

Abstract
Tinnitus is a vexing disorder of hearing characterized by sound sensations originating in the head without any external stimulation. The specific etiology of these sensations is uncertain but frequently associated with hearing loss. The “neurophysiogical” model of tinnitus has enhanced appreciation of central nervous system (CNS) contributions. The model assumes that plastic changes in the primary and non-primary auditory pathways contribute to tinnitus with the former perhaps sustaining them, and the latter contributing to perceived severity and emotionality. These plastic changes are triggered by peripheral injury, which results in new patterns of brain activity due to anatomic alterations in the connectivity of CNS neurons. These alterations may change the balance between excitatory and inhibitory brain processes, perhaps producing cascades of new neural activity flowing between brainstem and cortex in a self-sustaining manner that produces persistent perceptions of tinnitus. The bases of this model are explored with an attempt to distinguish phenomenological from mechanistic explanations.
Learning outcomes
(1) Readers will learn that the variables associated with the behavioral experience of tinnitus are as complex as the biological variables. (2) Readers will understand what the concept of neuroplastic brain change means, and how it is associated with tinnitus. (3) Readers will learn that there may be no one brain location associated with tinnitus, and it may result from interactions between multiple brain areas. (4) Readers will learn how disinhibition, spontaneous activity, neural synchronization, and tonotopic reorganization may contribute to tinnitus.
Keywords: Tinnitus, Neuroplasticity, Dorsal Cochlear Nucleus, Inferior Colliculus, Auditory Cortex, Noise Exposure, Salicylate, MRI, PET, Spontaneous Activity, Neural Synchrony, Neural Unmasking
1. Introduction
There is growing appreciation of the fact that the brain and its neural connections are not static throughout life. A Neuroplastic change in the ‘wiring diagram’ of the brain, dependent on experience, is a dynamic process due to input from our senses and the changing chemical environment of our bodies. While the majority of these neuroplastic alterations are positive, allowing us to adapt to a changing environment or recover completely from mild brain injuries, there are maladaptive neuroplastic events, and tinnitus may be one of these. In its broadest sketch, current thinking proposes that damage to the inner ear, which reduces the neural output from the cochlea, results in neural changes, initially in the earliest nuclei of the auditory brainstem, and then extending into the deeper regions of the auditory brain. One theory proposes that these changes upset the balance between excitatory and inhibitory brain processes, with the result being neural hyperactivity. The current focus is on hyperactivity of spontaneous neuron discharges, which appears to be sustained for long time periods. Additional neuroplastic events can extend the sphere of tinnitus brain changes to involve attentional, emotional, and non-auditory sensory regions of the brain. These ideas are encompassed in the neurophysiological model of tinnitus, and the essential points associated with this concept are developed in this essay.
2. Tinnitus background
The word tinnitus is interpreted to mean a ‘ringing in the ears,’ and is derived from the Latin tinnire which means ‘to ring.’ Tinnitus in its most common form presents as ‘phantom’ sensations unassociated with any physical stimulus arising in the ear canal. It is an enigmatic disorder manifest with mysterious symptoms and a perplexing neurobiology. Tinnitus may occur transiently for durations from seconds to hours and in this manifestation is almost universally experienced. Whether these transient episodes are totally benign, or serve some sort of adaptive function, or have a more sinister implication have not been explored. Long-term sustained tinnitus is a disorder that may last a lifetime, and is experienced as a roaring, buzzing, humming, cricket-like, whistling, hissing, tonal, or ringing sound that is a recognizable sensation received in the mind as a percept. This percept is localized to one ear or the other, both ears, or somewhere in the head (Heller, 2003; Stouffer and Tyler, 1990). As