

Abstract
The subunit orientation of a dimeric enzyme influences the mechanism of action and function. To determine the subunit arrangement of lipoprotein lipase (LPL), a molecular biology-based approach was initiated. An eight amino acid linker region was engineered between two LPL monomers and expressed in COS-7 cells. The resultant tandem-repeat molecule (LPLTR) was lipolytically active and had kinetic parameters, salt inhibition, cofactor-dependent activity, heparin-binding characteristics, and a functional unit size very similar to the expressed native human enzyme. By these criteria, LPLTR was the functional equivalent of native LPL. Considering the length of the linker peptide (no more than 24 Å), monomers in the tethered molecule were restricted to a head-to-tail subunit arrangement. Since LPLTR demonstrated native enzyme-like properties while constrained to this subunit arrangement, these results provide the first compelling evidence that native LPL monomers are arranged in a head-to-tail subunit orientation within the active dimer. Thus, LPL function in physiology, lipolysis, and binding to cell-surface components must now be addressed with this subunit orientation in mind. The utility of the tandem-repeat approach to resolve the subunit arrangement of an obligate dimer has been demonstrated with LPL and could be generalized for use with other oligomeric enzymes.
Lipoprotein lipase (LPL) plays a vital role in lipoprotein metabolism. The functional enzyme is bound to the capillary endothelium of numerous tissues and hydrolyzes chylomicron and VLDL triglycerides to free fatty acids for energy utilization and triglyceride synthesis (1–4). Also, a role for LPL, via a noncatalytic mechanism in remnant catabolism has been postulated, whereby lipoproteins are bridged by the lipase to cell-surface components for intracellular metabolism (5–10). The essential physiological role of this enzyme is underscored by observations that LPL-defective mice are not viable (11, 12) and that human LPL-deficiency is marked by hypertriglyceridemia, a risk factor for atherosclerosis (13, 14).
Both LPL hydrolysis of lipid substrates and lipoprotein bridging are dependent on a dimer subunit structure (5, 15). A detailed understanding of LPL subunit arrangement is an initial step in finding mechanisms involved in these important enzyme functions. However, subunit orientation within the native dimer is not known. Lipoprotein-binding studies and structure–function investigations using monoclonal antibodies (16) provide indirect evidence for a head-to-tail subunit arrangement. On the other hand, computer modeling studies have generated an LPL molecule more in accord with a head-to-head subunit orientation (17). Clarification of LPL subunit organization would assist in the evaluation of interdomain and intersubunit interactions, as well as enzyme with substrate and enzyme to cell-surface component interactions.
Methods previously used to determine protein subunit orientation have relied generally on physical observations, such as crystallography or electron microscopy. As an alternative, we have extended a molecular biology-based method (18) and used it to examine LPL subunit arrangement. The method assumes that LPL quaternary structure underlies overall activity, i.e., subunits must interact in a precise way to achieve proper functional juxtaposition, and in the absence of these crucial contacts the enzyme is inactive. It was considered that an engineered peptide joining LPL subunits could be used to differentiate between the various models proposed for the enzyme (16, 17). As a first test of this approach, a linker region was engineered between the end of the first LPL monomer and the start of the second. It was essential that the region joining the two subunits be too short to permit a head-to-head subunit organization, thereby constraining dimers into a head-to-tail arrangement. The retention of catalytic activity and other functional parameters in such a monomer–repeat LPL molecule would then provide strong evidence of a head-to-tail subunit orientation in the native enzyme.
This report presents findings on such an LPL tandem repeat molecule (LPLTR). LPL monomers, joined by an eight amino acid linker, were expressed in COS cells and characterized for lipolytic activity, kinetic parameters, heparin binding, and sucrose gradient centrifugation. By the criteria of lipase activity, kinetic constants, and heparin binding, LPLTR was the functional equivalent of native LPL. Sucrose gradient centrifugation indicated that LPLTR was a functional monomer, suggesting that in the native LPL dimer, two monomer subunits are arranged in a head-to-tail manner. To our knowledge, this is the first demonstration of LPL subunit structure orientation, resolved by a new method that could be used for other oligomeric proteins.
