipids in HD have been reported.
In this study, we used microarray analysis to explore gene expression deficits associated with glycobiology in R6/1 transgenic mice. These mice express the N-terminal portion of human huntingtin, containing 115 CAG repeats, driven by the human huntingtin promoter (Mangiarini et al., 1996). We validate abnormal expression of the genes encoding glycosyltransferases involved in the synthesis of gangliosides in the striatum of the R6/1 HD transgenic mice and in postmortem caudate samples from human HD subjects. We further demonstrate disturbed levels of glycolipids in the CNS of R6/1 HD transgenic mice and human HD caudate. These findings indicate that deficits in glycolipid metabolism, primarily gangliosides, represent a previously unrecognized feature of the pathological processes in HD.
Materials and Methods
Animals
HD R6/1 transgenic mice (‘B6CBACa-Tg(HDexon1)61Gpb/J’), generated in the laboratory of Professor Gillian Bates (Mangiarini et al., 1996), which express exon 1 of human HD gene carrying 116 CAG repeats, were obtained from Jackson Laboratories (Bar Harbor, Maine, USA). The mice were bred using original R6/1 breeders obtained from Jackson Laboratories and reared in a colony at The Scripps Research Institute. At the age of 4 weeks, mice were genotyped according to the Jackson Laboratories’ protocol to determine which of them were hemizygous for the HD transgene and to verify CAG repeat length. All procedures were in strict accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Human samples
Human postmortem caudate samples were obtained from Harvard Brain Tissue Resource Center (McLean Hospital, Belmont, MA, USA). Caudate samples consisted of three HD cases neuropathologically evaluated as Vonsattel grade 3, denoting a loss of > 80 % of medium spiny neurons (Vonsattel and DiFiglia, 1998): a female 53 years old, postmortem interval (PMI)= 16.90 h and two males 46 and 54 years old, PMI= 22.25 h and 15.92 h,respectively, and three controls, a female 58 years old, PMI= 24.25 h and two males 49 and 55 years old, PMI= 27.13 h and 14.50 h, respectively. The pH values of the samples ranged from 6.1 to 6.5. All samples were stored at -70°C until use.
RNA preparations
Total RNA was prepared from the striata of symptomatic (> 6 months) HD transgenic mice and age-matched wild-type littermate controls using NucleoSpin RNA Kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instructions. Three independent sets of RNA were isolated from each time point using n = 2-4 mice (50:50 ratio of males: females) per experiment. RNA from human postmortem caudate was isolated from each subject using the Versagene RNA Purification System (GENTRA Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. All the samples were treated with DNaseI to eliminate genomic DNA contamination. RNA quantification was determined by spectrophotometer readings. The ratio of OD260/OD280 was used to evaluate the purity of the nucleic acid samples and the quality of the extracted total RNA was determined using agarose gel electrophoresis and checked with an Agilent Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). RNA yields were comparable across all diseased and control samples.
Microarray analysis
RNA from the 3 replicate preparations of wild-type and transgenic striata was labeled and hybridized to the MOE430 2.0 Affymetrix array or GLYCOv2 chip (Consortium for Functional Glycomics). Arrays were scanned using Affymetrix ScanArray 3000 and standard Affymetrix protocols as described previously (Lockhart et al., 1996) (protocol available at http://affymetrix.com). Present (P) and Absent (A) calls were determined with the Affymetrix algorithm (GCOS, Affymetrix, Santa Clara, CA, USA). Conditions were divided into two groups: HD and wild-type control. Probe sets were considered present overall for a given condition if they had been assigned a present call in at least two of the three replicate samples for that condition. The resulting lists identified as present in at least one of the two conditions (‘Present list’) totaled 20381 genes in the MOE430 2.0 array. Expression signal values were generated using the RMA algorithm (Irizarry et al., 2003), which models the performance of the perfect match probe sets using all chips from this study. This model is then used to calculate corrected expression values for all probe sets. (For more information, see http://www.stat.berkeley.edu/bolstad/RMAExpress/RMAExpress.html). Using the ‘Present list’ and their respective RMA expression signal values, hierarchical clustering by sample was performed with BRB-ArrayTools software (http://linus.nci.nih.gov/BRB-ArrayTools.html) using default settings for correlation and linkage metrics. Class comparisons were performed in BRB-ArrayTools to identify specific genes whose expression differed between the two groups. This class comparison test was conducted using a randomized variance model, a univariate threshold of 0.05, and a multivariate permutation-based false discovery rate calculation. The predicted proportion of false discoveries was preset at 10% and the confidence level was set at 80%, resulting in lists consisting of: HD vs. wild-type, 2905 genes for Affymetrix array and 30 genes for GLYCOv2 chip. From these lists, ganglioside metabolism-related genes were recognized using Affymetrix probe sets, identified from UniGene cluster ID numbers. For genes that were represented by more than one probe set, all probe sets showed similar patterns of gene expression regulation.
For comparison of gene expression changes observed in R6/1 mice to changes reported in human HD brains, the log 2 -transformed expression values reported by Hodges et al,. 2006 in Supplementary Table S1 were converted to Fold change= 2 M(Tables 1 and 2).
Table 1.
Genes showing altered expression levels in the R6/1 Huntington’s disease transgenic mice and in human HD caudate samples with grades 0-2, in comparison to wild-type/controls.
| Accession # | Category | Common Name | Fold change R6/2 striatum | Fold Change Human Caudate* |
|---|---|---|---|---|
| NM_011062 | Affymetrix housekeeping | Pdpk1 | 0.59 | 0.73 |
| NM_139134 | CBP:C-Type Lectin | Chodl | 0.95 | 0.97 |
| NM_010494 | CBP:I-Type Lectin | Icam2 | 0.89 | 1.42 |
| NM_018886 | CBP:S-Type Lectin | Lgals8 | 1.47 | 0.74 |
| NM_008549 | Glycan Degradation | Man2a1 | 0.64 | 1.43 |
| NM_028749 | Glycan Degradation | Npl | 0.72 | 1.91 |
| NM_010299 | Glycan Degradation | Gm2a | 0.66 | 1.23 |
| NM_010498 | Glycan Degradation | Ids | 1.50 | 0.63 |
| NM_008080 | Glycan-transferase | B4galnt1 | 0.65 | 1.14 |
| NM_173739 | Glycan-transferase | Galntl4 | 0.43 | A |
| NM_017377 | Glycan-transferase | B4galt2 | 0.52 | NC |
| NM_175383 | Glycan-transferase | B3gnt1 | 0.67 | 0.59 |
| NM_016888 | Glycan-transferase | B3gnt2 | 0.48 | A |
| NM_019578 | Glycan-transferase | Extl1 | 0.70 | NC |
| NM_145926 | Glycan-transferase | Mgat4b | 0.51 | 0.95 |
| NM_172948 | Glycan-transferase | Mgat5b | 0.55 | 0.84 |
| NM_178048 | Glycan-transferase | St3gal2 | 0.79 | A |
| NM_011375 | Glycan-transferase | St3gal5 | 0.69 | 0.83 |
| NM_012028 | Glycan-transferase | St6galnac5 | 0.81 | 0.62 |
| NM_011374 | Glycan-transferase | St8sia1 | A | 1.11 |
| NM_009181 | Glycan-transferase | St8sia2 | 0.42 | 0.93 |
| NM_009182 | Glycan-transferase | St8sia3 | 0.35 | 0.58 |
| NM_023850 | Glycan-transferase | Chst1 | 0.59 | 0.83 |
| NM_013822 | Notch pathway | Jag1 | 0.46 | 1.2 |
| NM_009142 | Chemokine | Cx3cl1 | 0.51 | 0.58 |
| NM_008107 | Growth Fact & Receptor | Gdf1 | 0.68 | 1.33 |
| NM_008046 | Growth Fact & Receptor | Fst | 0.62 | NC |
| NM_023653 | Growth Fact & Receptor | Wnt2 | 0.77 | NC |
| NM_016696 | Proteoglycan | Gpc1 | 0.72 | 0.86 |
Expression fold-changes represent the fold difference of geometric means between HD mice and wild-type controls at a p<0.05 significance level in either Affymetrix mouse 430 2.0 array or GLYCOv2 chip.
Changes for human caudate samples represent the fold difference of geometric means between HD mice and wild-type controls at a p<0.05 significance level, as reported in Supplementary Table S1 by Hodges et al. (Hodges et al., 2006). Accession numbers show Reference Sequence Collection IDs.
Table 2.