many as 42 million Americans experience some form of sustained or chronic tinnitus (~14%), with perhaps 10 million having sought medical help. Tinnitus becomes more acute with stress, occurs more frequently in the elderly, and men are affected more frequently than women (Lockwood, 2005). The severity of the tinnitus sensations vary, and many sufferers are capable of adapting to them in the sense that, “yes, I can hear the ringing in my head but it does not bother me.” In others, the severity of the sensations can increase resulting in the emergence of psychological problems. Overall capacity for attention can deteriorate as focus shifts to the tinnitus with a concomitant loss in the ability to concentrate on other things. Emotional aspects may emerge advancing in severity from annoyance, frustration, and anger, to anxiousness, and even depression (see Kaltenbach, 2006 for a review). These emotional reactions to tinnitus lead to an increasing inability to cope and an ever more severe impact on life quality (Dobie, 2003). In its worst manifestation, tinnitus can drive patients to suicide. There are other socio-economic and geographic factors that play in the incidence of tinnitus, but it is a disorder that exhibits little discrimination, and down the ages documented evidence suggests that luminaries such as Pliney the Elder (CE 23-79), Michelangelo Buonarroti (1475–1564), Martin Luther (1483–1546), Sir Francis Bacon (1561–1626), Jean-Jacques Rousseau (1712–1778), Francisco Goya y Lucientes (1746–1828), Ludwig von Beethoven, (1770–1827), Robert Schumann (1810–1856), Charles Darwin (1809–1882), and Bedrich Smetana (1824–1884) all experienced sustained tinnitus (Morgenstern, 2005).
Two forms of tinnitus are identified; objective and subjective, with the former associated with the detection of real sounds “inside the body’ (Schleuning, 1991). These sounds may arise from turbulent blood flow due to vascular malformations, heart disease, or skull base vascular tumors, all of which produce pulsatile-like sensations. Also, middle-ear muscle spasms, or other muscle contractions, can cause ‘clicking’ sounds, and while spontaneous otoacoustic emissions are real acoustic signals in the ear canal, they are only rarely heard by patients (Lockwood, 2005). Subjective tinnitus, which is the focus of this presentation, is far more prevalent and represents the ‘phantom’ sensation of sound that varies in its percept from unobtrusively mild to unpleasantly loud. It is the auditory equivalent of an array of phantom sensations perceived in the somatosensory (phantom limb pain), visual (phosphene), olfactory (phantosmia), gustatory (metallic taste), and vestibular (vertigo) systems (Møller, 2006). Indeed, the similarities between the phantom sensations of severe tinnitus and the phantom pain associated with a missing limb provide important insight into the concept of central nervous system (CNS) plasticity as a process contributing to tinnitus (Moller, 2000). Despite a huge array of information concerning the epidemiology, etiology, and clinical manifestations of tinnitus there has yet to emerge an effective treatment for subjective tinnitus. Finally, excellent critical literature summaries on the neurobiology of tinnitus have emerged which detail the aspects touched upon below (e.g. Baguley, 2002; Bauer, 2004; Eggermont, 2005; Eggermont & Roberts, 2004; Kaltenbach, 2006; Kaltenbach, Zhang, & Afman, 2000; Lockwood et al., 1998, 2005; Møller, 2006; Salvi, Lockwood, & Burkard, 2000).
3. Peripheral ear contributions
Most (but not all) cases of tinnitus are associated with hearing loss arising from intense sound exposure, aging (presbycusis), or exposure to drugs like salicylates, aminoglycoside antibiotics, quinine, or cisplatin (König, Schaette, Kempter, & Gross, 2006; Sindhusake et al., 2004). These ototraumatic agents result in damaged or destroyed cochlear structures giving rise to abnormal inner-ear function and a diminution of signal output from the cochlea. There is universal agreement that tinnitus is induced or triggered by abnormal events in the cochlea. However, there is little support in the literature for a purely cochlear mechanism sustaining the perception of tinnitus (Baguley, 2002). Nevertheless, this section looks at several suggestions for how the peripheral ear might contribute to tinnitus.
