

Abstract
The mouse RNA-binding protein, TB-RBP, suppresses translation in vitro and attaches mRNAs to microtubules by binding to conserved elements in the 3′ untranslated regions of specific mRNAs. We have now purified TB-RBP from testicular and brain cytoplasmic extracts and cloned its cDNA. We find that the mouse TB-RBP cDNAs contain an open reading frame of 228 amino acids with a leucine zipper domain within its C terminus, a transmembrane helix, and a group of putative phosphorylation sites. TB-RBP shows 99% identity to the human protein, translin, a recombination hotspot-binding protein associated with chromosomal translocations [Aoki, K., Suzuki, K., Sugano, T., Tasaka, T., Nakahara, K., Kuge, O., Omori, A. & Kasai, M. (1995) Nat. Genet. 10, 167–174]. As shown for translin, TB-RBP also binds to single-stranded DNAs containing a broad range of consensus sequences, many of which are similar to the Y and H RNA-binding sequences. Recombinant TB-RBP was synthesized and an antiserum was prepared against the recombinant protein. The identity between translin and TB-RBP was confirmed by demonstrating that immunoprecipitation of TB-RBP from testicular extracts abolished formation of the RNA-TB–RBP complex. Based upon its DNA binding to target sequences in clustered breakpoint regions, we propose that TB-RBP may be involved in DNA recombination or DNA repair in male germ cells.
RNA–protein interactions modulate gene expression at many different posttranscriptional levels (reviewed in refs. 1–3). In addition to mRNA processing and transport from the nucleus to the cytoplasm, RNA-binding proteins facilitate mRNA translation, stability, and subcellular localization. In many cases, these regulatory proteins recognize cis-acting sequences within the 5′ or 3′ untranslated regions (UTRs) of mRNAs (4–8). Such interactions are especially common during oogenesis, where RNA-binding proteins help mask, accumulate, localize, and ultimately translate many “maternal” mRNAs (reviewed in refs. 4, 9, and 10).
In the testis, an analogous situation exists for “paternal” mRNAs in the differentiating male germ cells. This occurs because many sperm-specific proteins are synthesized after cessation of transcription in the haploid phase of spermatogenesis. This necessitates posttranscriptional regulation for many mRNAs, which are transcribed in meiotic or postmeiotic cells and undergo spatial and temporal translation at specific developmental times (reviewed in refs. 4 and 11–14). The importance of the 3′ UTR for translational control in mRNAs encoding proteins such as the sperm nuclear protein, protamine 1, has been well documented in “in vitro” studies and in transgenic mice (15–18).
We have previously identified a phosphoprotein, testis brain–RNA-binding protein (TB-RBP), that specifically binds to highly conserved cis-acting sequences in the 3′ UTRs of a number of testicular and brain mRNAs, including protamines 1 and 2 (15, 19). TB-RBP represses the in vitro translation of mRNA constructs containing specific conserved sequences of protamine 2 (15) and also binds mRNAs encoding proteins, such as tau and myelin basic protein, to microtubules (16, 20).
To begin to define the mechanisms of action of TB-RBP, we have isolated TB-RBP from testicular and brain cytoplasmic extracts and cloned its cDNA from a mouse testis cDNA library. We report here that TB-RBP is identical to the DNA-binding protein, translin, a protein that binds to a group of single-stranded DNA sequences present at breakpoint junctions of chromosomal translocations (21).
MATERIALS AND METHODS
Preparation of Testicular and Brain Extracts.
Testicular and brain cytoplasmic extracts were prepared from sexually mature male CD-1 mice (Charles River Breeding Laboratories) by a modified procedure of Kwon and Hecht (15) and Han et al. (20). The testes were decapsulated, washed three times with buffer A (10 mM Hepes, pH 7.6/1.5 mM MgCl2/10 mM KCl/0.5 mM DTT/0.5 mM phenylmethylsulfonyl flouride (PMSF)/0.5 μg/ml leupeptin/0.7 μg/ml pepstatin/2 μg/ml aprotinin) and resuspended in buffer A to a final volume of 0.5–1 g/ml. Testes were minced and homogenized in a Teflon glass homogenizer on ice until most of the cells were lysed. The homogenates were centrifuged for 15 min at 5,000 rpm in a Sorvall SS-34 rotor to remove tissue debris, unbroken cells, and nuclei. After 0.11 vol of buffer B (100 mM Hepes, pH 7.6/30 mM MgCl2/100 mM KCl) was added to the supernatants, the solution was centrifuged for 1 hr at 34,000 rpm in a Beckman SW 50.1 rotor. The high-speed supernatants were either used immediately or stored at −70°C. A similar protocol was followed to prepare brain cytosol extracts. The protein concentration of supernatants ranged from 10 to 20 mg/ml.
Preparation of RNA Transcripts.
For RNA gel retardation assays, [32P]-labeled RNAs were transcribed from a pGem 3Z plasmid containing transcript c (a 67 nucleotide insert containing the Y and H elements of the 3′ UTR of mP2) (19). Transcriptions were carried out with 20 units of SP6 polymerase, 60 μCi of [32P]CTP (3000 Ci/mmol; 1 Ci = 37 GBq; Amersham)/19 μm unlabeled CTP, and 495 μm UTP, TTP, and ATP in a reaction volume of 20 μl. RNA transcripts were purified as previously described (20).
For RNA affinity chromatography, transcript c was subcloned into the HindIII and AvaI sites of the pSP64 poly(A) vector (Promega), generating the construct pSP64-C. After linearizing pSP64-C with EcoRI, SP6 polymerase was used to synthesize RNA containing transcript c and 30 nucleotides of poly(A).