Functional annotation clustering of genes differentially expressed in R6/1 Huntington’s disease transgenic mice at a significance level of p<0.05
| Probe ID | Gene Description | Gene ID | Fold change R6/1 | Fold change Human* |
|---|---|---|---|---|
| Glycosyltransferases | ||||
| 1423305_at | exostoses (multiple)-like 1 | Extl1 | 0.776 | NC |
| 1434703_at | exostoses (multiple)-like 3 | Extl3 | 0.725 | 0.89 |
| 1421415_s_at | glucosaminyl (n-acetyl) transferase 2, i-branching enzyme | Gcnt2 | 0.661 | 0.94 |
| 1459522_s_at | glycogenin | Gyg1 | 1.314 | 1.22 |
| 1417435_at | like-glycosyltransferase | Large | 0.632 | 0.65 |
| 1424720_at | mannoside acetylglucosaminyltransferase 4, isoenzyme b | Mgat4b | 0.571 | 0.95 |
| 1428644_at | mannoside acetylglucosaminyltransferase 5 | Mgat5 | 0.418 | NC |
| 1434531_at | mannoside acetylglucosaminyltransferase 5, isoenzyme b | Mgat5b | 0.72 | 0.84 |
| 1451738_at | o-linked n-acetylglucosamine (glcnac) transferase | Ogt | 1.36 | 0.85 |
| 1437999_x_at | phosphatidylinositol glycan, class q | Pigq | 0.717 | NC |
| 1422502_at | poly (adp-ribose) polymerase family, member 1 | Parp1 | 0.776 | 1.10 |
| 1428931_a_at | poly (adp-ribose) polymerase family, member 6 | Parp6 | 0.627 | 1.39 |
| 1426618_a_at | protein o-linked mannose beta1,2-n-acetylglucosaminyltransferase | Pomgntl | 0.781 | 1.12 |
| 1460704_at | radical fringe gene homolog (drosophila) | Rfng | 0.799 | 1.05 |
| 1419063_at | udp galactosyltransferase 8a | Ugt8a | 0.425 | NC |
| 1418080_at | udp-gal:betaglcnac beta 1,4- galactosyltransferase, polypeptide 2 | B4galt2 | 0.568 | NC |
| 1434135_at | udp-galnac:betaglcnac beta 1,3-galactosaminyltransferase, polypeptide 2 | B3galnt2 | 0.738 | A |
| 1420852_a_at | udp-glcnac:betagal beta-1,3-n-acetylglucosaminyltransferase 1 | B3gnt1 | 0.495 | 0.59 |
| 1418195_at | udp-n-acetyl-alpha-d-galactosamine acetylgalactosaminyltransferase 10 | Galnt10 | 0.807 | 1.31 |
| 1454780_at | udp-n-acetyl-alpha-d-galactosamine acetylgalactosaminyltransferase-like 4 | Galntl4 | 0.489 | A |
| Ganglioside Biosynthesis / Sialyltransferases | ||||
| 1449468_at | sialyltransferase 7 | St6galnac5 | 0.806 | 0.62 |
| 1421891_at | st3 beta-galactoside alpha-2,3-sialyltransferase 2 | St3gal2 | 0.595 | 0.88 |
| 1449198_a_at | st3 beta-galactoside alpha-2,3-sialyltransferase 5 | St3gal5 | 0.688 | 0.83 |
| 1420377_at | st8 alpha-n-acetyl-neuraminide alpha-2,8-sialyltransferase 2 | St8sia2 | 0.419 | 0.93 |
| 1451008_at | st8 alpha-n-acetyl-neuraminide alpha-2,8-sialyltransferase 3 | St8sia3 | 0.324 | 0.58 |
| Sphingolipid Metabolism | ||||
| 1434034_at | ceramide kinase | Cerk | 0.793 | 1.07 |
| 1454078_a_at | galactose-3-o-sulfotransferase 1 | Gal3st1 | 0.758 | 1.05 |
| 1422341_s_at | lysophospholipase 3 | Lypla3 | 1.208 | NC |
| 1437513_a_at | serine incorporator 1 | Serinc1 | 1.383 | 0.81 |
| 1422779_at | sphingomyelin phosphodiesterase 3, neutral | Smpd3 | 0.489 | 0.92 |
| Sulfotransferase Activity | ||||
| 1428140_at | 3-oxoacid coA transferase 1 | Oxct1 | 0.645 | 0.73 |
| 1449147_at | carbohydrate (keratan sulfate gal-6) sulfotransferase 1 | Chst1 | 0.567 | 0.83 |
| 1428902_at | carbohydrate sulfotransferase 11 | Chst11 | 0.654 | 0.87 |
| 1422758_at | carbohydrate sulfotransferase 2 | Chst2 | 0.557 | 0.84 |
| 1422739_at | heparan sulfate 2-o-sulfotransferase 1 | Hs2st1 | 0.711 | 0.82 |
| 1417293_at | heparan sulfate 6-o-sulfotransferase 1 | Hs6st1 | 0.648 | 1.09 |
| 1428367_at | n-deacetylase/n-sulfotransferase (heparan glucosaminyl) 1 | Ndst1 | 0.567 | 1.10 |
| 1421733_a_at | protein-tyrosine sulfotransferase 1 | Tpst1 | 0.573 | 1.33 |
| 1416086_at | protein-tyrosine sulfotransferase 2 | Tpst2 | 0.695 | 1.35 |
| 1448609_at | thiosulfate sulfurtransferase, mitochondrial | Tst | 0.731 | 1.58 |
Expression fold-changes represent the fold difference of geometric means between HD mice and wild-type controls at a p<0.05 significance level in the Affymetrix mouse 430 2.0 array. Functional annotation clustering was made applying the 2.1 program version of DAVID, the Database for Annotation, Visualization and Integrated Discovery, from the National Institute of Allergy and Infectious Diseases (NIAID), NIH (http://david.abcc.ncifcrf.gov).Shade boxes represent sequences reported as changed in the GLYCOv2 chip as well.
Changes for human caudate samples represent the fold difference of geometric means between HD mice and wild-type controls at a p<0.05 significance level, as reported in Supplementary Table S1 by Hodges et al. (Hodges et al., 2006).
Quantification of mRNA by real-time PCR analysis
For cDNA synthesis, 1 μg total RNA from mice striatum or human caudate samples was reverse transcribed with the SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA), using an oligodT primer per the manufacturer’s protocol. Specific primers for each studied sequence and for mouse and human endogenous controls were designed using Primer Express 1.5 software (Applied Biosystems, Foster City, CA, USA) and their specificity for binding to the desired sequences was searched against NCBI database (Supp. Table 1). Standard curves were generated for each gene of interest using serial dilutions of mouse or human cDNAs. All primers used showed efficiencies between 90% and 118% and r2 values greater than 0.97, parameters calculated by linear regression analysis of the threshold cycle (Ct) vs. log[template] blots using Prism 3.0 software (Graph Pad, San Diego, CA, USA). Realtime PCR experiments were performed using the ABI PRISMs 7900HT Sequence Detection System (Applied Biosystems). Amplification was performed on a cDNA amount equivalent to 25 ng total RNA with 1 × SYBR® Green universal PCR Master mix (Applied Biosystems) containing deoxyribonucleotide triphosphates, MgCl2, AmpliTaq Gold DNA polymerase, and forward and reverse primers. PCR reactions were performed on two independent sets of template (n= 6 mice or human samples per condition). Experimental samples and no-template controls were all run in duplicate. The PCR cycling parameters were: 50°C for 2 min, 95°C for 10 min, and 40 cycles of 94°C for 15 s, 60°C for 1 min. Finally, a dissociation protocol was also performed at the end of each run to verify the presence of a single product with the appropriate melting point temperature for each amplicon. To further ascertain the specificity and size of the PCR products, the products were run alongside molecular weight markers on a 2% agarose gel in 1 X trisacetate EDTA (TAE). The amount of studied cDNA in each sample was calculated using SDS2.1 software (Applied Biosystems) by the comparative threshold cycle (Ct) method and expressed as 2exp(Ct) using hypoxanthine guanine phosphoribosyl transferase (HPRT) as an internal control for mice sequences, whereas β-2-microglobulin (B2M) was used for the human sequences. Expression of both genes was reported to be unchanged in mice and human microarray studies (Desplats et al., 2006, Hodges et al., 2006). For calculations applying the Ct method, age-matched wild-type littermate control mice, or control, non-affected human individuals were used as calibrator samples.
In situ hybridization analysis
Perfused brains were postfixed in 4% paraformaldehyde for 12 h, cryoprotected in 30% sucrose/4% paraformaldehyde overnight before being rapidly frozen on dry ice. In situ hybridization was performed on coronal free-floating sections (25-μm thick) from brains of HD transgenic mice and wild-type controls. Brain sections were hybridized at 55°C for 16 h with a 35S-labeled, single-stranded antisense cRNA probe against each cDNA clone of interest at 107 cpm/mL (Supp. Table 2). Excess probe was removed by washing with 2 × saline-sodium citrate buffer (SSC; 1 × SSC= 0.15 m NaCl/0.015 m sodium citrate) containing 14 mm β-mercaptoethanol (30 min), followed by incubation with 4 μg/mL ribonuclease in 0.5 m NaCl/0.05 m EDTA/0.05 m Tris-HCl (pH 7.5) for 1 h at 37°C. High-stringency washes were carried out at 55°C for 2 h in 0.5 × SSC/50% formamide/0.01 m β-mercaptoethanol, and then at 68°C for 1 h in 0.1 × SSC/0.01 m β-mercaptoethanol/0.5% sarkosyl. Slices were mounted onto gelatin-coated slides and dehydrated with ethanol and chloroform before autoradiography. Slides were exposed for 1-4 days to Kodak X-AR film and then dipped in Ilford K-5 emulsion. After 4 weeks, slides were developed with Kodak D19 developer, fixed, and counterstained with Richardson’s blue. Sense strand probes gave only background hybridization signals.