EXPERIMENTAL PROCEDURES
Construction of a Tandem Repeat of LPL.
Two human LPL cDNAs (19) were amplified separately by the polymerase chain reaction (PCR) and contained engineered restriction endonuclease sites to facilitate assembly and subcloning into the expression plasmid pcDNA3 (Invitrogen).
Primers.
The NH2 terminal primer of the first LPL cDNA (TGACAAGCTTGCCACCATGGAGAGCAAAGCCCTGCTC) contained an HindIII restriction site and the first 21 coding nucleotides of the LPL cDNA. The reverse primer (CGATGGATCCGCCTGACTTCTTATTCAG) contained a BamHI restriction site replacing the stop codon followed by the reverse complement to the last six codons of LPL. The second LPL cDNA was amplified using the forward primer (CGATGGATCCATCGAAGGTCGTCTCGAGGCCGACCAAAGAAGAG), which contained a BamHI restriction site, a Factor Xa site, an XhoI restriction site, and the first 16 nucleotides of the mature LPL protein. The COOH-terminal primer (ACGTTCTAGAGAATTCACATGCCGTTCTTTG) contained an XbaI restriction site and the reverse complement to 20 bases of the 3′ untranslated region of the LPL cDNA.
PCR Conditions.
Each amplification reaction (50 μl) contained 50 ng of plasmid DNA template, 800 ng of each primer, 200 μM of each deoxynucleotide, 1× reaction buffer (New England Biolabs) supplemented with MgSO4 (2 mM final concentration), 1% dimethyl sulfoxide (DMSO), 100 μg/ml bovine serum albumin, and 1 unit of VENT polymerase. Reactions proceeded for 45 s at 95°C, 45 s at 58°C, and 90 s at 72°C for a total of 25 cycles. Products were purified, digested, and inserted sequentially into the pcDNA3 expression vector using the HindIII, BamHI, and XbaI restriction endonuclease sites. The final construct contained sequences coding for the LPL signal peptide, residues 1–448, an 8 amino acid peptide linker containing a Factor Xa site, residues 1–448, and 20 bases of the LPL 3′ untranslated region.
DNA Transfection and Expression.
COS-7 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum and antibiotics. To mediate the transfection of COS cells, coprecipitates of plasmid DNA and CaPO4 were prepared according to the manufacturer’s instructions (Invitrogen) and applied to 60–70% confluent COS cells in a 10-cm dish. After an overnight incubation, cells were rinsed with PBS before they were treated with 3 ml of 10% (vol/vol) DMSO in PBS solution for 2.5 min. After removal of the DMSO solution, fresh DMEM supplemented with 1% serum substitute (Nutridoma) and 20 units/ml heparin was added to each dish. The medium was harvested and replaced every 24 h for a 3-day period. After centrifugation at 1000 × g for 10 min to remove cellular debris, the harvested medium was stored at −80°C.
Heparin-Sepharose Chromatography.
Ten milliliters of medium from COS cells transfected with either the wild-type LPL cDNA or LPLTR construct were applied to a heparin-Sepharose column (5 × 40 mm) equilibrated with 5 mM sodium barbital (pH 7.2), containing 0.35 M NaCl/10% (vol/vol) glycerol. The column was extensively washed with equilibration buffer and then eluted with a linear NaCl gradient (0.35–2.0 M) in 5 mM sodium barbital (pH 7.2), containing 10% (vol/vol) glycerol. Fractions were assayed for lipase activity and the salt concentration monitored by conductivity measurements.
Sucrose Gradient Ultracentrifugation.