UV Crosslinking of RNA–Protein Complexes.
Gel retardation assays of RNA–protein complexes were carried out by the method of Kwon and Hecht (19). For UV-crosslinking, RNA–protein complexes were irradiated on ice in a UV Stratalinker 1800 (Stratagene) with 254-nm, 8-W UV bulbs for 40 min, and resolved in 4% native polyacrylamide gels. The RNA–protein complexes, identified by autoradiography, were cut from the gels and digested with RNase T1 for 30 min at room temperature. After mixing with SDS buffer (125 mM Tris⋅HCl, pH 6.8/0.1% SDS/1 mM EDTA), the gel slices were placed on top of an SDS/15% polyacrylamide gel and electrophoresed to resolve the UV-crosslinked RNA–protein complex (20).
Affinity Purification of TB-RBP.
TB-RBP was purified by a modification of the method of Gu and Hecht (22). Polyadenylylated transcript c RNA was generated in vitro with the SP6 MEGAscript kit (Ambion) using the EcoRI linearized pSP64-C vector. To allow quantitation of RNA, a trace amount of [32P]CTP was incorporated into the RNA transcripts. One milliliter of poly(U) agarose beads (Pharmacia, type 6) was suspended in RNA-binding buffer (25 mM Hepes, pH 7.5/100 mM KCl) and packed into a 15-ml column. The polyadenylylated transcript c (200 μg in 3 ml of RNA-binding buffer) was loaded over the column at 4°C and recycled five times. The binding efficiency of the poly(A)+ RNA to the poly(U) agarose beads was determined by monitoring the [32P]-labeled RNA in the washes. After the RNA bound to the poly(U) agarose beads was equilibrated with the extract buffer (1 vol buffer A plus 0.11 vol buffer B), 25 ml of testicular or brain extracts (10–20 mg/ml) containing 60 units/ml RNasin and 5 mg/ml heparin was mixed with the affinity beads at room temperature. To maximize RNA-protein binding, the extract and RNA affinity beads were incubated for 1 hr with gentle shaking. The beads were then pelleted by centrifugation at 1,000 rpm (Beckman J2-21) for 5 min, resuspended in 20 ml of washing buffer A (20 mM Hepes, pH 7.5/40 mM KCl/5 mg/ml heparin) and packed into a 15-ml column. The column was washed with 20 ml washing buffer B (20 mM Hepes, pH 7.5/40 mM KCl/0.5% Nonidet P-40), 20 ml of washing buffer C (20 mM Hepes, pH 7.5/40 mM KCl/40 μg/ml poly(ACU)/0.5% Nonidet P-40), and 20 ml of washing buffer D (20 mM Hepes, pH 7.5/40 mM KCl/5 mg/ml heparin). Bound protein fractions were step eluted with 2 ml of 0.5 M, 1.0 M, and 1.5 M KCl in 20 mM Hepes (pH 7.5). The eluted protein fractions were dialyzed against buffer E (10 mM Hepes, pH 7.5/40 mM KCl/3 mM MgCl2/1 mM DTT/5% glycerol/0.5 mM PMSF) at 4°C before being analyzed by SDS/PAGE and RNA-binding assays.
Purification of TB-RBP by DEAE-Sepharose Chromatography.
The dialyzed protein fraction, containing the enriched TB-RBP from the RNA affinity column, was applied to a column packed with 1.5 ml DEAE-Sepharose 6B (Pharmacia) equilibrated with buffer E. After the protein was loaded, the column was washed with 20 ml of buffer E and then eluted with a step gradient of 100 mM, 300 mM, 500 mM, and 1,000 mM KCl in 10 mM Hepes (pH 7.5). After dialysis against buffer E, aliquots of fractions were assayed for RNA-binding activity and for protein purity by SDS/PAGE.
Determination of Partial Protein Sequence.
The protein fraction from the DEAE-Sepharose 6B column, containing the highly enriched TB-RBP, was concentrated with a Centricon-30 filter (Amicon) and aliquots were electrophoresed in SDS/12.5% PAGE. Proteins were transferred from the polyacrylamide gel to a polyvinylidene diflouride membrane with a Semi-Dry trans-Blot SD (Bio-Rad) at 20 V for 1 hr at 4°C. One protein band, corresponding to TB-RBP, was visualized by Ponceau S staining, cut out, and microsequenced by the Department of Microchemistry of Harvard University.
Isolation and Analysis of TB-RBP cDNAs.
Two oligonucleotide primers, A (5′-GTTGTACAGAGTTTAGAACA-3′) and B (5′-GAGATGATCTTCCACATCCAGA-3′), predicted from two peptide sequences of TB-RBP, were synthesized and used for PCR amplification to generate a 306-bp TB-RBP cDNA fragment. After confirming by sequencing that the cDNA encoded the predicted amino acid sequence, the 306-bp cDNA was used as a probe for cDNA library screening. Two mouse testis cDNA libraries, an oligo(dT) primed λZAP II cDNA library (Stratagene) and a 5′-stretch λgt11 cDNA library (CLONTECH), were screened by plaque hybridization with the 306-bp [32P]-labeled cDNA probe. Ten positive clones were isolated and sequenced yielding most of the TB-RBP sequence. To obtain the 5′ terminus, a 280-bp TB-RBP cDNA was generated from a testicular cDNA library constructed with the Marathon Race kit (CLONTECH) by PCR amplification using an internal TB-RBP primer (5′-CCAATGCTCATGAAACCTGTAAT-3′) and an adaptor primer.
Preparation of Recombinant TB-RBP.