One suggestion is that tinnitus may be caused by spontaneous otoacoustic emissions (SPOAEs) that arise from nonlinear behavior of the cochlear partition (due in large measure to outer hair cell motility), and which can be objectively measured as an acoustic signal in the ear canal. Patients, however, are generally unaware of this sound, and when it occurs concurrently with tinnitus, there is little correspondence between the tinnitus sensation and the acoustic emission (Penner and Burns, 1987). In addition, spontaneous emissions can be abolished by salicylate intake, whereas the detection of tinnitus is rarely lessened by taking aspirin (Long and Tubis, 1988; Penner and Coles, 1992). Another suggestion is the relation between the loss of inner and/or outer hair cells (IHC, OHC) and the production of abnormal signals coming out of the cochlea. It is known that the IHCs are more resistant to ototrauma than OHCs, and intact IHCs can exist in cochlear regions where there is complete OHC loss (Stypulkowski, 1990). In these areas of the cochlea the OHC support of the tectorial membrane might be altered sufficiently so that the membrane is in direct contact with the IHC hair bundles. This could cause them to depolarize more readily (Jastreboff, 1990), because of stimulation by the thermal or Brownian motion of endolymph. The result would be an increase in neural spontaneous activity from the cochlea and this might engender tinnitus. Yet another possibility is the loss of lateral efferent connectivity, due to hair cell loss at this boundary, either directly to outer hair cells, or via en passant efferent innervation to inner hair cell dendrites. Such efferent damage could alter inhibitory feedback and upset the cochlear balance between inhibitory and excitatory events (Chery-Croze, Truy, & Morgan, 1994). The resulting imbalance might enhance spontaneous activity in the auditory nerve. Yet another suggestion is loss of OHC motility, which might enhance the sensitivity of the IHCs, causing an increase in the level of spontaneous input to the brain (LePage, 1995). It is also possible that a permanent OHC injury causes a self-sustaining level of contraction, giving rise to a sustained neural event signaling tinnitus. To these ideas should be added notions about the excessive release of glutamate by the IHCs, arising from OHC dysfunction (Patuzzi, 2002), again producing some form of sustained cochlear output. Other cochlear neurochemical alterations might potentate the effects of glutamate release on synaptic auditory nerve receptors increasing auditory nerve spontaneous activity (Sahey & Nodar, 2001).
An idea not yet explored arises from the effects of overstimulation on the protein structures of the hair cell stereocilia. The stereocilia or sensory hairs form a hair bundle at the apical pole of the hair cell, and the deflection of these hairs lead to the gating of transduction channels via tip-links, depolarization of the hair cell membrane, the release of neurotransmitter (Glutamate), and activation of neural discharges in auditory nerve fibers.
It is now known that the cytoskeletal core, the rootlet and the extracellular links of stereocilia can be damaged by traumatic overstimulation (Husbands, Steinberg, Kurian, & Saunders, 1999; Kurian, Krupp, & Saunders, 2003; Saunders, Cohen, & Szymko 1991; Saunders, Schneider, & Dear, 1985). Fig. 1 shows aspects of this injury. A protein matrix consisting of vertical threads of filamentous actin, bound together into a precise paracrystalline array by cross bridging proteins fimbrin and espin, form the core of a stereocilium (Drenckhahan et al., 1991; Zheng et al., 2000). This cytoskeletal matrix is important because it imparts rigidity to the sensory hair. Fig. 1A is an electronmicrograph showing a single stereocilium with its vertical actin filaments and fimbrin cross bridges (Saunders et al., 1991). In addition, the membrane sheath of each stereocilium tapers at its base to an ankle where the membrane becomes contiguous with the top of the hair cell. The actin filaments in the center of the lower third of the stereocilium form a dense and tightly knit bundle that projects into the cuticular plate at the top of the hair cell. This bundle is called the rootlet, and it binds with additional proteins in the cuticular plate to tightly anchor the stereocilia to the hair cell. The spring properties of the rootlet (as the sensory hair bends), the rigidity imparted by the intracellular actin matrix, the length of the sensory hair, and the extracellular “side”, “ankle”, and “tip” links between individual hairs, allow the bundle to move as a whole, they impart stiffness to the bundle, and they determine its overall mechanical property (Bashtanov, Goodyear, Richardson, & Russel, 2004).