Sucrose gradients were prepared as described (20). Briefly, 5–20% sucrose gradients containing 50 mM ammonium hydroxide (pH 8.0), 0.2% sodium deoxycholate, and 10 units/ml heparin were prepared. Glucose-6-phosphate dehydrogenase (G6PDH) (114 kDa) and malate dehydrogenase (MDH) (74 kDa) were added to each sample to serve as internal standards. Cytochrome c (12.5 kDa), ovalbumin (45 kDa), and catalase (240 kDa) were simultaneously centrifuged in a separate tube and acted as external standards. After centrifugation (22 h at 202,000 × g at 4°C), 480-μl fractions were collected and assayed for enzyme activity and protein concentration.
Enzyme Assays.
LPL activity was monitored using radiolabeled triolein substrate (21). Apolipoprotein CII-dependent lipase activity was determined by assaying medium in the presence or absence of rat serum (3% vol/vol). The effect of salt concentration on LPL activity was determined by assaying in the presence of either 0.15 or 1 M NaCl. Vmax and Km(apparent) of triolein hydrolysis for LPL and LPLTR were calculated after varying substrate concentration from 0.028–2.8 mM. Antibody inhibition of enzyme activity was performed by incubating 20 μl of medium from transfected COS cells with increasing amounts of anti-LPL chicken serum at 4°C for 1 h prior to lipase activity determinations. Nonimmune chicken serum was used as a negative control. G6PDH (22) and MDH (23) activities were assayed by established methods. Protein concentration was determined by the Bradford method using bovine serum albumin as a standard protein (24).
RESULTS
To maximize LPLTR expression, ensure a short linker region, and simplify product identification, the following steps were incorporated in construct generation by PCR. First, the leader peptide coding sequences were included only for the first LPL monomer and deleted from the second. Second, the linker region was limited to eight amino acids, to preclude head-to-head subunit association. Finally, sequences coding for the linker region included those that specified a Factor Xa cleavage site (IEGR) to facilitate the identification of Factor Xa-susceptible expression products. The completed LPLTR construct consisted of sequences coding for residues −20–448 (first monomer), GSIEGRLE (linker), and 1–448 (second monomer) (Fig. 1).
Figure 1.
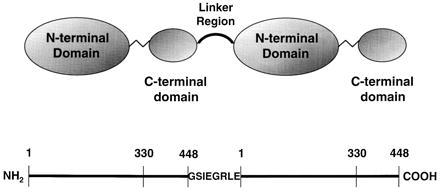
Schematic diagram of the LPLTR molecule. The top portion indicates two LPL monomers, with domain designations (16), joined by an eight amino acid linker region (boldface type). The linker region sequence is shown below and contains a Factor Xa cleavage site (IEGR). In the lower portion, approximate boundaries of LPL domains and the linker region are displayed on a linear representation of the molecule. The numbers correspond to amino acid residues in the mature, secreted molecule.
COS cells were transfected by the vector lacking insert, LPLTR, and native LPL constructs. Only cells transfected with the LPLTR and LPL constructs had detectable lipase activity in the culture medium (Fig. 2). Lipase activity of LPLTR and LPL was stimulated by serum and inhibited by 1 M NaCl, both diagnostic of the native enzyme (Fig. 2 A and B). Salt inhibition of LPLTR was less pronounced then that for LPL, suggesting that tethered monomers were slightly more resistant to ionic perturbation. In addition, the lipolytic activity of both enzymes was inhibited to the same extent by monospecific polyclonal antiserum raised against purified LPL (Fig. 2C). Furthermore, Km and kcat/Km for both LPLTR and LPL were very similar, indicating that substrate affinity and catalytic efficiency were not compromised by covalent linkage of two LPL monomers (Table 1). By these activity-based criteria, LPLTR was the functional equivalent of the native enzyme.
Figure 2.