To prepare recombinant TB-RBP, a cDNA encoding the entire open reading frame of TB-RBP was amplified by PCR with the primers 5′-GTGAATTCGCCACCATGTCTGTGAGCGA-3′ and 5′-GCCCCGGGTAACTACAGAAAGAGGCA-3′. The PCR product was cloned into the EcoRI and SmaI sites of the prokaryotic expression vector pGEX-4T-1 (Pharmacia), fidelity of amplification was confirmed by DNA sequencing, and TB-RBP was expressed as a glutathione S-transferase (GST)-TB-RBP fusion protein in Escherichia coli strain BL21. Transformants were grown in 2×YT medium at 25°C for 3–4 hr to an optical density of 1.0, and the fusion protein was induced with 0.1 mM isopropyl β-d-thiogalactoside for 4 hr. The cells were harvested and the GST-TB-RBP was purified with the Bulk GST Purification Module (Pharmacia). After thrombin cleavage, TB-RBP was separated from GST with the Bulk GST Purification Module (Pharmacia). From a one-liter culture, about 1 mg of TB-RBP was obtained.
Production of Anti-TB-RBP Antibodies.
An affinity-purified rabbit anti-TB-RBP peptide antibody was prepared by Quality Controlled Biochemicals (Hopkinton, MA) using the TB-RBP peptide sequence of KNDSLRKRYDGLKYDV. A rabbit polyclonal anti-TB-RBP antibody was raised against the recombinant TB-RBP protein by HRP (Denver, PA).
Immunodepletion of TB-RBP and Western Blot Assay.
BSA-blocked protein A agarose beads (150 μl, 17% slurry) in 50 μl Tris-buffered saline (TBS) were incubated with 50 μl pre-immune serum or 50 μl anti-TB-RBP serum for 1 hr at 4°C. The beads were washed four times with 1 ml of TBS containing 0.1% Nonidet P-40 and 0.25% BSA and then incubated with 50 μl of testicular cytosolic extract (14 mg/ml) in a total volume of 150 μl diluted with TBS containing 0.1% Nonidet P-40. Incubations were carried out for 4 hr at 4°C. After centrifugation for 2 min at 1,500 rpm, the supernatants were collected and used for RNA gel shift and Western blot assays. The pellets were assayed for TB-RBP by Western blotting.
For Western blotting, equal volumes (14 μl) of supernatant were electrophoresed in SDS/10% polyacrylamide gels and the proteins were transferred onto nitrocellulose membranes. The membranes were blotted overnight with TBS containing 5% dry milk at 4°C, and then incubated with the anti-TB-RBP peptide antibody (1:1,000) in TBS containing 0.25% milk for 1.5 hr at room temperature (RT). After washing, the membranes were incubated with protein A conjugated with horseradish peroxidase and the TB-RBP was detected with the enhanced chemiluminescence protocol (Amersham).
DNA Gel Shift Analysis.
One hundred nanograms of the oligonucleotide, Bcl-CL1 (GCCCTCCTGCCCTCCTTCCGCGGG) (21), was labeled at its 5′ end with [32P]ATP and T4 polynucleotide kinase. Unincorporated [32P]ATP was removed by passage through a Chromaspin-10 column (CLONTECH). For the DNA-binding assay, recombinant TB-RBP (50 ng) was incubated with 4 μg of poly(dI⋅dC) in 20 μl of binding buffer (20 mM Hepes, pH 7.6/3 mM MgCl2/40 mM KCl/2 mM DTT/5% glycerol) at RT for 10 min. The [32P]labeled Bcl-CL1 (0.14 pmol in 1 μl) was then added to the extract and incubated at RT for 10 additional min. The DNA–protein complex was resolved by electrophoresis in a 4% polyacrylamide gel (60:1) run at RT and 10 V/cm for 2–3 hr. Following drying of the gel, radiolabeled DNA–protein complexes were detected by autoradiography.
RESULTS
Size Estimation of TB-RBP from UV Crosslinking.
We have reported that a protein we have previously called p18, and now designate TB-RBP, forms an RNA–protein complex with transcript c of the 3′ UTR of mP2 (15, 20). This change in protein name and estimated size (see below) is based on our previous incorrect assumption that the RNA component of the UV-crosslinked RNA–protein complex of about 30 kDa contributed significantly to the migration of the complex in SDS/PAGE (19).
To more precisely estimate the molecular weight of the protein component of this RNA–protein complex, we have performed an exhaustive RNase T1 digestion of UV-crosslinked RNA–protein complexes. After UV crosslinking, we detect transcript c–protein complexes of about 32 kDa with testicular or brain extracts (Fig. 1, lane 1) (20). Increased digestion of the UV-crosslinked RNA–protein complexes with RNase T1 (up to 50 units) reduces the molecular weight of the complex estimated from electrophoresis in a SDS/15% polyacrylamide gel to about 30 kDa (Fig. 1, lanes 2 and 3). Since most of the RNA is digested from the RNA–protein complex under these incubation conditions, we conclude that the molecular weight of TB-RBP, the protein component of the RNA–protein complex, is near 30 kDa.
Figure 1.