Lipid Isolation, Purification, and Quantification
Total lipids were isolated and purified from lyophilized brain tissue by using modifications of previously described procedures (Seyfried et al., 1978a, Seyfried et al., 1978b, Hauser et al., 2004, Kasperzyk et al., 2005, Denny et al., 2006). Neutral and acidic lipids were separated by using DEAE-Sephadex (A-25; Pharmacia Biotech, Upsala, Sweden) column chromatography as previously described (Macala et al., 1983, Seyfried et al., 1984, Kasperzyk et al., 2005). The total lipid extract, suspended in solvent A (CHCl3:CH3OH:H2O, 30:60:8 by vol), was then applied to a DEAE-Sephadex column (1.2 ml bed volume) that had been equilibrated with solvent A. The column was washed twice with 20 ml of solvent A, and the entire neutral lipid fraction, consisting of the initial eluent plus washes, was collected. This fraction contained cholesterol, phosphatidylcholine, phosphatidylethanolamine, plasmologens, ceramide, sphingomyelin, and cerebrosides. The acidic lipids were then eluted from the column with 30 ml of solvent B (CHCl3:CH3OH:0.8 m Na acetate, 30:60:8 by vol). This fraction contained the gangliosides and other less hydrophilic acidic lipids, including free fatty acids, cardiolipin, phosphatidylserine, phosphatidylinositol, phosphatidic acid, and sulfatides. The gangliosides were isolated and purified from other acidic lipids and analyzed by using the resorcinol assay as previously described (Kasperzyk et al., 2005).
High-Performance Thin-Layer Chromatography
All lipids were analyzed qualitatively by high-performance thin-layer chromatography (HPTLC) according to previously described methods (Ando et al., 1978, Seyfried et al., 1978b, Macala et al., 1983, Kasperzyk et al., 2005, Denny et al., 2006). Lipids were spotted on 10 × 20 cm Silica gel 60 HPTLC plates (E. Merck, Darmstadt, Germany) using a Camag Linomat V auto-TLC spotter (Camag Scientific Inc., Wilmington, NC). The amount of ganglioside sialic acid spotted per lane was equivalent to approximately 1.5 μg for mice forebrain or human caudate samples. For the analysis of neutral lipids, 70 μg of mouse tissue or 35 μg of human tissue (dry weight) were spotted per lane. For acidic lipids analysis, 100 μg of mouse tissue or 60 μg of human tissue (dry weight) were spotted per lane. To enhance precision, an internal standard (oleoyl alcohol) was added to the neutral and acidic lipid standards and samples as previously described (Macala et al., 1983, Kasperzyk et al., 2005). Purified lipid standards were purchased from Matreya Inc. (Pleasant Gap, PA, USA) or Sigma (St. Louis, MO, USA) or were a gift from Dr. Robert Yu (Medical College of Georgia, Augusta, GA, USA). The HPTLC plates were sprayed with the resorcinol-HCl reagent and heated at 95°C for 30 min to visualize gangliosides (Kasperzyk et al., 2005). For neutral or acidic phospholipids, the plates were developed to a height of either 4.5 cm or 6 cm, respectively, with chloroform:methanol:acetic acid:formic acid:water (35:15:6:2:1 by vol), then developed to the top with hexanes:diisopropyl ether:acetic acid (65:35:2 by vol) as previously described (Macala et al., 1983, Seyfried et al., 1984). Neutral and acidic lipids were visualized by charring with 3% cupric acetate in 8% phosphoric acid solution, followed by heating in an oven at 160-170°C for 7 min. The percentage distribution of the individual lipid bands was determined by scanning the plates on a Personal Densitometer SI with ImageQuant software (Molecular Dynamics, Sunnyvale, CA, USA) for gangliosides, acidic lipids, and neutral lipids.
Statistics
Statistical analyses were performed using Prism software (Graph Pad, San Diego, CA, USA). A two-way ANOVA and two-tailed Student’s t-test were used to evaluate the significance of differences in lipid content in wild-type vs. transgenic mice and control vs. HD human samples. For real-time PCR data, a one-sample t test (confidence interval 95%) was performed against a hypothetical value of 1 to assess significant differences between expression of each gene in wild-type vs. transgenic mice and control vs. HD diseased human samples. Pearson correlation calculations were used to determine correlations between gene expression levels of ganglioside-related genes and ganglioside levels in human HD samples. For these analyses r2 reflects the Pearson correlation coefficient squared.
Results
Expression of “glycobiology-related” genes in the R6/1 transgenic mice
Glycolipid metabolism abnormalities have been reported to occur in several neurodegenerative diseases (Kolter and Sandhoff, 2005). Nevertheless, a comprehensive study to determine the relevance of this imbalance to the HD pathology has not been reported. Therefore, we investigated the expression of genes related to glycobiology using the GLYCOv2 chip, a customized array built by the Consortium for Functional Glycomics, that contains 2,174 probe sets targeting 942 mouse transcripts, including transcripts encoding glycosyltransferases, glycan-binding and degradation proteins, transporters and N-glycan biosynthesis-related proteins, among many other categories (www.functionalglycomics.org/static/consortium/resources/resourcecoreh.shtml). Arrays were hybridized with three biological replicates for each condition, using total RNA extracted from the striata of R6/1 transgenic mice and wild-type littermate controls ( >6 months old). Class comparisons were performed in BRB-ArrayTools to identify specific genes differentially expressed between classes (see methods for details).
We found 29 sequences to be differentially expressed (p<0.05) in the striata of the R6/1 transgenic mice when compared to wild-type animals (Table 1), representing 3.1 % of the total transcripts in the array. Fifty-five percent of the sequences (16 genes) fell into two related functional annotation clusters: glycan-transferases and glycan-degradation-related proteins (according to Gene Ontology). While these categories represent 21.5 % and 4.6 % of the total probe IDs contained on the chip respectively, these proportions were shifted in our study, such that 40 % of all the glycomics-related sequences whose expression was altered in the R6/1 transgenic mice were glycan-transferases and 13.3 % were glycan-degradation-related proteins (Fig. 1). Whereas several types of gene expression alterations have been shown to occur in HD (Thomas, 2006), the overrepresentation of two main clusters suggests that the abnormalities observed in the expression of these genes reflect specific mechanisms, rather than general changes in gene expression operating in R6/1 HD transgenic mice.
Figure 1.

Relative distribution of the functional annotation clusters of the genes whose expression was reported as changed in the R6/1 transgenic HD mice (open bars) compared to the distribution of the functional annotation clusters of the total genes included in the GLYCOv2 array (shaded bars).
To extend this analysis we also hybridized the Affymetrix mouse 430 2.0 array, which contains 39,000 mouse probe sets, with the same RNA samples from the R6/1 transgenic mice and wild-type controls. Functional annotation clustering of the 2,905 sequences reported to be differentially expressed in the R6/1 transgenic mice (p<0.05) expanded our previous results to include additional genes encoding glycosyltransferases, sulfotransferases, sialyltransferases and proteins related to sphingolipid metabolism (Table 2). We then compared our lists of differentially expressed genes to the microarray data output reported from post-mortem caudate samples (grades 0-2 pathology) from HD subjects (Hodges et al., 2006). We found that 85% of the expression differences identified in R6/1 transgenic mice using the Glycov2 chip, and 84% of the differences using the Affymetrix array, were also significantly (p<0.05) altered in the caudate from post-mortem HD subjects with early pathology (Tables 1 and 2). The combined analysis of both arrays at the gene function level highlighted one particular pathway of sphingolipid metabolism, that involved in ganglioside biosynthesis, as severely impaired in both R6/1 transgenic mice and human HD caudate samples. Expression of all of the genes encoding the enzymes acting on their catabolism was reduced. Hence, we further focused our study to investigate disruptions in this pathway in HD.