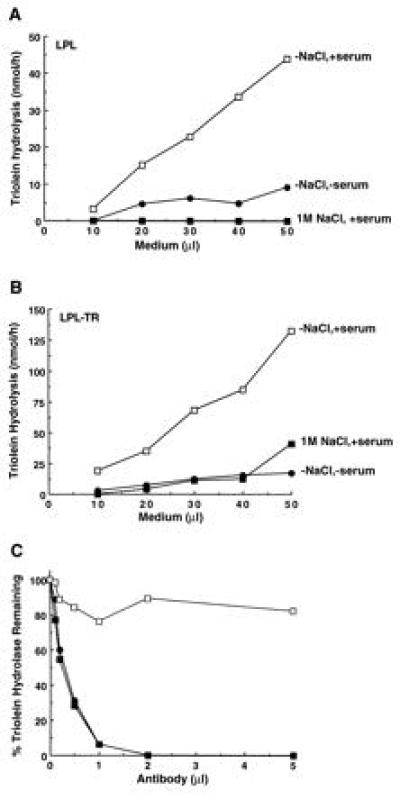
LPL and LPLTR lipolytic activity: susceptibility to NaCl, serum, and monospecific antibodies. In A (for LPL) and B (for LPLTR), the triolein hydrolase activity of varying amounts of medium was determined under normal assay conditions (open symbols), or in the presence of 1 M NaCl (▪) or absence of serum (•). (C) Inhibition of LPL (•) and LPLTR (▪) by monospecific anti-LPL antiserum. Also shown is the effect of nonimmune serum on LPLTR lipase activity (□). The 100% values for LPL and LPLTR were 36 and 44 nmol/min per ml, respectively.
Table 1.
LPL and LPLTR kinetic constants
| kcat nmol/min per ml | Km, mM | Catalytic efficiency, kcat/Km | |
|---|---|---|---|
| LPL | 8,390 | 0.32 | 26,200 |
| LPLTR | 4,620 | 0.20 | 23,110 |
Medium from COS cells transfected with LPL or LPLTR was assayed for triolein hydrolase activity. kcat/Km was derived from values calculated by the parameter fit program from Elsevier Biosoft. Representative data from two experiments are shown.
Western blot analyses of medium showed an ≈110-kDa protein specifically expressed by cells transfected with the LPLTR construct (Fig. 3, lane 3). The 110-kDa band was absent when the medium was treated with Factor Xa, presumably the consequence of the engineered cleavage site in LPLTR (data not shown). In addition, a band of ≈55 kDa was detected in the medium from cells transfected with LPL (Fig. 3, lane 2), whereas neither band was seen in cells transfected with the vector lacking an insert, although a nonspecific 66-kDa band was observed (Fig. 3, lane 1). These findings indicated specific expression and secretion of LPLTR and LPL and suggested that these products were responsible for the lipase activity observed in the culture medium.
Figure 3.
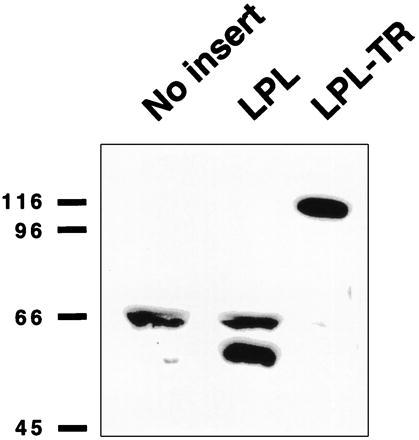
Western blot analysis of transfected cell medium. Medium from cells transfected with vector lacking an insert (lane 1; No insert), LPL (lane 2), or LPLTR (lane 3; LPL-TR) was separated by SDS/PAGE, electroblotted, and probed with monospecific antiserum to LPL. For lanes 2 and 3, the amount of medium loaded was adjusted to display the same amount of activity units (≈150 nmol/h; 150 and 50 μl, respectively); lane 1 received the volume equivalent of lane 2. The migration positions of molecular weight standards (in kDa) are indicated on the left. LPL-transfected cell medium demonstrated a specific band at ≈55 kDa; that of LPLTR at ≈110 kDa.