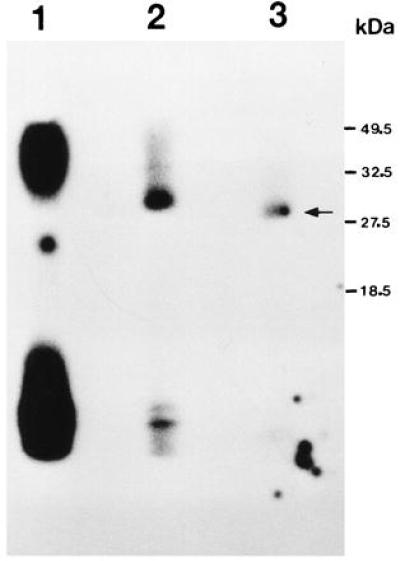
Saturation digestion of UV-crosslinked RNA–protein complexes with RNase T1. [32P]-Labeled transcript c–protein complexes were crosslinked with UV irradiation, and the complexes were isolated from a native polyacrylamide gel, subjected to RNase T1 digestion, and resolved in an SDS/15% PAGE. Lane 1, control UV-crosslinked RNA–protein complex without RNase T1 digestion. Lanes 2 and 3, UV–crosslinked complex after digestion with 5 and 50 units of RNase T1, respectively. Arrow points to the fastest migrating RNA–protein complex obtained after digestion with 50 units of RNase T1. Sizes of prestained protein markers are indicated to the right of the gel.
Purification of TB-RBP.
Based upon the binding affinity between TB-RBP and the Y and H sequence elements of the 3′ UTR of mP2, we have purified TB-RBP to near homogeneity in a two-step procedure using RNA affinity and DEAE-Sepharose ion exchange chromatographies. Testicular and brain extracts were passed over a RNA affinity column constructed with a polyadenylylated transcript c. After extensive washing, bound proteins were step eluted with increasing salt and RNA gel shift assays were performed to detect TB-RBP. The majority of TB-RBP was eluted from the RNA affinity column in the 0.5 M KCl fraction (Fig. 2A, lane E1). Small amounts of TB-RBP were observed in the flowthrough, first wash, and 1.0 M KCl eluted fractions (Fig. 2A, lanes FT, W1, and E2), and no TB-RBP was detected in the later wash fractions or the 2.0 M KCl elution fraction (Fig. 2A, lanes W2, W3, W4, and E3).
Figure 2.
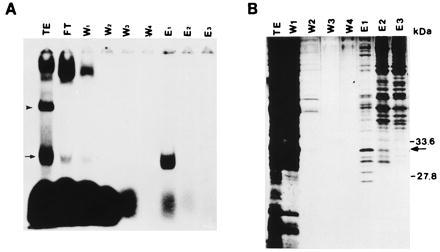
RNA gel retardation assay and SDS/PAGE analysis of the protein fractions obtained by RNA affinity chromatography. (A) After dialysis, aliquots (5 μl) of protein from each purification step were analyzed for RNA binding by a RNA gel retardation assay using [32P]-labeled transcript c. The RNA–protein complexes were then resolved in 4% native polyacrylamide gel. TE, crude testicular extract; FT, flowthrough fraction; W1-W4, wash fractions; E1-E3, 0.5, 1.0, and 2.0 M KCl step elution fractions, respectively. (B) Aliquots (30 μl) of the protein fractions analyzed in A were electrophoresed in SDS/12.5% PAGE and silver stained. Sizes of the prestained protein markers are indicated at right. Arrow in A indicates the location of the TB-RBP–transcript c complex in crude extracts. Arrowhead indicates the RNA–protein complex formed with transcript c and another testis RNA-binding protein (23).
To determine the level of purification obtained by the RNA affinity procedure, we silver stained aliquots of the same RNA affinity purified protein fractions after electrophoresis in SDS/12.5% polyacrylamide gels (Fig. 2B). In contrast to the heterogenous population of proteins in the crude testicular extract (Fig. 2B, lane TE), a reduced number of proteins were eluted from the RNA affinity column after the extensive washes (Fig. 2B, lanes E1, E2, and E3). A group of proteins of about 25–35 kDa eluted in the E1 fraction (Fig. 2B, lane E1), the fraction containing most of the TB-RBP-binding activity (Fig. 2A, lane E1). One protein band of about 30 kDa closely matched the molecular weight of TB-RBP estimated from the RNase T1 digestions (Figs. 1, lane 3 and 2B, arrow). The absence of RNA-binding activity in the E2 fraction, which contained a small amount of a similar size 30-kDa protein (Fig. 2B, lane E2), is likely due to the loss of TB-RBP activity as a result of the higher salt concentration of the E2 fraction. These data suggest that after RNA affinity chromatography, TB-RBP is highly enriched in a fraction that elutes at 0.5 M KCl.
To further purify TB-RBP, DEAE-Sepharose chromatography was used (Fig. 3). RNA gel shift assays reveal that when the E1 fraction of the RNA affinity column was loaded onto a DEAE-Sepharose column, the majority of TB-RBP was step eluted in the 300 mM KCl fraction as previously seen with crude extracts (15) (Fig. 3A). When aliquots of the same protein fractions are silver stained following electrophoresis in SDS/12.5% polyacrylamide gels, one predominant protein of about 30 kDa is seen in the 300 mM KCl fraction (Fig. 3A). Based on its size, elution, and RNA-binding properties, we conclude that this protein of about 30 kDa is TB-RBP, the protein that binds to the 3′ UTR of protamine mRNAs (15).
Figure 3.