To validate the array findings, we performed quantitative PCR (qPCR) on striatal RNA isolated from two independent sets of wild-type and R6/1 transgenic mice, paired for their analysis. Significant differences in expression of St8sia2 and St8sia3, genes encoding proteins homologous to GD3 synthase, and B4galnt1 (gene encoding GM2/GD2 synthase) were found between wild-type and R6/1 transgenic mice correlating well with the initial array data (Fig. 2). The signal for St8sia1 (encoding GD3 synthase), was called as “Absent” on the array, indicating low confidence in the hybridization signal. However, considering that this enzyme catalyzes an important step in ganglioside biosynthesis, we included St8sia1 in the qPCR analysis. We found St8sia1 mRNA expression to be significantly elevated in the R6/1 transgenic mice (Fig. 2), perhaps reflecting a compensatory mechanism to maintain the overall physiological levels of gangliosides. The expression levels of St3gal5 (encoding GM3 synthase), Gm2a, St3gal2 (encoding GM1b/GD1a/GT1b synthase) and St6galnac5 (SiaT7e) were not found to be significantly different in the R6/1 transgenic mice vs. wild-type controls (Fig. 2).
Figure 2.

Gene expression levels of ganglioside-metabolism enzymes in striatum of symptomatic R6/1 transgenic HD mice compared to wild-type littermate controls. qPCR amplification of the indicated transcripts from striatum of symptomatic R6/1 transgenic animals and wild-type controls was performed as described in Methods. Values are expressed as the averages of the 2exp(Ct) ± SEM of duplicate determinations from two independent experiments (n = 4 mice per condition). Amplification of the hypoxanthine guanine phosphoribosyl transferease (HPRT) housekeeping gene was used as an internal reference and the expression level from the age-matched wild-type controls was used as calibrator in the ΔCt method and showed as a basal line at point 1 on the y axis. Asterisks denote significant differences in gene expression as determined by one-sample t-test (confidence interval 95%) with a p-value <0.05.
Regional distribution of glycosyltransferase transcripts in CNS
We analyzed the CNS expression patterns of the genes encoding putative ganglioside biosynthetic enzymes in wild-type mice using in situ hybridization. St3gal5 and St3gal2 transcripts showed moderately abundant expression levels in cortex and hippocampus, with lower levels throughout the rest of the brain (Fig. 3). In contrast, St8sia1 and St8sia3 probes showed more remarkable patterns of hybridization. St8sia1 is expressed in discrete cortical layers (mainly layer V), hippocampus (mainly the CA1 subregion), septum and in several different thalamic nuclei (Fig. 3). The St8sia3 isoform exhibited abundant expression in the striatum, the region that undergoes specific degeneration in HD. Hybridization signals were also detected in the olfactory tubercle, cortex, hippocampus and several hypothalamic nuclei (Fig. 3). The remaining genes tested showed very low levels of expression throughout the brain (data not shown).
Figure 3.

CNS regional expression of transcripts of genes encoding ganglioside biosynthetic enzymes. In situ hybridization analysis was performed on free-floating coronal sections of wild-type mice brains. Antisense 35S-labeled riboprobes against the indicated genes were used as described in Methods. CP, caudate putamen; cx, cortex; hipp, hippocampus; hyp, hypothalamus; MS, medial septal nucleus; thal, thalamus.
Ganglioside content and distribution in the R6/1 transgenic mice forebrain
To determine how the observed changes in gene expression would in turn affect the ganglioside content and distribution in the mouse brain, we analyzed the qualitative and quantitative distribution of individual gangliosides in the forebrain of wild-type and the R6/1 transgenic mice by means of high-performance thin-layer chromatography (HPTLC) (Fig. 4). Total ganglioside content remained unchanged in the R6/1 transgenic mice when compared to wild-type controls, however the most prominent difference among individual gangliosides was a 38 % reduction in GM1 content in the R6/1 transgenic mice (Table 3). GD3 ganglioside levels in both, wild-type and transgenic mice, were found in trace amounts, coincident with the findings reported for adult mice cerebellum (Seyfried and Yu, 1984, Seyfried and Yu, 1985, Denny et al., 2006).
Figure 4.
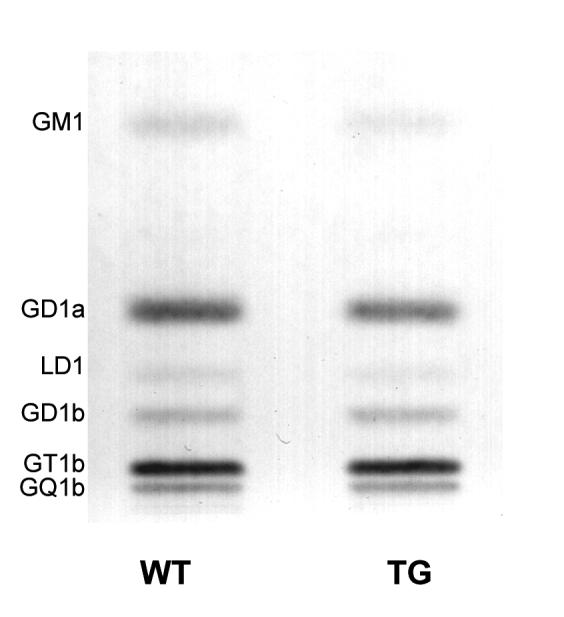
HPTLC of gangliosides in the R6/1 transgenic mice forebrain. Quantitative analysis of the individual gangliosides in transgenic and wild-type mice forebrain (cortex and striata pooled together). The amount of ganglioside sialic acid spotted per lane was equivalent to approximately 1.5 μg. The plate was developed by a single ascending run with CHCl3:CH3OH:dH2O (55:45:10 by vol) containing 0.02% CaCl2 · 2H2O. The bands were visualized with resorcinol-HCl spray.
Table 3.
Ganglioside Content and Distribution in the R6/1 Transgenic Mice
| Ganglioside content (μg sialic acid / 100 mg dry weight)a | ||||||
|---|---|---|---|---|---|---|
| WT | R6/1 | |||||
| Total | 528.0 ± 48.0 | 531.0 ± 43.0 | ||||
| GM1 | 60.1 ± 6.7 | 37.1 ± 3.8* | ||||
| GD1a | 186.1 ± 20.1 | 167.6 ± 14.9 | ||||
| LD1 | 17.7 ± 1.1 | 19.0 ± 1.2 | ||||
| GD1b | 42.8 ± 4.5 | 51.2 ± 6.0 | ||||
| GT1b | 161.7 ± 15.6 | 186.4 ± 19.2 | ||||
| GQ1b | 59.4 ± 6.7 | 69.5 ± 3.0 | ||||
Values represent the mean ± S.E.M. of n =3 animals per group.
Determined from densitometric scanning of HPTLC as shown in Figure 4.
Indicates significant difference between R6/1 transgenic and wilt type mice at p < 0.05 as determined by Student’s t-test (unpaired, two-tailed).
Neutral and acidic lipid content and distribution in the R6/1 transgenic mice forebrain
To complete the study of glycosphingolipids, we analyzed the content and distribution of neutral and acidic lipids in the forebrain of the R6/1 transgenic mice and wild type controls. We found a significant decrease in cerebrosides and sulfatides in the forebrains of the R6/1 transgenic mice (Table 4; HPTLC plates not shown). Interestingly, these are myelin-enriched lipids (Sandhoff et al., 1971, Seyfried and Yu, 1980, Kaye et al., 1992) and this decrease may be suggestive of abnormalities in myelin content in the R6/1 transgenic mouse model of HD. Our detection method did not show differences in cholesterol content between the R6/1 transgenic mice and wild type controls, as previously suggested in other models of HD (Sipione et al., 2002, Valenza et al., 2005).
Table 4.
Neutral and Acidic Lipid Distribution in Forebrain of the R6/1 Transgenic Micea
| (mg / 100 mg dry weight)b | ||
|---|---|---|
| Lipids | WT | TG |
| Neutral | ||
| Cholesterol Esters | ND | ND |
| Triglycerides | ND | ND |
| Cholesterol | 6.0 ± 0.12 | 5.9 ± 0.07 |
| Ceramide | trace | trace |
| Cerebrosides | 3.1 ± 0.03 | 2.5 ± 0.11* |
| Phosphatidylethanolamine | 7.3 ± 0.23 | 7.0 ± 0.18 |
| Phosphatidylcholine | 4.4 ± 0.21 | 5.0 ± 0.21 |
| Sphingomyelin | 0.8 ± 0.01 | 0.8 ± 0.04 |
| Acidic | ||
| Cardolipin | 0.53 ± 0.01 | 0.63 ± 0.03 |
| Phosphatic Acid | ND | ND |
| Sulfatides | 0.81 ± 0.02 | 0.67 ± 0.03* |
| Phosphatidylserine | 2.69 ± 0.09 | 2.64 ± 0.26 |
| Phosphatidylinositol | 0.72 ± 0.00 | 0.71 ± 0.06 |
Values represent the mean ± SEM of n = 3 animals per group.
Determined from densitometric scanning of HPLTC plates (not shown).
ND represents non-detectable.
Indicates that the value is significantly different from that of the wild-type mice at p < 0.05 as determined by Student’s t-test (unpaired, two-tailed).