Fig. 1.

A. A transmission electron micrograph through the shaft of a normal stereocilium showing the vertical actin filaments with a hint of the horizontal cross bridges. B. and C. Micrographs of overstimulated stereocilia with B showing mild disassembly of the actin/cross bridge matrix and C showing more severe damage (modified from Saunders et al., 1991). D. A micrograph of the stereocilia ankle region where it articulates with the top of the hair cell. The left and right hairs show actin filament deterioration in this area, while the hair to the right has broken from its rootlet (arrow) (from Tilney et al., 1982).
Excessive hair displacements during overstimulation produce mechanical stress on the proteins in the shaft and rootlet of the stereocilia, the extracellular linkages that maintain bundle coherence, and can cause structural damage. Fig. 1B and C reveals that the protein structures in the core of the stereocilia (Fig. 1A) can be disassembled, and this can occur with different levels of severity. Panel D shows that actin filament damage can also occur in the region of the rootlet, and the rootlet itself can be severed after exposure to intense stimuli (Liberman, 1987; Tilney, Saunders, Egelman, & DeRosier, 1982; Wang, Hirose, & Liberman, 2002). The injuries in Fig. 1 would mechanically decouple the hair from its mechanical input and isolate it from the top of the hair cell. Overstimulation can also sever the tip links (Husbands et al., 1999; Kurian et al., 2003; Pickles, Osborne, & Comis, 1987).
A given stereocilium may not exhibit all these injuries, but the consequence of any combination of them is a reduction in hair bundle stiffness, and this reduction causes the hairs, when stimulated at a given level, to move through a greater displacement than when stiffness is normal (Duncan and Saunders, 2000; Saunders and Flock, 1986; Szymko and Nelson, 1995). The author speculates that reduced hair stiffness would enhance hair displacement in the Brownian noise of the endolymphatic fluid environment. An increased displacement in these less stiff hairs from ambient thermal noise may be sufficient to gate surviving transduction channels, depolarize the hair cell, and release neurotransmitter. This could increase spontaneous activity in afferent fibers attached to affected hair cells. An increase in auditory nerve spontaneous activity might be difficult to detect, especially if focal lesions of damaged stereocilia occurred on relative few hair cells. Such a scenario would enhance spontaneous activity in relatively few auditory nerve fibers.
All of these possibilities for generating a signal that might be interpreted by the CNS as tinnitus are entirely speculative, and represents phenomenological evidence rather than direct evidence of a mechanistic process that triggers or sustains tinnitus. Eggermont and Roberts (2004) recently summarized the literature and with exception of the case where high doses of salicylate were administered (Müller, Klinke, Arnold, & Oestreicher, 2003), the universal outcome in auditory nerve fibers from overstimulation or ototoxic treatment is reduced sound-driven activity and a reduction (or no change) in spontaneous activity (Kiang, Moxon, & Levine, 1970; Liberman and Kiang, 1978; Mulheran, 1999; Stypulkowski, 1990). Even the suggestion that stereocilia cytoskeletal damage is a contributing factor becomes less compelling in the light of recent evidence that the crystalline matrix of stereocilia undergo a continuous renewal (Grati et al., 2006; Rzadzinska et al., 2004; Schneider, Belyantseva, Azevedo, & Kachar, 2002), and that losses in stereocilia stiffness can rapidly recover (Saunders and Flock, 1986; Szymko et al et al., 1995; Duncan and Saunders, 2000). Perhaps the most important observation that tinnitus has a retrocochlear origin is the observation that tinnitus persists after surgical resection of the auditory nerve (House and Brackman, 1981). Nevertheless, it is widely held that abnormal output from the damaged cochlea, in whatever form it might take, serves as the trigger for neuroplastic and permanent changes in the CNS that lead to the percepts of tinnitus. These changes are now considered.