To compare additional functional parameters between the enzymes, heparin-binding studies were performed (Fig. 4). LPLTR and LPL elution positions from heparin-Sepharose columns were determined and established to be nearly identical; both enzymes eluted from the columns at 1.2 M NaCl (Fig. 4), a concentration characteristic of active, dimeric native LPL (25). This finding demonstrates that no gross misfolding of elements critical for heparin binding had occurred as a result of tethering two monomers together in the LPLTR molecule.
Figure 4.
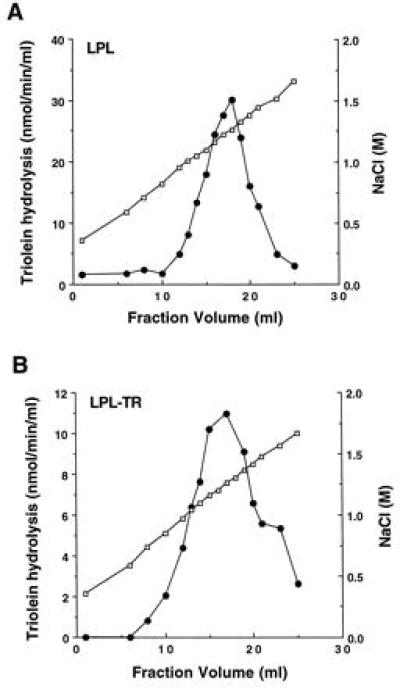
LPL (A) and LPLTR (B) heparin binding. LPL and LPLTR medium was loaded on separate heparin-Sepharose columns, washed, and eluted with an NaCl gradient. Fractions were assayed for salt content by conductivity measurements (open symbols) and for lipase activity (solid symbols). Total activity recovered for both enzyme preparations was >70%.
Both kinetic characteristics and heparin-binding behavior indicated that LPLTR was near-identical to the native enzyme. However, a possibility existed that LPLTR was not functioning as a monomer, but as a larger size oligomer. Therefore, sucrose gradient centrifugation was used to determine the size of the functional unit. Lipase activity of LPLTR sedimented as a distinct species about the same size as G6PDH (Fig. 5A). LPL also sedimented near that position, although slightly smaller in size (Fig. 5B). A linear relationship was observed when the molecular weight of standard proteins versus sucrose concentrations was plotted (Fig. 5C); the molecular mass of the LPL functional unit was ≈100 kDa, that of LPLTR ≈110 kDa. These data clearly indicate the functional size of LPLTR to be a monomeric protein or, equivalently, a tethered dimer. LPLTR is not an oligomer, rather its functional size closely corresponds to that of native LPL. In view of the restricted length of the linker region (8 amino acids versus 448 for the LPL monomer), LPLTR likely folds back on itself in a head-to-tail manner and, significantly, assumes a lipolytically active conformation. In turn, these findings suggest that the monomer arrangement in native LPL is also in a head-to-tail orientation.
Figure 5.