Expression of genes encoding putative ganglioside biosynthetic enzymes in human caudate from HD patients
To determine whether the changes observed in the R6/1 transgenic mouse model represent a yet undiscovered feature of pathology in human HD, we quantified the transcript levels of the corresponding human genes encoding putative ganglioside biosynthetic enzymes using real-time PCR analysis on postmortem caudate samples from HD subjects and controls. The expression of St3gal5, the enzyme that initiates the synthesis of the ganglio-series of gangliosides, St8sia3, B4galnt1 and St3gal2 was significantly decreased in the caudate from HD patients, while expression of the other ganglioside genes tested was not significantly changed between HD and control samples (Fig. 5). The changes in these key enzymes might be expected to cause an important imbalance in ganglioside metabolism in the pathology of HD.
Figure 5.

Gene expression levels of ganglioside-metabolism enzymes in postmortem caudate of HD subjects compared to control individuals. qPCR amplification of the indicated transcripts was performed on human postmortem caudate samples as described in Methods. Values are expressed as the averages of the 2exp(Ct) ± SEM of duplicate determinations from two independent experiments (n = 3 samples per condition). HD brains were a pathological grade 3. Amplification of the beta-2-microglobulin (B2M) housekeeping gene was used as an internal reference and the expression level from the control brains was used as calibrator in the ΔCt method and showed as a basal line at point 1 on the y axis. Asterisks denote significant differences in gene expression as determined by one-sample t-test (confidence interval 95%) with a p-value <0.05.
Ganglioside content and distribution in human caudate from HD patients
We next analyzed the qualitative and quantitative distribution of individual gangliosides in human postmortem caudate samples from HD and control subjects by HPTLC (Fig. 6). We processed three HD caudate samples, pathology grade 3, which were age and sex matched to three control samples. A two-way ANOVA used to compare all the ganglioside measurements in the caudate of HD vs. control subjects, revealed a significant difference in HD caudate compared to controls, despite high variability among individual subjects (p<0.05). Comparing levels of individual gangliosides using a two-tailed Student’s t-test showed a significant elevation of GD3 in HD caudate when compared to controls (Table 5). This is indicative of an overall disruption in ganglioside balance in HD. A further within-subject comparison revealed significant correlations between decreased B4galnt1 mRNA expression levels and lower GD1a, GD1b and GT1b content in individual HD subjects, with r2 values, determined from Pearson correlation coefficients, of 0.996; 0.921 and 0.862, respectively. Additional correlations were also found between decreased St3gal2 mRNA expression and lower levels of GD1a and GT1b gangliosides in HD caudate, with r2 values of 0.944 and 0.866, respectively.
Figure 6.
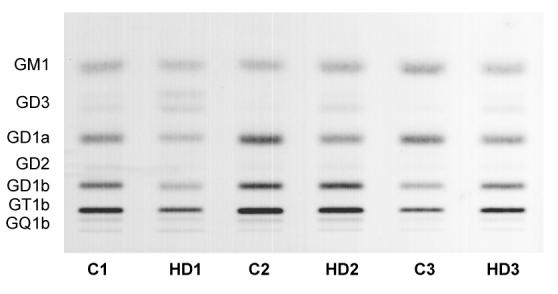
HPTLC of gangliosides in HD human caudate. Quantitative analysis of the individual gangliosides in postmortem caudate samples from HD patients (pathology grade 3) and control subjects, age and sex matched (n = 3 samples/group). The amount of ganglioside sialic acid spotted per lane was equivalent to approximately 1.5 μg. The plate was developed by a single ascending run with CHCl3:CH3OH:dH2O (55:45:10 by vol) containing 0.02% CaCl2 · 2H2O. The bands were visualized with resorcinol-HCl spray.
Table 5.
Ganglioside Content and Distribution in Caudate of Huntington Disease Subjects
| Control | HD | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | Average | 1 | 2 | 3 | Average | ||||||
| Age | 49 | 55 | 22 | 42 | 46 | 53 | 54 | 51 | |||||
| Sex | M | M | M | - | M | F | M | - | |||||
|
Ganglioside content (μg sialic acid / 100 mg dry weight)a |
Difference % |
||||||||||||
| Total | 205 | 393 | 276 | 291 ± 55 | 172 | 260 | 93 | 175 ± 48 | -40 | ||||
| GM1 | 37.4 | 58.6 | 82.0 | 59.3 ± 12.9 | 36.6 | 53.5 | 18.8 | 36.3 ± 10.0 | -39 | ||||
| GD3 | 8.8 | 3.5 | 3.8 | 5.4 ± 1.7 | 9.9 | 16.9 | 15.5 | 14.1 ± 2.2 * | 162 | ||||
| GD1a | 45.9 | 115.2 | 88.1 | 83.1 ± 20.2 | 42.9 | 52.5 | 16.2 | 37.2 ± 10.9 | -55 | ||||
| GD2 | 7.1 | 3.8 | 2.0 | 4.3 ± 1.5 | 2.4 | 4.1 | 1.6 | 2.7 ± 0.7 | -37 | ||||
| GD1b | 46.3 | 85.4 | 43.7 | 58.5 ± 13.5 | 36.4 | 58.4 | 14.7 | 36.5 ± 12.6 | -38 | ||||
| GT1b | 52.5 | 112.2 | 51.5 | 72.0 ± 20.1 | 39.7 | 64.4 | 22.7 | 42.2 ± 12.1 | -41 | ||||
| GQ1b | 7.4 | 13.8 | 5.2 | 8.8 ± 2.6 | 4.5 | 10.6 | 3.4 | 6.2 ± 2.2 | -30 | ||||
Determined from densitometric scanning of HPTLC as shown in Figure 6.
Indicates that the value is significantly different from that of the control caudate at p < 0.05 as determined by the two-tailed Student’s t-test.
Analysis of neutral and acidic lipids was also performed on human post-mortem caudate samples. No significant differences in the levels of neutral or acidic lipids, including cholesterol, were detected in the caudate of HD subjects when compared to controls (data not shown).
Discussion
In the present study, we demonstrate differences in the levels of mRNAs encoding enzymes involved in glycoprotein and glycolipid metabolism in the R6/1 transgenic mouse model of HD and in postmortem human caudate samples from HD patients when compared to wild-type/controls. Our findings uncover a novel abnormality in the synthesis of glycosphingolipids: the decreased accumulation of transcripts for glycosyl- and sialyl-transferases, the enzymes contributing to ganglioside synthesis. Our microarray results revealed many apparent isoforms of common ganglioside synthesis-related genes, which we found differentially expressed within the CNS. We further show abnormal distribution of select gangliosides and neutral and acidic lipids in R6/1 transgenic mice and human HD caudate samples.
Gangliosides are important constituents of cell-membranes and are associated with a plethora of biological functions, including cellular recognition and adhesion, signal transduction, growth regulation and differentiation (Yu et al., 2004, d’Azzo et al., 2006). Structurally, gangliosides are glycosphingolipids that contain one or more sialic acid residues and are biosynthetically derived from lactosylceramide (Svennerholm and Raal, 1961, Ledeen, 1966, Svennerholm, 1980, Yu et al., 2004). While they are present in most vertebrate cells and tissues, gangliosides are particularly abundant in the nervous system where they are expressed most frequently as components of the outer leaflet of the plasma membranes of neural and glial cells (Ledeen, 1978, Seyfried et al., 1984, Schwarz and Futerman, 1996). Imbalance of ganglioside levels can result in apoptosis and disruption in Ca+2 signaling, both of which have been associated with HD (Ross, 2002).
Abnormalities in gene expression of ganglioside-related enzymes were observed in this study in both the striatum of the R6/1 transgenic mice and the caudate of HD human brains. However, how these changes in expression translate into expected alterations of ganglioside levels is not straightforward, indicating complex regulation of this system in the CNS. We did observe correlations in human brain between the decrease in gene expression of GM2/GD2 synthase (B4galnt1) and the decreases in downstream products GD1a, GD1b and GT1b, and the decreased expression of GM1b/GD1a/GT1b synthase (St3gal2) and the decreased levels of their direct products GD1a and GT1b, when analyzed in individual subjects. Hence, transcriptional dysregulation of genes encoding particular ganglioside synthetic enzymes appears to translate into altered levels of some downstream gangliosides. However, these correlations were not observed R6/1 transgenic mouse brain. Further, altered levels of GD3 and GM1, in human HD and R6/1 transgenic mouse brains, respectively, were not correlated with altered mRNA levels of associated biosynthetic enzymes. Unfortunately, the mRNAs encoding two additional enzymes in this pathway, B3galt4 and St8sia5, were found at too low abundance to give reliable hybridization signals, hence direct correlations between their levels and ganglioside products was not possible in mouse or human.
While expression of many genes was found to be altered at the transcriptional level, we did not measure protein levels or activity of the biosynthetic enzymes, which can also regulate intracellular ganglioside levels. Feedback control of several glycosyltransferases has been observed in vitro (Kolter et al., 2002). Their activities also depend on phosphorylation state, pH of the environment (Kolter et al., 2002) and physical association with other glycosyltransferases (Yu et al., 2004). It is possible that the enzyme activity of many of these proteins, or alternatively the protein expression levels themselves, compensate for alterations in mRNA levels, in attempts to prevent disruption of ganglioside levels, which may be very detrimental to the CNS, as manifested by numerous mouse models and human disorders.