4. Neuroplasticity
Neuroplasticity is a term with many meanings, but essentially refers to the capacity of the brain to reorganize its neural networks on the basis of new experience. This is accomplished, in large measure, by rearranging the connections between neurons. The most obvious of plastic functions is the persistent brain changes that must occur with memory formation and learning. This reorganization begins with alterations in gene expression that cause neuronal or even glial and vascular changes at the molecular, cellular, and tissue levels. Ultimately new brain structures form or are eliminated (synapses for example), and the process can be punctate, or it can affect whole areas of the CNS, leading to modifications in the location of specific information processing functions. Brain reorganization may emerge quickly or slowly, may be permanent or labile, and may reflects a shift in the influence of excitatory or inhibitory events. Often they are associated with the synaptic communication between neurons, but could also occur as changes in cellular membrane properties. The plastic change may be positive and adaptive as with learning or memory, or in the compensation after brain injury that results in new neuronal networks that restore normal function. Alternatively, the neuroplastic changes might be maladaptive leading perhaps to an imbalance in excitatory and inhibitory events in the brain. Indeed, tinnitus may be the consequence of such maladaptive neuroplastic brain alterations (Eggermont, 2005). Kaltenbach, Zhang, and Finlayson (2005) has even gone a step further and described tinnitus as the perceptual manifestation of plastic brain changes that result in neural hyperactivity. The quest for where neural hyperactivity may reside, how it can be identified, what initiates it, and how it is sustained, constitutes much of the driving force behind current efforts to uncover the neural basis of tinnitus.
5. Brainstem plasticity and tinnitus
Hyperactivity in brainstem auditory nuclei can be seen in enhanced sound-driven activity or increased spontaneous neural activity. Fig. 2 shows evoked response amplitude at different intensities elicited by a click stimulus in the cochlear nucleus (CN) and inferior colliculus (IC) of a group of young BALB/c mice measured nine days after exposure to a short duration wide band noise at 105 dB SPL (Saunders, Bock, James, & Chen, 1972). Non-exposed control data are seen for another group of animals. The exposed mice showed a threshold shift and significant hyperactive responses above 95 dB in the CN and above 85 dB in the IC. Similar hyperactivity for sound driven single-unit activity after overstimulation in the IC of C57BL/6J mice has also been reported (Willott and Lu, 1982).
Fig. 2.

Click stimulus input/output evoked response functions in young mice recorded from the CN and IC. Data obtained from a group of overstimulated and control animals nine days post exposure. Hyperactivity in the exposed animals is apparent at high stimulus levels (redrawn from Saunders et al., 1972).
More detailed evoked response and single-unit data from the CN, IC and auditory cortex (AC) of the chinchilla have been obtained for tone burst stimuli (McFadden et al., 1998; Salvi et al., 2000; Salvi, Powers, Saunders, Boettcher, & Clock, 1992; Salvi, Saunders, Gratton, Arehole & Powers, 1990; Wang et al., 1996). Overstimulated or carboplatin (a cisplatin derivative that destroys OHCs but not IHCs) treated animals both showed a reduction in cochlear output, but hyperactive input-output at higher brain centers (as in Fig. 2) when the tone burst evoking the response was a lower frequency than the region of hearing loss. However, the hyperactivity occurred only in the IC for ears with acoustic trauma and in the AC for carboplatin damaged ears. Interestingly, this hyperactivity in the IC emerged within several hours post-exposure. While hyperactive sound-driven activity is a sign of plastic brain changes, by itself it is unlikely the cause of tinnitus. This is because sound-driven activity, even if elicited by ambient levels of sound stimulation, is derived from a dynamic input signal that is ever changing as the organism moves through its environment.