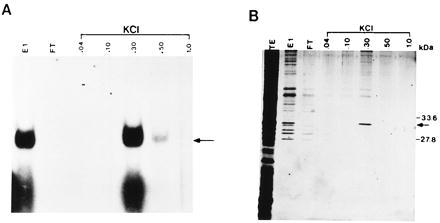
RNA gel retardation assay and SDS/PAGE analysis of the protein fractions obtained by DEAE-Sepharose chromatography. (A) Aliquots (5 μl) of protein from each purification step were assayed for RNA–protein complex formation as described in Fig. 2. E1, fraction E1 from the RNA affinity column of Fig. 2; FT, flowthrough fraction; 0.04, 0.1, 0.3, 0.5, and 1.0 (M) represent the concentrations of KCl used for step elution, respectively. Arrow indicates the TB-RBP–transcript c complex. (B) Aliquots (30 μl) of the protein fractions analyzed in A were electrophoresed in SDS/12.5% PAGE and silver stained. The sizes of the prestained protein markers are indicated at right. Arrow indicates position of TB-RBP.
Partial Amino Acid Sequence of TB-RBP.
To facilitate sequence analysis, the proteins in the 300 mM KCl fraction of the DEAE-Sepharose column were concentrated, resolved in SDS/12.5% PAGE, and transferred to a polyvinylidene diflouride membrane. The predicted protein of 30 kDa was visualized by Ponceau S staining and excised for microsequencing. After trypsin digestion, sequences were obtained from 2 peptides containing 10 amino acids (Val-Val-Gln-Ser-Leu-Glu-Gln-Thr-Ala-Arg) and 13 amino acids (Glu-Ala-Val-Thr-Glu-Ile-Leu-Gly-Ile-Glu-Pro-Asp-Arg). Using the two peptide sequences of TB-RBP, a computer search against the genembl database revealed that the two sequenced peptides were identical to amino acids 27–36 and 117–129 of a human DNA-binding protein, translin, proposed to be associated with chromosomal translocations in lymphoid cells (21).
Isolation of TB-RBP cDNAs.
The complete cDNA encoding TB-RBP was obtained by a combination of screening two mouse testis cDNA libraries and PCR amplification of the 5′ terminus. As reported for translin, the TB-RBP cDNAs encode a protein of 228 amino acids (including the initiation methionine) with a molecular weight of 26.2 kDa (21). Although no common RNA-binding motifs are detected in TB-RBP, a heptad repeat of hydrophobic amino acids characteristic of a leucine zipper from amino acids 177–212 and a transmembrane helix from amino acids 93–114 are seen. The mouse TB-RBP shows 90% identity in nucleotide sequence and 99% amino acid identity to the human protein, translin (21). The three amino acids that differ between human translin and mouse TB-RBP at amino acids 49 (alanine to threonine), 66 (glycine to serine), and 226 (valine to glycine) represent neutral changes (Fig. 4, vertical lines). TB-RBP contains five potential phosphorylation sites for protein kinase C located at amino acids 34, 67, 84, 190, and 213, (24) and three potential sites for tyrosine kinase at amino acids 85, 200, and 213 (25). Domains of TB-RBP also share significant homology to a number of other known proteins. Amino acids 9–35 and 14–47 of TB-RBP show 62% and 55% similarity to amino acids 473–499 and 768–801 of a human kinesin heavy chain, respectively (26). A 44 amino acid sequence from residues 82–126 exhibits a 51% similarity to amino acids 18–62 of the human mitochondrial protein, cytochrome c oxidase polypeptide II (27).
Figure 4.

Amino acid sequence comparison of TB-RBP and translin. The underlined sequence denotes the putative leucine-zipper domain. The double underlined sequence denotes the transmembrane helix. The three amino acids that differ between TB-RBP and translin are denoted by vertical lines. The amino acid sequence of mouse TB-RBP is 99% identical to human translin.
Recombinant TB-RBP Binds to Single-Stranded DNA Oligonucleotides That Contain Chromosomal Breakpoint Consensus Sequences.
Recombinant TB-RBP synthesized in E. coli and purified as a GST fusion protein was used in DNA gel shift assays to confirm that TB-RBP binds to specific single-stranded DNA sequences. As reported by Aoki et al. (21), the recombinant protein binds to the oligonucleotide Bcl-CL1, a target sequence within the clustered breakpoint region of the Bcl-2 oncogene in follicular lymphoma patients (Fig. 5, lane 1). These complexes were sequence specific, since complete competition was seen when unlabeled Bcl-CL1 was used as a competitor (Fig. 5, lanes 5–7), whereas no significant diminution in DNA–protein complex was seen with a nonspecific oligonucleotide, MPR2PRI (Fig. 5, lanes 2–4). Similar DNA–protein complexes were seen with TB-RBP present in testis and brain extracts (data not shown).
Figure 5.
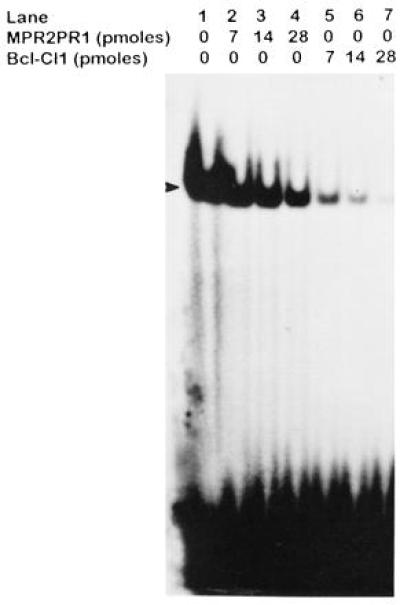
Recombinant TB-RBP binds to single-stranded DNA oligonucleotides containing specific chromosomal breakpoint sequences. Fifty nanograms of recombinant TB-RBP was incubated with increasing amounts of nonspecific competitor MPR2PRI (5′-CTATAGAATTCTCAAGCTTGC-3′) (lanes 2–4) or specific competitor Bcl-CL1 (lanes 5–7) for 10 min at RT. [32P]-labeled Bcl-CL1 (0.14 pmol) was then added and incubated for 10 min. The TB-RBP–DNA complex (arrowhead) was detected by a mobility shift assay on a 4% polyacrylamide gel.