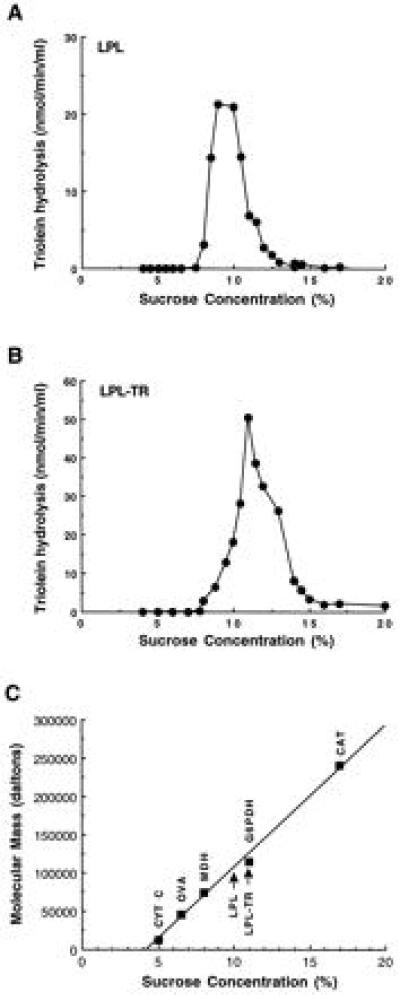
Size of the LPL and LPLTR functional unit: determination by sucrose gradient centrifugation. Medium from LPL (A) and LPLTR (B) transfected cells was centrifuged into a 5–20% sucrose gradient and fractionated for lipase activity determinations. Sucrose concentration for each fraction was determined by refractive index measurements. (C) Peak sedimentation position of standard proteins performed in the same experiment. LPL and LPLTR activity peaks are indicated by arrows. cyt C, cytochrome c (12.5 kDa); OVA, ovalbumin (45 kDa); MDH, (74 kDa); G6PDH, (114 kDa); CAT, catalase (240 kDa).
DISCUSSION
Dimeric enzymes are frequently found in nature and knowledge of their subunit orientation is important when investigating structure–function relationships. Previous ways to determine subunit orientation have mostly relied on physical methods to examine proteins in a static state. Our studies are based on a new approach that used cloning techniques to generate molecules in a head-to-tail subunit arrangement. Sucrose gradient results indicated that LPLTR was the same size as dimeric native enzyme (≈100 kDa) and was lipolytically active. However, since gradient and assay conditions necessarily differed due to detergent effects, a possibility remains that LPLTR changed subunit structure from monomer to oligomer during the course of the assay. We consider this possibility remote, because the native enzyme does not convert from monomer to oligomer (26). Rather, it is more likely that LPLTR was active as a monomeric molecule (or equivalently, a tethered dimer) while constrained to a head-to-tail subunit arrangement by the short linker region. Additional findings established that the tandem linkage of LPL monomers resulted in the production of a lipase with kinetic constants (Table 1), catalytic properties (Fig. 2), and heparin binding (Fig. 4) similar to those of LPL. Interpretation of these results within the context of the short linker region leads to the conclusion that one-half of the molecule folded back on itself in a head-to-tail subunit arrangement. Therefore, because LPLTR is virtually indistinguishable from native LPL by several criteria, the subunit arrangement in the native enzyme is also likely to be in a head-to-tail manner.
The other possible LPL subunit arrangement, head-to-head, could have been examined by other strategies. For example, one study engineered a leucine zipper moiety and lone sulfhydryl group onto the 3′ end of a construct to form a dimeric enzyme in a head-to-head arrangement (27). Conclusions reached were credible because the engineered enzyme retained catalytic activity, implying native or near-native conformation. This approach was unsuitable for examining LPL subunit orientation because sulfhydryl substitution itself renders the enzyme inactive. More importantly, pursuit of alternate strategies was deemed unnecessary given the results of the tandem-repeat experiments and the likelihood that a dimeric enzyme would form an active species in only one subunit arrangement. However, a combination of these two methods affords a way to determine the subunit orientation of other obligate dimers, providing enzymatic activity is preserved following the engineered change.
Investigation of LPL structure–function relationships by the use of genetic engineering techniques is motivated and underscored by the lack of crystal structure information for this critical enzyme of lipid metabolism. In this void, alternative methods have been employed to better understand the molecular interactions underlying lipase function. Site-directed mutagenesis is one example used to examine LPL functional groups (28–30). However, since this method generally results in the production of inactive protein, it is not known whether loss of function is a direct result of the change, or due to a global effect at a remote site. On the other hand, the domain-exchange strategy can create active chimeric lipases, which retain parental characteristics, thereby revealing the relative location of specific enzyme functions (31–33). However, neither technique directly addresses the question of subunit orientation and motivated the development of another approach for LPL subunit orientation determination.