Another form of neural response is spontaneous activity (SA), and this is more interesting with regard to tinnitus since it occurs in the absence of specific stimulation. Spontaneous activity in the nervous system occurs because of the stochastic or random release of neurotransmitter at synaptic junctions. This random release is assumed to be an inherent property of the pre-synaptic cell and its membrane, and in the absence of other factors occurs at relatively constant rates that are unique to different neurons. Inner ear ototrauma can lead to increases in the levels of SA in the dorsal cochlear nucleus (DCN), the IC, and the auditory cortex (AC). These changes occur over variable time scales, may exhibit tonotopic specificity, are robust, and are sustained for long times (Kaltenbach et al., 2000, 2005). Fig. 3 shows SA in groups of young adult hamsters, measured from multi-unit activity as an electrode was advanced along the tonotopic axis of the DCN. The original data were recorded as events per second (Kaltenbach et al., 2000), but are replotted here as a percent of the maximum response. Distance along the DCN surface is in millimeters (from low to high frequency) unreferenced to any specific frequency location. The exposure was a 10.0 kHz tone at 127 dB SPL for 4 h, and the parameter in the figure is days post exposure. At two days, the level of SA was reduced by about 10 percent from control levels (control data not plotted). At five days post exposure SA increased substantially with the maximum activity occurring in the 9.0 kHz region of the DCN. By 14 days there was some additional increase in activity, and at 30 days the spatial extent of the hyperactive SA narrowed with the peak location shifting into the 12.0 kHz location. At 180 days post exposure the SA showed some additional increase with the maximum shifted to the 18.0 kHz region of the DCN. A comparison of the SA levels at 18.0 kHz revealed that it was approximately 20 events/second in control animals, but by 180 days it was around 85 per second in the exposed animals; a greater than four-fold change.
Fig. 3.

Spontaneous activity along the surface of the hamster DCN in groups of overstimulated animals between 2 and 180 days post exposure. After 2 days the activity increased considerably, exhibited some selectivity with regard to DCN location, and was sustained for at least 180 days (redrawn from Kaltenbach et al., 2000).
Kaltenbach and his colleagues (2005 colleagues (2006) have investigated hyperactive SA in the DCN thoroughly, and there is little doubt that it is a valid phenomenon. Is this increase in SA a neuroplastic event that represents the sustained neural activity hypothesized as the basis of tinnitus? While the observations at the DCN are compelling it needs to be recognized that they may simply trigger similar or additional activity more centrally. Indeed, ablating the DCN in rats exhibiting tinnitus behavior failed to alter their psychophysically measured tinnitus perception. This observation suggests that the sustained DCN hyperactivity may not be the generator feeding forward neural activity that activates the sensations of chronic tinnitus (Brozoski and Bauer, 2005). It is worth mentioning a recent study examining spontaneous activity in the IC of CBA/J mice following unilateral or bilateral overstimulation with noise, or salicylate treatment. These animals showed no pervasive increase in SA at this brain level (Ma, Hidake, & May, 2006).
There is now ample evidence that cochlear damage causes substantial structural, neurochemical and physiological changes throughout the auditory system (see Salvi et al., 2000 for a thorough literature review). These include such plastic responses as axonal pruning and new growth, trans-synaptic degeneration and new synapse formation, neuron shrinkage, and changes in the tonotopic distribution of neurons within nuclei. There are indications of an increase in the number of neurons projecting from the CN to the IC. The growth associated protein, GAP-43, is again expressed in the adult IC after cochlear damage, and may assist in the emergence of these new axons and their synapses. Neurochemical changes in central auditory nuclei occur, and the down-regulation of GAD (the synthesizing enzyme for the inhibitory neurotransmitter GABA) in the IC is an example. There are general reductions in the release and binding of GABA throughout the auditory brainstem, while in the CN, protein synthesis is reduced along with glucose metabolism.
It is frequently assumed that enhanced SA in the DCN results from a reduction in inhibitory activity in the complex neural circuitry of this nucleus. Based on current understanding of DCN neuronal circuitry and its functional aspects, a model for disinhibition has been proposed (for a most recent summary see Kaltenbach et al., 2005). Briefly, there are well characterized interneurons in the DCN called granule and cartwheel cells (see Fig. 4). Cochlear damage may lead to a reduction in spontaneous activity on the Type II afferent fibers (from the OHCs), and through the granule-cartwheel cell circuitry produce a reduction in the inhibition applied to the output cell of the DCN, the fusiform cell. The fusiform cell is normally spontaneously active and when this cell is disinhibited by reduced input from the cartwheel cell, the level of SA should increase.