When TB-RBP Is Immunodepleted from a Testicular Extract, No RNA–Protein Complex Is Detected.
To confirm that the RNA-binding protein TB-RBP in crude extracts and translin are the same protein, antibody prepared against recombinant TB-RBP was used to immunoprecipitate TB-RBP from testicular extracts, and RNA gel shifts were performed with transcript c. When RNA gel shifts were performed with the supernatant from a control immunoprecipitation in which pre-immune serum was used, no reduction in the amount of expected RNA–protein complex was seen (Fig. 6A, lane 2). In contrast, following immunoprecipitation with antiserum to TB-RBP, no RNA–protein complex was detected (Fig. 6A, lane 3). This precipitation appears specific since no diminution in the amount of RNA binding was seen with the mouse germ-cell specific protein p48/52 present in the same extract (Fig. 6, open arrowhead). Western blots of the supernatant fractions of the immunoprecipitated testis extract confirmed that TB-RBP was removed from the supernatant by immunoprecipitation with the antiserum to TB-RBP (Fig. 6B, lane 3). No precipitation was seen with the preimmune serum (Fig. 6B, lane 2). Western blots of the precipitates confirmed that TB-RBP was specifically precipitated by its antiserum (data not shown).
Figure 6.
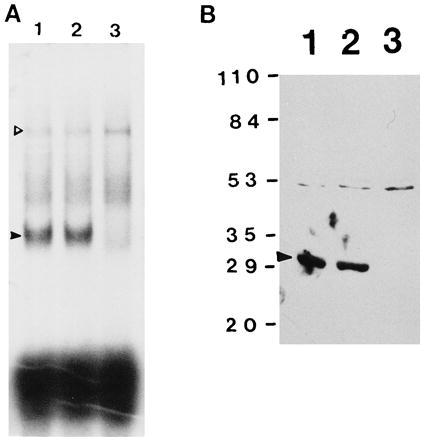
Immunoprecipitation of TB-RBP inhibits the formation of TB-RBP–RNA complexes. (A) Equal amounts of the supernatant from testicular cytoplasmic extracts (20 μg) incubated with TBS (lane 1), pre-immune serum (lane 2), or anti-TB-RBP antibody (lane 3) were incubated with [32P]-labeled transcript c and the RNA–protein complexes were resolved on 4% native polyacrylamide gels. The TB-RBP–RNA complex is indicated by a solid arrowhead. The p48/52–RNA complex is indicated by an open arrowhead. (B) Western blot assays. Equal volumes (14 μl) of the supernatants used in A were resolved on an SDS/10% polyacrylamide gel, transferred to a nitrocellulose filter, and TB-RBP was detected with antibody against a TB-RBP peptide. TB-RBP is indicated by a solid arrowhead.
DISCUSSION
We have purified TB-RBP, an RNA-binding protein in testis and brain, whose binding specificities and cellular locations have suggested a cytoplasmic involvement in translational control and cellular transport of specific mRNAs (15, 16, 19, 20). Based on the amino acid sequence predicted from cDNAs encoding TB-RBP, we conclude that mouse TB-RBP and the human DNA-binding protein, translin, are about 90% similar at the nucleotide level and nearly identical (differing in 3 amino acids out of 228) at the amino acid level. Translin is proposed to function as a DNA-binding protein that binds to specific DNA sequences at breakpoint junctions of chromosomal translocations of lymphoid malignancies (21). The DNA-binding ability of translin is dependent upon the formation of a multimeric structure (21) probably mediated through the leucine zipper in the carboxyl terminus. The leucine zipper may also play an important role for protein–protein interactions of TB-RBP in the cytoplasm as has been shown for a LINE 1 RNA-binding protein (28). TB-RBP also contains a putative transmembrane helix at amino acids 93–114, which could facilitate interactions between TB-RBP and microtubule proteins (16).
Although not common, there is a good precedent for germ-cell nucleic acid-binding proteins that bind both DNA and RNA. Proteins such as the Xenopus 5S RNA-binding protein (29), the Xenopus FRG Y2/p54/p56 proteins (30–32), and the mouse germ-cell specific homologue to FRG Y2/p54/p56 (23, 33) all possess both RNA- and DNA-binding activities. It has been proposed that translin recognizes two types of DNA target sequences: a combination of the consensus sequences ATGCAG and GCCC [A/T] [G/C] [G/C] [A/T] or a tandem repeat of GCCC [A/T] [G/C] [G/C] [A/T] with gaps of a few nucleotides intervening (21). As reported by Aoki et al. (21), we have confirmed that Bcl-CL1, a single-stranded DNA oligonucleotide from a clustered breakpoint in the Bcl-2 oncogene in lymphoma patients, specifically binds recombinant TB-RBP (Fig. 5) and TB-RBP in testis and brain extracts (data not shown), whereas one of the reported nonspecific DNA sequences does not (Fig. 5). The TB-RBP-binding sites in many translationally regulated testicular mRNAs such as protamine 1 and 2 and transition protein 1 and brain mRNAs such as tau and myelin basic protein recognize RNA sequences similar to the DNA-binding consensus sequences identified for translin (15, 16). Since translin is abundant in the cytoplasm of many cell lines while its nuclear location has been restricted to lymphoid cell lines with rearranged Ig and T cell antigen receptor loci, translin may also have additional cytoplasmic functions in lymphoid and nonlymphoid cells.