In the brains of both mouse and humans, significant decreases in expression were observed for B4galnt1, which encodes GM2/GD2 synthase, and St8Sia3, whose protein product shows sequence homology to GD3 synthase. Additionally, we detected a significant decrease in St3gal5 (encoding GM3 synthase) and St3gal2 (encoding GM1b/GD1a/GT1b synthase) in human HD caudate, while St8sia1 (encoding GD3 synthase) was increased and St8sia2, which encodes another sialyltransferase related to GD3 synthase, was decreased in the R6/1 mouse striatum. The metabolic pathway by which the protein products of these genes control ganglioside biosynthesis is shown in Fig. 7. GM2/GD2 synthase is responsible for transfer of GalNAc from UDP-GalNAc of lactosylceramide, GM3 and GD3 to produce GA2, GM2 and GD2, respectively (Fig. 7). GD3 synthase (St8sia1) catalyzes the addition of a second sialic acid group to GM3, generating GD3 and initiating the synthesis of the b-series of gangliosides (Fig. 7). This enzyme is member of a family of sialyltransferases composed of five proteins that vary in sequence homology and substrate specificity. St8sia2 and St8sia3 encode enzymes belonging to the same family and are annotated as ganglioside biosynthetic enzymes in the DAVID database. While its specific role in ganglioside metabolism is unclear, the St8sia2 gene product is involved in the transfer of sialic acid residues to specific N-CAM isoforms yielding PSA N-CAM (Kojima et al., 1996).
Figure 7.
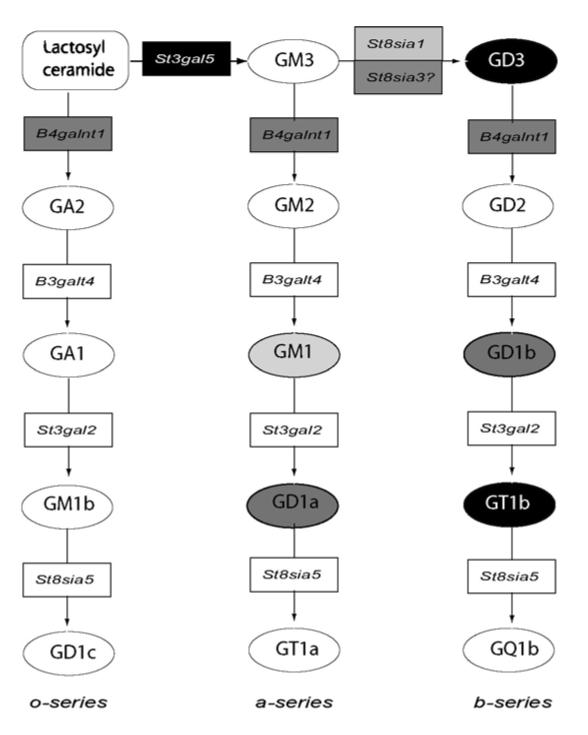
Biosynthetic pathway for the ganglio-series of glycosphingolipids. Gangliosides are named according to the simplified nomenclature of Svennerholm (Svennerholm, 1994): the “ganglio” core containing four saccharide residues is designated by the letter G, followed by a letter designating the total number of sialic acids (M, mono; D, di; T, tri; Q, tetra; P, penta; A, asilo, none). The following number represents the length of the ganglio core, with 1 representing the full four-saccharide core, and shorter structures having higher numbers. Lower case letters designate the number of sialic acids linked to the internal Gal residue (a=1, b=2) and Greek letters indicate number of sialic acids linked to the GalNAc residue (α=1, β=2). Glycosyltransferase genes are represented by boxes. Gangliosides are represented by circles. Shade code represent gene expression and ganglioside level changes found in the present study: light grey indicates changes in the R6/1 transgenic mouse; black indicates changes in human HD caudate; dark grey, indicates changes in both, human and mouse. Based on KEGG Pathway Database, reference pathway mim00604 (www.kegg.com/kegg/pathway.html)
St8sia3, which shares 36 % homology in its amino acid sequence with GD3 synthase is able to catalyze the conversion of GM3 to GD3 in vitro, but may catalyze the transfer of sialic acid to alternative acceptors in vivo, such as 2,3- sialosylparagloboside (Yoshida et al., 1995). Our results favor this later hypothesis, since decreases in expression of St8sia3 mRNA do not correlate with the levels of GD3, which were found to be elevated in human caudate but not detected in mouse striatum. Upregulation in the expression of GD3 synthase (St8sia1) was observed in R6/1 mouse striatum, but not in human HD caudate, consistent with previous microarray studies showing only a 1.1-fold increase in St8sia1 in human HD caudate compared to controls (Hodges et al., 2006). The apparent multiple isoforms of GD3 synthase suggest that each performs region- and/or context-specific functions involving sialylation of lipids and/or proteins.
Accordingly, we demonstrated that the expression patterns of St8sia1 and St8sia3 within the brain varies widely, with St8sia3 transcripts exhibiting enriched expression in the striatum and St8sia1 showing very low levels of expression in that region. Isoform-specific expression of these and other ganglioside-related genes may explain differences in ganglioside profiles in different brain regions. This may partly explain the lack of overt changes in ganglioside levels observed in the forebrain of R6/1 transgenic mice. While our gene expression studies were performed in striatum, the lipid analyses were performed in forebrain, hence any changes in lipid content that might be specific to striatum may have been masked or compensated by cortical levels. Nonetheless, we hypothesize that deficits in transcription in this particular isoform, St8sia3, could have important consequences in the striatum, whose selective deficits are the hallmark of HD.
In R6/1 HD transgenic mice, we also observed a significant decrease in GM1, a myelin-enriched ganglioside (Sandhoff et al., 1971, Kaye et al., 1992, Vyas and Schnaar, 2001). This decrease may result from lower myelin levels as indicated by decreases in the myelin-enriched lipids, cerebrosides and sulfatides, rather than as a result of decreased levels of its synthesis enzyme. Hence, the R6/1 mice may mimic some aspects of myelin-related dysfunction, which has been observed in human HD (Fennema-Notestine et al., 2004, Beglinger et al., 2005, Ciarmiello et al., 2006), although further studies are needed to explore this possibility.
In human HD caudate we observed an overall decrease in ganglioside levels compared to control subjects. Reductions in individual gangliosides could be related to the loss of neurons in which they are enriched. Therefore we can not exclude the possibility that altered expression of ganglioside biosynthetic enzymes is secondary to changes in neuronal content, however given that expression differences in several of these genes also occur in human HD subjects with grades 0-2 pathology, we argue against this reasoning. In contrast to the decrease observed for most ganglioside species, we detected an increase in the amount of GD3 in human HD caudate. Abnormally high levels of GD3 have been previously associated with HD (Bernheimer H., 1979, Higatsberger et al., 1981) and with other neurodegenerative disorders (Ledeen et al., 1968, Tamai et al., 1978, Bernheimer H., 1979) and are thought to represent an astroglial response to neurodegeneration (Seyfried and Yu, 1985, Levine et al., 1986). While the increases in GD3 were not correlated with changes in expression of St8sia1 or its isoforms, it may be associated with the observed decrease in expression levels of B4galnt1. In a single case report, biochemical studies showed accumulation of GM3 and GD3 associated with reduced activity of GM2/GD2 synthase, resulting in severe impairment of physical and motor development, extensive neurological dysfunction and death in infancy (Max et al., 1974, Fishman et al., 1975). GD3 is known to stimulate reactive oxygen species production, leading to apoptosis (Scorrano et al., 1999). Hence, the high levels of GD3 in human HD caudate may be indicative of apoptotic neurodegeneration.
A major difference between the mouse models for HD and the human disease itself, is that none of the mouse models, including R6/1 transgenic mice, mimic the severe neurodegenerative changes observed in humans, although some models show slight neuronal loss in the striatum (Mangiarini et al., 1996, Hodgson et al., 1999). Nonetheless, we have shown that gene expression deficits in ganglioside-related genes are also found in HD caudate samples with early pathology, as well as those with end-stage pathology. These findings highlight the relevance of glycolipid deficits to the human disease and they may be part of the pathological process, not merely a result of neuronal death. However, further quantitative analyses to validate microarray findings are necessary to assess in which extent disrupted glycolipid metabolism impact on HD pathology.