The tandem-repeat strategy described here augments existing approaches of lipase engineering. LPL monomers, artificially restricted to a head-to-tail dimer orientation, displayed catalytic activity. This finding implies that LPLTR secondary, tertiary, and quaternary structures were not significantly altered, and that folding of regions into domains, interdomain interactions, and crucial intersubunit associations were maintained in a native, or near-native state. In conjunction with other techniques, such as crystallography and NMR spectroscopy, this strategy could reveal new insights into the mechanism by which catalysis is accomplished. These studies represent initial attempts to determine subunit orientation by using a molecular biology-based approach; more sophisticated studies will be used in the future to better understand the underlying principles of lipolysis in this physiologically important class of enzymes.
Acknowledgments
We would like to thank Osnat Ben-Zeev and Mark Doolittle for advice with sucrose gradients and Western blots. We also thank Ron Kaback for critical reading of the manuscript. This work was supported by Veterans Affairs Merit Review and National Institutes of Health Grant HL28481. J.S.H. was supported by a Medical Research Council of Canada fellowship.
ABBREVIATIONS
- LPL
lipoprotein lipase
- LPLTR
LPL monomer tandem repeat
- G6PDH
glucose-6-phosphate dehydrogenase
- MDH
malate dehydrogenase
References
- 1.Jackson R L. In: The Enzymes. Boyer P D, editor. Vol. 16. New York: Academic; 1983. pp. 141–180. [Google Scholar]
- 2.Semenkovich C F, Luo C-C, Nakanishi M K, Chen S-H, Smith L C, Chan L. J Biol Chem. 1990;265:5429–5433. [PubMed] [Google Scholar]
- 3.Olivecrona T, Bengtsson-Olivecrona G. Curr Opin Lipidol. 1990;1:222–230. [Google Scholar]
- 4.Santamarina-Fojo S, Dugi K A. Curr Opin Lipidol. 1994;5:117–125. doi: 10.1097/00041433-199404000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Beisiegel U, Weber W, Bengtsson-Olivecrona G. Proc Natl Acad Sci USA. 1991;88:8342–8346. doi: 10.1073/pnas.88.19.8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chappell D A, Fry G L, Waknitz M A, Iverius P-H, Williams S E, Strickland D K. J Biol Chem. 1992;267:25764–25767. [PubMed] [Google Scholar]
- 7.Nykjaer A, Bengtsson-Olivecrona G, Lookene A, Moestrup S K, Peterson C M, Weber W, Beisiegel U, Gliemann J. J Biol Chem. 1993;268:15048–15055. [PubMed] [Google Scholar]
- 8.Williams S E, Inoue I, Tran H, Fry G L, Pladet M W, Iverius P-H, Lalouel J-M, Chappell D A, Strickland D K. J Biol Chem. 1994;269:8653–8658. [PubMed] [Google Scholar]
- 9.Nykjaer A, Nielson M, Lookene A, Meyer N, Roigaard H, Etzerodt M, Beisiegel U, Olivecrona G, Gliemann J. J Biol Chem. 1994;269:31747–31755. [PubMed] [Google Scholar]
- 10.Chappell D A, Fry G L, Waknitz M A, Muhonen L E, Pladet M W, Iverius P H, Strickland D K. J Biol Chem. 1993;268:14168–14175. [PubMed] [Google Scholar]
- 11.Coleman T, Seip R L, Gimble J M, Lee D, Maeda N, Semenkovich C F. J Biol Chem. 1995;270:12518–12525. doi: 10.1074/jbc.270.21.12518. [DOI] [PubMed] [Google Scholar]