The absence of common RNA recognition motifs in TB-RBP sets it apart from many RNA-binding proteins (34). However, RNA-binding proteins, such as the human teratocarcinoma protein p40, which binds to LINE-1 RNA (28), an AU-rich sequence-binding protein (35), thymidylate synthase (36), and one of the iron responsive element-binding proteins (IRE-BP1) (37), all lack common RNA-binding domains. Some of these RNA-binding proteins perform additional functions, i.e., IRE-BP1 possesses aconitase activity (38) and the AU-rich sequence-binding protein has enoyl-CoA hydratase activity (35). Although we do not know whether TB-RBP has any enzymatic activity, it shows significant homology to a mitochondrial cytochrome c oxidase polypeptide II and to a kinesin heavy chain protein. Interestingly, cytochrome c oxidase, the kinesin heavy chain, and the mitochondrial enoyl-CoA hydratase all bind to components of the cytoskeleton as shown “in vitro” for TB-RBP (16). It is likely that TB-RBP interacts with kinesin-type motor molecules as it transports mRNAs along microtubules.
The identity between TB-RBP and translin suggests an apparent contradiction between the widespread expression of translin in somatic tissues (21) and our reports of TB-RBP RNA-binding activity being restricted to testis and brain (20). Using RNA gel shift assays, we have detected TB-RBP in extracts of brain and testis but not in extracts of spleen, kidney, liver, lung, or heart (20). In fact, TB-RBP mRNAs and DNA-binding activities are widely expressed in somatic tissues and recombinant translin can bind to a wide range of single-stranded DNA junction sequences (ref. 21, X.-Q.W. and N.B.H., unpublished work). We propose that posttranslational modifications of TB-RBP in specific tissues such as testis and brain allow TB-RBP to selectively bind RNA.
The activities of many proteins are regulated by kinases and phosphatases. For instance, phosphorylation of a 60-kDa Xenopus oocyte RNA-binding protein suppresses assembly of mRNPs (39) and phosphorylation modifies the RNA binding of an 82-kDa clam oocyte protein, thereby activating masked mRNAs (9). Although treatment of testicular extracts with potato acid-phosphatase abolishes the RNA-binding activity of TB-RBP (15), we do not know that this is solely a direct effect. In fact, recombinant TB-RBP expressed in E. coli can bind RNA (X.-Q.W. and N.B.H., unpublished data), suggesting that phosphorylation of TB-RBP may change its binding specificity. Preliminary data indicate that the nucleic acid-binding properties of TB-RBP vary, dependent upon their cellular location and developmental stage (X.-Q.W. and N.B.H., unpublished data). The testis and brain enzymes involved in the activation/deactivation of TB-RBP have yet to be identified. It will be important to determine whether the translins present in the cytosol and nuclei of lymphoid cell lines also undergo posttranslational modifications and how nuclear and cytoplasmic translins differ.
In conclusion, we have purified a protein and cloned the mouse cDNA of a highly conserved protein, TB-RBP/translin. In the cytosol, TB-RBP/translin has been proposed to translationally regulate stored mRNAs during spermiogenesis and facilitate transport of specific mRNAs in the nervous system (15, 16). TB-RBP/translin also binds to specific conserved DNA sequences often involved in chromosomal translocations in lymphoid cells (21). Based upon the specificity of translin binding to consensus sequences of breakpoints in chromosome translocations, we propose that in the testis TB-RBP functions in the nuclei of germ cells in meiotic recombination or DNA repair in addition to serving as an RNA- and microtubule-binding protein in the cytoplasm of testicular cells.
Acknowledgments
We thank Dr. E. Goldberg of Northwestern University for bringing the transmembrane helix region of TB-RBP to our attention, Dr. E. M. Eddy of the National Institute on Environmental Health Sciences for generously sharing mouse testis cDNA libraries, and Ms. T. Clements for her outstanding secretarial assistance. This research was supported by National Institutes of Health Grant HD28832.
ABBREVIATIONS
- TB-RBP
testis brain–RNA-binding protein
- UTR
untranslated region
- GST
glutathione S-transferase
- RT
room temperature
References
- 1.Izaurralde E, Mattaj I W. Cell. 1995;81:153–159. doi: 10.1016/0092-8674(95)90323-2. [DOI] [PubMed] [Google Scholar]
- 2.Curtis D, Lehmann R, Zamore P D. Cell. 1995;81:171–178. doi: 10.1016/0092-8674(95)90325-9. [DOI] [PubMed] [Google Scholar]
- 3.St. Johnston D. Cell. 1995;81:161–170. doi: 10.1016/0092-8674(95)90324-0. [DOI] [PubMed] [Google Scholar]
- 4.Richter J D. Bioassays. 1991;13:179–183. doi: 10.1002/bies.950130406. [DOI] [PubMed] [Google Scholar]
- 5.Melefors O, Hentze M W. Bioassays. 1993;15:531–541. doi: 10.1002/bies.950150203. [DOI] [PubMed] [Google Scholar]