Several mice have been generated to date carrying disruptions in key genes that control the synthesis of gangliosides. Mice in which B4galnt1 gene is disrupted remain viable despite a lack of complex gangliosides, although they show evidence of dysmyelination and axonal degeneration and exhibit certain motor functions defects (Takamiya et al., 1996, Liu et al., 1999, Sheikh et al., 1999). Mice that carry mutations in St3gal5 (GM3 synthase) and B4galnt1 (GM2/GD2 synthase) genes are not able to form any ganglioside in the CNS. These mice exhibit rapid and profound neurodegeneration with severe pathology in white matter, with axonal degeneration and disrupted axon-glial interactions (Yamashita et al., 2005). These findings demonstrate that gangliosides play important roles in stabilizing the maturing CNS. Noteworthy, we found here that both genes have decreased expression in human HD brains.
Recent studies have suggested that ganglioside treatment is beneficial in some rodent models exhibiting functional impairment. It has been shown that exogenously applied GM1 in rats following bilateral lesions of the caudate nucleus with quinolinic acid (QA) showed improved performance in the Morris water maze, suggesting that GM1 treatment can attenuate QA-induced learning deficits in this rodent model for HD (Dunbar et al., 2006). It is not known whether treatment with other gangliosides would also show beneficial effects.
To summarize, we report abnormal expression of the genes encoding several enzymes related to glycosphingolipid metabolism in the R6/1 transgenic mouse model of HD and in human caudate from end-stage HD patients when compared to controls. Further, we demonstrate altered glycosphingolipid and ganglioside levels, most noticeably in human brains, which may contribute, at least in part, to the presentation of symptoms observed in the R6/1 transgenic mouse model of HD and in human HD subjects. These findings implicate disruptions in glycolipid metabolism in the pathology of HD and, while further studies are necessary to asses if this is an early event in HD pathology, or an end-stage symptom, restoration of glycosphingolipid balance may constitute an alternative therapeutic target for HD.
Supplementary Material
Acknowledgements
This work was supported by NIH grants #NS44169 (EAT), MH069696 (EAT) and HD39722 (TNS); Boston College Research Expense Fund (TNS); National Tay-Sachs and Allied Disease Association, Inc. (NTSAD) (TNS). The gene microarray analysis was conducted by the Gene Microarray Core of The Consortium for Functional Glycomics funded by the National Institute of General Medical Sciences grant GM62116. Human brain tissue was provided by Harvard Brain Tissue Resource Center, which is supported by PHS grant #R24-MH 068855.
Abbreviations used
- 2,3-SPG
2,3-sialosylparagloboside
- B2M
β-2-microglobulin
- CBP
carbohydrate binding protein
- Ct
threshold cycle
- GalNAc
N-acetylgalactosamine
- HD
Huntington’s disease
- HPRT
hypoxanthine guanine phosphoribosyl transferase
- LD1
9-O-acetyl disialolactoneotetraosyl ceramide
- N-CAM
neural cell adhesion molecule
- PMI
postmortem interval
- polyQ
polyglutamine
- PSA N-CAM
polysialylated neural cell adhesion molecule
- QA
quinolic acid
- qPCR
quantitative real-time PCR
- SSC
saline-sodium citrate buffer
- UDP-GalNAc
UDP-N-acetylgalactosamine
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ando S, Chang NC, Yu RK. High-performance thin-layer chromatography and densitometric determination of brain ganglioside compositions of several species. Anal Biochem. 1978;89:437–450. doi: 10.1016/0003-2697(78)90373-1. [DOI] [PubMed] [Google Scholar]
- Beglinger LJ, Nopoulos PC, Jorge RE, Langbehn DR, Mikos AE, Moser DJ, Duff K, Robinson RG, Paulsen JS. White matter volume and cognitive dysfunction in early Huntington’s disease. Cogn Behav Neurol. 2005;18:102–107. doi: 10.1097/01.wnn.0000152205.79033.73. [DOI] [PubMed] [Google Scholar]
- Bernheimer H. SG, Price KS, Hornykiewicz O. Brain Gangliosides in Huntington’s Disease. Adv Neurol. 1979;23:463–471. [Google Scholar]
- Borri PF, Op den Velde WM, Hooghwinkel GJ, Bruyn GW. Biochemical studies in Huntington’s chorea. VI. Composition of striatal neutral lipids, phospholipids, glycolipids, fatty acids, and amino acids. Neurology. 1967;17:172–178. doi: 10.1212/wnl.17.2.172. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nat Rev Neurosci. 2005;6:919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- Ciarmiello A, Cannella M, Lastoria S, Simonelli M, Frati L, Rubinsztein DC, Squitieri F. Brain White-Matter Volume Loss and Glucose Hypometabolism Precede the Clinical Symptoms of Huntington’s Disease. J Nucl Med. 2006;47:215–222. [PubMed] [Google Scholar]
- d’Azzo A, Tessitore A, Sano R. Gangliosides as apoptotic signals in ER stress response. Cell Death Differ. 2006;13:404–414. doi: 10.1038/sj.cdd.4401834. [DOI] [PubMed] [Google Scholar]
- Denny CA, Kasperzyk JL, Gorham KN, Bronson R. T. a., Seyfried TN. Influence of caloric restriction on motor behavior, longevity, and brain lipid composition in Sandhoff disease mice. J Neurosci Res. 2006;83:1028–1038. doi: 10.1002/jnr.20798. [DOI] [PubMed] [Google Scholar]
- Desplats PA, Kass KE, Gilmartin T, Stanwood GD, Woodward EL, Head SR, Sutcliffe JG, Thomas EA. Selective deficits in the expression of striatal-enriched mRNAs in Huntington’s disease. J Neurochem. 2006;96:743–757. doi: 10.1111/j.1471-4159.2005.03588.x. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA, et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- Dunbar GL, Sandstrom MI, Rossignol J, Lescaudron L. Neurotrophic enhancers as therapy for behavioral deficits in rodent models of Huntington’s disease: use of gangliosides, substituted pyrimidines, and mesenchymal stem cells. Behav Cogn Neurosci Rev. 2006;5:63–79. doi: 10.1177/1534582306289367. [DOI] [PubMed] [Google Scholar]
- Fennema-Notestine C, Archibald SL, Jacobson MW, Corey-Bloom J, Paulsen JS, Peavy GM, Gamst AC, Hamilton JM, Salmon DP, Jernigan TL. In vivo evidence of cerebellar atrophy and cerebral white matter loss in Huntington disease. Neurology. 2004;63:989–995. doi: 10.1212/01.wnl.0000138434.68093.67. [DOI] [PubMed] [Google Scholar]
- Fishman PH, Max SR, Tallman JF, Brady RO, Maclaren NK, Cornblath M. Deficient Ganglioside Biosynthesis: a novel human sphingolipidosis. Science. 1975;187:68–70. doi: 10.1126/science.803227. [DOI] [PubMed] [Google Scholar]
- Hauser EC, Kasperzyk JL, d’Azzo A, Seyfried TN. Inheritance of lysosomal acid beta-galactosidase activity and gangliosides in crosses of DBA/2J and knockout mice. Biochem Genet. 2004;42:241–257. doi: 10.1023/b:bigi.0000034429.55418.71. [DOI] [PubMed] [Google Scholar]
- Higatsberger MR, Sperk G, Bernheimer H, Shannak KS, Hornykiewicz O. Striatal ganglioside levels in the rat following kainic acid lesions: comparison with Huntington’s disease. Exp Brain Res. 1981;44:93–96. doi: 10.1007/BF00238752. [DOI] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, Hollingsworth ZR, Collin F, Synek B, Holmans PA, Young AB, Wexler NS, Delorenzi M, Kooperberg C, Augood SJ, Faull RL, Olson JM, Jones L, Luthi Carter R. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- Hooghwinkel GJ, Bruyn GW, de Rooy RE. Biochemical studies in Huntington’s chorea. VII. The lipid composition of the cerebral white and gray matter. Neurology. 1968;18:408–412. doi: 10.1212/wnl.18.4.408. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasperzyk JL, d’Azzo A, Platt FM, Alroy J, Seyfried TN. Substrate reduction reduces gangliosides in postnatal cerebrum-brainstem and cerebellum in GM1 gangliosidosis mice. J Lipid Res. 2005;46:744–751. doi: 10.1194/jlr.M400411-JLR200. [DOI] [PubMed] [Google Scholar]
- Kaye EM, Alroy J, Raghavan SS, Schwarting GA, Adelman LS, Runge V, Gelblum D, Thalhammer JG, Zuniga G. Dysmyelinogenesis in animal model of GM1 gangliosidosis. Pediatr Neurol. 1992;8:255–261. doi: 10.1016/0887-8994(92)90361-2. [DOI] [PubMed] [Google Scholar]