- 12.Weinstock P H, Bisgaier C L, Aaltosetala K, Radner H, Ramakrishnan R, Levakfrank S, Essenburg A D, Zechner R, Breslow J L. J Clin Invest. 1995;96:2555–2568. doi: 10.1172/JCI118319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hegele R A, Vezna C, Moorjani S, Lupien P J, Gagne C, Brun L-D, Little J A, Connelly P W. J Clin Endocrinol Metab. 1991;72:730–732. doi: 10.1210/jcem-72-3-730. [DOI] [PubMed] [Google Scholar]
- 14.Connelly P W, Maguire G F, Lee M, Little J A. Am J Hum Genet. 1990;46:470–477. [Google Scholar]
- 15.Garfinkel A S, Kempner E S, Ben-Zeev O, Nikazy J, James S J, Schotz M C. J Lipid Res. 1983;24:775–780. [PubMed] [Google Scholar]
- 16.Wong H, Davis R C, Thuren T, Goers J W, Nikazy J, Waite M, Schotz M C. J Biol Chem. 1994;269:10319–10323. [PubMed] [Google Scholar]
- 17.Van Tilbeurgh H, Roussel A, Lalouel J-M, Cambillau C. J Biol Chem. 1994;269:4626–4633. [PubMed] [Google Scholar]
- 18.Sahin-Toth M, Lawrence M C, Kaback H R. Proc Natl Acad Sci USA. 1994;91:5421–5425. doi: 10.1073/pnas.91.12.5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wion K L, Kirchgessner T G, Lusis A J, Schotz M C, Lawn R M. Science. 1987;235:1638–1641. doi: 10.1126/science.3823907. [DOI] [PubMed] [Google Scholar]
- 20.Ashby P, Tolson A M, Robinson D S. Biochem J. 1978;171:305–311. doi: 10.1042/bj1710305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nilsson-Ehle P, Schotz M C. J Lipid Res. 1976;17:536–541. [PubMed] [Google Scholar]
- 22.Olive C, Levy R. Methods Enzymol. 1975;41:196–201. doi: 10.1016/s0076-6879(75)41046-1. [DOI] [PubMed] [Google Scholar]
- 23.Akira Y. Methods Enzymol. 1969;13:141–142. [Google Scholar]
- 24.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 25.Peterson J, Fujimoto W Y, Brunzell J D. J Lipid Res. 1992;33:1165–1170. [PubMed] [Google Scholar]
- 26.Osborne J C, Jr, Bengtsson-Olivecrona G, Lee N S, Olivecrona T. Biochemistry. 1985;24:5606–5611. doi: 10.1021/bi00341a048. [DOI] [PubMed] [Google Scholar]
- 27.Lindsley J E. Proc Natl Acad Sci USA. 1996;93:2975–2980. doi: 10.1073/pnas.93.7.2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emmerich J, Beg O U, Peterson J, Previato L, Brunzell J D, Brewer H B, Jr, Santamarina-Fojo S. J Biol Chem. 1992;267:4161–4165. [PubMed] [Google Scholar]
- 29.Hata A, Ridinger D N, Sutherland S, Emi M, Shuhua Z, Myers R L, Ren K, Cheng T, Inoue I, Wilson D E, Iverius P-H, Lalouel J-M. J Biol Chem. 1993;268:8447–8457. [PubMed] [Google Scholar]
- 30.Ma Y, Henderson H E, Liu M-S, Zhang H, Forsythe I J, Clarke-Lewis I, Hayden M R, Brunzell J D. J Lipid Res. 1994;35:2049–2059. [PubMed] [Google Scholar]
- 31.Wong H, Davis R C, Nikazy J, Seebart K E, Schotz M C. Proc Natl Acad Sci USA. 1991;88:11290–11294. doi: 10.1073/pnas.88.24.11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis R C, Wong H, Nikazy J, Wang K, Han Q, Schotz M C. J Biol Chem. 1992;267:21499–21504. [PubMed] [Google Scholar]
- 33.Dichek H L, Parrott C, Ronan R, Brunzell J D, Brewer H B, Santamarina-Fojo S. J Lipid Res. 1993;34:1394–1401. [PubMed] [Google Scholar]