- 6.Decker C, Parker R. Curr Opin Cell Biol. 1995;7:386–392. doi: 10.1016/0955-0674(95)80094-8. [DOI] [PubMed] [Google Scholar]
- 7.Wormington M. BioEssays. 1994;16:533–535. doi: 10.1002/bies.950160804. [DOI] [PubMed] [Google Scholar]
- 8.Henderson B R. BioEssays. 1996;18:739–746. doi: 10.1002/bies.950180909. [DOI] [PubMed] [Google Scholar]
- 9.Walker J, Dale M, Standart N. Dev Biol. 1996;173:292–305. doi: 10.1006/dbio.1996.0024. [DOI] [PubMed] [Google Scholar]
- 10.Spirin A S. Mol Reprod Dev. 1994;38:107–117. doi: 10.1002/mrd.1080380117. [DOI] [PubMed] [Google Scholar]
- 11.Hecht N B. In: Cell and Molecular Biology of the Testis. Desjardins C, Ewing L, editors. Oxford: Oxford Univ. Press; 1993. pp. 400–432. [Google Scholar]
- 12.Hecht N B. Dev Genet. 1995;16:95–103. doi: 10.1002/dvg.1020160202. [DOI] [PubMed] [Google Scholar]
- 13.Schäfer M, Nayernia K, Engel W, Schäfer U. Dev Biol. 1995;172:344–352. doi: 10.1006/dbio.1995.8049. [DOI] [PubMed] [Google Scholar]
- 14.Kleene K C. Mol Reprod Dev. 1996;43:268–281. doi: 10.1002/(SICI)1098-2795(199602)43:2<268::AID-MRD17>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 15.Kwon K Y, Hecht N B. Mol Cell Biol. 1993;13:6547–6557. doi: 10.1128/mcb.13.10.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han J R, Yiu K C, Hecht N B. Proc Natl Acad Sci USA. 1995;92:9550–9554. doi: 10.1073/pnas.92.21.9550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braun R E, Peschon R R, Behringer R R, Brinster R L, Palmiter R D. Genes Dev. 1989;3:793–802. doi: 10.1101/gad.3.6.793. [DOI] [PubMed] [Google Scholar]
- 18.Lee K, Haugen H S, Clegg C H, Braun R E. Proc Natl Acad Sci USA. 1995;92:12451–12455. doi: 10.1073/pnas.92.26.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwon K Y, Hecht N B. Proc Natl Acad Sci USA. 1991;88:3584–3588. doi: 10.1073/pnas.88.9.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han J R, Gu W, Hecht N B. Biol Reprod. 1995;53:707–717. doi: 10.1095/biolreprod53.3.707. [DOI] [PubMed] [Google Scholar]
- 21.Aoki K, Suzuki K, Sugano T, Tasaka T, Nakahara K, Kuge O, Omori A, Kasai M. Nat Genet. 1995;10:167–174. doi: 10.1038/ng0695-167. [DOI] [PubMed] [Google Scholar]
- 22.Gu W, Hecht N B. Mol Cell Biol. 1996;16:4535–4543. doi: 10.1128/mcb.16.8.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwon K Y, Murray M T, Hecht N B. Dev Biol. 1993;158:90–100. doi: 10.1006/dbio.1993.1170. [DOI] [PubMed] [Google Scholar]
- 24.Kishimoto A, Nishiyama K, Nakanishi H, Uratsuji Y, Nomura H, Takeyama Y, Nishizuka Y. J Biol Chem. 1985;260:12492–12499. [PubMed] [Google Scholar]
- 25.Cooper J A, Esch F S, Taylor S S, Hunter T. J Biol Chem. 1984;259:7835–7841. [PubMed] [Google Scholar]
- 26.Navone F, Niclas J, Hom-Booher N, Sparks L, Bernstein H D, Mccaffrey G, Vale R D. J Cell Biol. 1992;117:1263–1275. doi: 10.1083/jcb.117.6.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruvolo M, Disotell T R, Allard M W, Brown W M, Honeycutt R L. Proc Natl Acad Sci USA. 1991;88:1570–1574. doi: 10.1073/pnas.88.4.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hohjon H, Singer M F. EMBO J. 1996;15:630–639. [PMC free article] [PubMed] [Google Scholar]
- 29.Pelham H R B, Brown D D. Proc Natl Acad Sci USA. 1980;77:4170–4174. doi: 10.1073/pnas.77.7.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murray M T, Krohne G, Franke W W. J Cell Biol. 1991;112:1–11. doi: 10.1083/jcb.112.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murray M T, Schiller D L, Franke W W. Proc Natl Acad Sci USA. 1992;89:11–15. doi: 10.1073/pnas.89.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tafuri S R, Wolffe A P. Trends Cell Biol. 1993;3:94–98. doi: 10.1016/0962-8924(93)90080-k. [DOI] [PubMed] [Google Scholar]
- 33.Nikolajczyk B S, Murray M T, Hecht N B. Biol Reprod. 1995;52:524–530. doi: 10.1095/biolreprod52.3.524. [DOI] [PubMed] [Google Scholar]
- 34.Burd C G, Dreyfuss G. Science. 1994;265:615–621. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- 35.Nakagawa J, Waldner H, Meyer-Monard S, Hofsteenge J, Jeno P, Moroni C. Proc Natl Acad Sci USA. 1995;92:2051–2055. doi: 10.1073/pnas.92.6.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu E, Koeller D M, Casey J L, Drake B A, Chabner B A, Elwood P C, Zinn S, Allegra C J. Proc Natl Acad Sci USA. 1991;88:8977–8981. doi: 10.1073/pnas.88.20.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klausner R D, Roualt T A, Harford J B. Cell. 1993;72:19–28. doi: 10.1016/0092-8674(93)90046-s. [DOI] [PubMed] [Google Scholar]
- 38.Kennedy M C, Lione M M, Blondin G A, Beinert H. Proc Natl Acad Sci USA. 1992;89:11730–11734. doi: 10.1073/pnas.89.24.11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kick D, Barrett P, Cummings A, Sommerville J. Nucleic Acids Res. 1987;15:4099–4109. doi: 10.1093/nar/15.10.4099. [DOI] [PMC free article] [PubMed] [Google Scholar]