- Kegel KB, Sapp E, Yoder J, Cuiffo B, Sobin L, Kim YJ, Qin ZH, Hayden MR, Aronin N, Scott DL, Isenberg G, Goldmann WH, DiFiglia M. Huntingtin associates with acidic phospholipids at the plasma membrane. J Biol Chem. 2005;280:36464–36473. doi: 10.1074/jbc.M503672200. [DOI] [PubMed] [Google Scholar]
- Kojima N, Tachida Y, Yoshida Y, Tsuji S. Characterization of mouse ST8Sia II (STX) as a neural cell adhesion molecule-specific polysialic acid synthase. Requirement of core alpha1,6-linked fucose and a polypeptide chain for polysialylation. J Biol Chem. 1996;271:19457–19463. doi: 10.1074/jbc.271.32.19457. [DOI] [PubMed] [Google Scholar]
- Kolter T, Proia RL, Sandhoff K. Combinatorial ganglioside biosynthesis. J Biol Chem. 2002;277:25859–25862. doi: 10.1074/jbc.R200001200. [DOI] [PubMed] [Google Scholar]
- Kolter T, Sandhoff K. Lysosomal glycosphingolipid storage diseases. Oxford University Press; Oxford: 2005. [Google Scholar]
- Ledeen R. The chemistry of gangliosides: a review. J Am Oil Chem Soc. 1966;43:57–66. doi: 10.1007/BF02641015. [DOI] [PubMed] [Google Scholar]
- Ledeen R, Salsman K, Cabrera M. Gangliosides in subacute sclerosing leukoencephalitis: isolation and fatty acid composition of nine fractions. J Lipid Res. 1968;9:129–136. [PubMed] [Google Scholar]
- Ledeen RW. Ganglioside structures and distribution: are they localized at the nerve ending? J Supramol Struct. 1978;8:1–17. doi: 10.1002/jss.400080102. [DOI] [PubMed] [Google Scholar]
- Levine SM, Seyfried TN, Yu RK, Goldman JE. Immunocytochemical localization of GD3 ganglioside to astrocytes in murine cerebellar mutants. Brain Res. 1986;374:260–269. doi: 10.1016/0006-8993(86)90420-8. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wada R, Kawai H, Sango K, Deng C, Tai T, McDonald MP, Araujo K, Crawley JN, Bierfreund U, Sandhoff K, Suzuki K, Proia RL. A genetic model of substrate deprivation therapy for a glycosphingolipid storage disorder. J Clin Invest. 1999;103:497–505. doi: 10.1172/JCI5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- Macala LJ, Yu R. K. a., S A. Analysis of brain lipids by high performance thin-layer chromatography and densitometry. J Lipid Res. 1983;24:1243–1250. [PubMed] [Google Scholar]
- MacDonald ME, Gusella JF. Huntington’s disease: translating a CAG repeat into a pathogenic mechanism. Curr Opin Neurobiol. 1996;6:638–643. doi: 10.1016/s0959-4388(96)80097-3. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Max SR, Maclaren NK, Brady RO, Bradley RM, Rennels MB, Tanaka J, Garcia JH, Cornblath M. GM3 (hematoside) sphingolipodystrophy. N Engl J Med. 1974;291:929–931. doi: 10.1056/NEJM197410312911802. [DOI] [PubMed] [Google Scholar]
- Ross CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron. 2002;35:819–822. doi: 10.1016/s0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Sandhoff K, Harzer K, Wassle W, Jatzkewitz H. Enzyme alterations and lipid storage in three variants of Tay-Sachs disease. J Neurochem. 1971;18:2469–2489. doi: 10.1111/j.1471-4159.1971.tb00204.x. [DOI] [PubMed] [Google Scholar]
- Schwarz A, Futerman AH. The localization of gangliosides in neurons of the central nervous system: the use of anti-ganglioside antibodies. Biochim Biophys Acta. 1996;1286:247–267. doi: 10.1016/s0304-4157(96)00011-1. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Petronilli V, Di Lisa F, Bernardi P. Commitment to apoptosis by GD3 ganglioside depends on opening of the mitochondrial permeability transition pore. J Biol Chem. 1999;274:22581–22585. doi: 10.1074/jbc.274.32.22581. [DOI] [PubMed] [Google Scholar]
- Seyfried TN, Ando S, Yu RK. Isolation and characterization of human liver hematoside. J Lipid Res. 1978a;19:538–543. [PubMed] [Google Scholar]
- Seyfried TN, Bernard DJ, Yu RK. Cellular distribution of gangliosides in the developing mouse cerebellum: analysis using the staggerer mutant. J Neurochem. 1984;43:1152–1162. doi: 10.1111/j.1471-4159.1984.tb12856.x. [DOI] [PubMed] [Google Scholar]
- Seyfried TN, Glaser GH, Yu RK. Cerebral, cerebellar, and brain stem gangliosides in mice susceptible to audiogenic seizures. J Neurochem. 1978b;31:21–27. doi: 10.1111/j.1471-4159.1978.tb12428.x. [DOI] [PubMed] [Google Scholar]
- Seyfried TN, Yu RK. Heterosis for brain myelin content in mice. Biochem Genet. 1980;18:1229–1237. doi: 10.1007/BF00484350. [DOI] [PubMed] [Google Scholar]
- Seyfried TN, Yu RK. Cellular localization of gangliosides in the mouse cerebellum: analysis using neurological mutants. Adv Exp Med Biol. 1984;174:169–181. doi: 10.1007/978-1-4684-1200-0_15. [DOI] [PubMed] [Google Scholar]
- Seyfried TN, Yu RK. Ganglioside GD3: structure, cellular distribution, and possible function. Mol Cell Biochem. 1985;68:3–10. doi: 10.1007/BF00219383. [DOI] [PubMed] [Google Scholar]
- Sheikh KA, Sun J, Liu Y, Kawai H, Crawford TO, Proia RL, Griffin JW, Schnaar RL. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc Natl Acad Sci U S A. 1999;96:7532–7537. doi: 10.1073/pnas.96.13.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J, Kooperberg C, Olson JM, Cattaneo E. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum Mol Genet. 2002;11:1953–1965. doi: 10.1093/hmg/11.17.1953. [DOI] [PubMed] [Google Scholar]
- Svennerholm L. Ganglioside designation. Adv Exp Med Biol. 1980;125:11. doi: 10.1007/978-1-4684-7844-0_2. [DOI] [PubMed] [Google Scholar]
- Svennerholm L. Designation and schematic structure of gangliosides and allied glycosphingolipids. Prog Brain Res. 1994;101:XI–XIV. doi: 10.1016/S0079-6123(08)61935-4. [DOI] [PubMed] [Google Scholar]
- Svennerholm L, Raal A. Composition of brain ganglio-sides. Biochim Biophys Acta. 1961;53:422–424. doi: 10.1016/0006-3002(61)90460-7. [DOI] [PubMed] [Google Scholar]
- Takamiya K, Yamamoto A, Furukawa K, Yamashiro S, Shin M, Okada M, Fukumoto S, Haraguchi M, Takeda N, Fujimura K, Sakae M, Kishikawa M, Shiku H, Furukawa K, Aizawa S. Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc Natl Acad Sci U S A. 1996;93:10662–10667. doi: 10.1073/pnas.93.20.10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai Y, Kojima H, Ikuta F, Kumanishi T. Alterations in the composition of brain lipids in patients with Creutzfeldt-Jakob disease. J Neurol Sci. 1978;35:59–76. doi: 10.1016/0022-510x(78)90102-8. [DOI] [PubMed] [Google Scholar]
- Thomas EA.Striatal Specificity of Gene Expression Dysregulation in Huntington’s disease J Neurosci Res 2006. in press [DOI] [PubMed] [Google Scholar]
- Valenza M, Rigamonti D, Goffredo D, Zuccato C, Fenu S, Jamot L, Strand A, Tarditi A, Woodman B, Racchi M, Mariotti C, Di Donato S, Corsini A, Bates G, Pruss R, Olson JM, Sipione S, Tartari M, Cattaneo E. Dysfunction of the cholesterol biosynthetic pathway in Huntington’s disease. J Neurosci. 2005;25:9932–9939. doi: 10.1523/JNEUROSCI.3355-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Vyas AA, Schnaar RL. Brain gangliosides: functional ligands for myelin stability and the control of nerve rege neration. Biochimie. 2001;83:677–682. doi: 10.1016/s0300-9084(01)01308-6. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Wu YP, Sandhoff R, Werth N, Mizukami H, Ellis JM, Dupree JL, Geyer R, Sandhoff K, Proia RL. Interruption of ganglioside synthesis produces central nervous system degeneration and altered axon-glial interactions. Proc Natl Acad Sci U S A. 2005;102:2725–2730. doi: 10.1073/pnas.0407785102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Kojima N, Kurosawa N, Hamamoto T, Tsuji S. Molecular cloning of Sia alpha 2,3Gal beta 1,4GlcNAc alpha 2,8-sialyltransferase from mouse brain. J Biol Chem. 1995;270:14628–14633. doi: 10.1074/jbc.270.24.14628. [DOI] [PubMed] [Google Scholar]
- Yu RK, Bieberich E, Xia T, Zeng G. Regulation of ganglioside biosynthesis in the nervous system. J Lipid Res. 2004;45:783–793. doi: 10.1194/jlr.R300020-JLR200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.