Fig. 4.
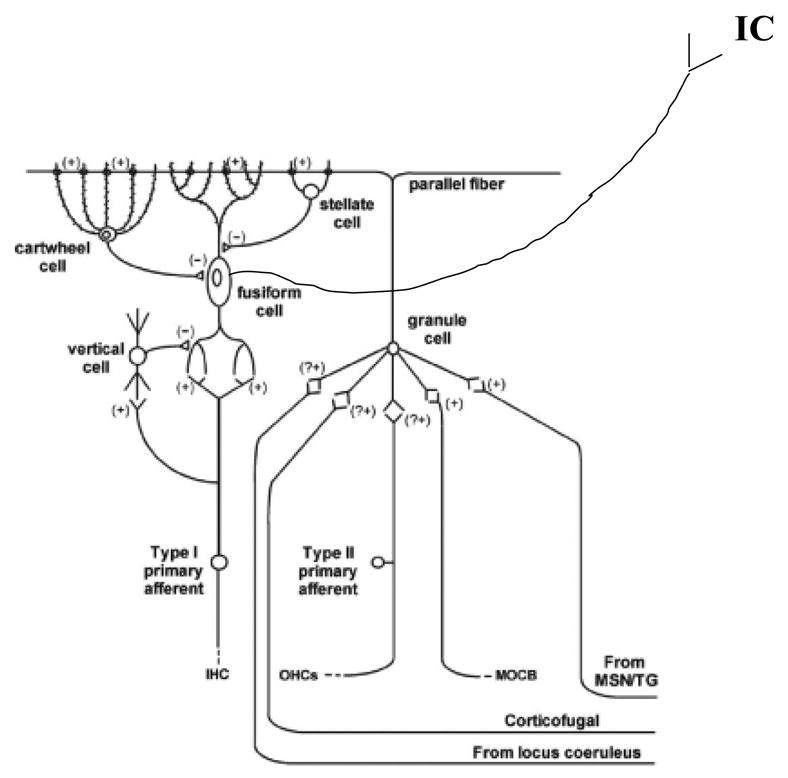
Neural wiring diagram of the dorsal cochlear nucleus showing inhibitory and excitatory inputs, and the interaction of interneurons. See text for explanation of inhibitory influences (permission from Kaltenbach et al., 2005).
Damage to IHCs would decrease activity in Type I auditory nerve fibers thus reducing the input to the fusiform cells (see Fig. 4). A branch of the Type I afferent fiber communicates with another DCN interneuron, the vertical cell, and this cell also provides an inhibitory input to the fusiform cell. Reduced activation of the vertical cell from cochlear injury attenuates its inhibitory influence, resulting in additional spontaneous activity from the fusiform cell. As noted above, the increased hyperactivity in the fusiform cell, whose axons project to the IC, may constitute an important sustained signal for tinnitus.
Disinhibition resulting in SA hyperactivity occurs with different time courses depending on the agent causing cochlear damage (loud sound or ototoxic drugs), the anesthetic and arousal state of the animal being tested (Yang et al., 2006), and the level of the auditory system from which it is measured (see Eggermont, 2005; Eggermont & Roberts, 2004). This disinhibition is mirrored in reduced levels of GABA in central auditory nuclei (at least as reported for the IC) following acoustic overstimulation (Bauer, 2000; Abbott, Hughes, Bauer, Salvi, & Caspary, 1999; Milbrandt, Holder, Wilson, Salvi, & Caspary, 2000). Plastic changes in other auditory brain areas involving not only the “classical” auditory pathway but the so called ‘non-classical’ pathway (the polysensory inputs to the auditory system, the limbic and the extralemniscal paths consisting of the DCN, the external nucleus of the IC, and the secondary auditory cortex AII) could cause a further reduction in inhibitory influences on the DCN via corticofugal feedback fibers onto the granule cells (see Fig. 4). Additional inhibitory inputs on the granule cells from the locus coeruleus and the limbic system may further serve to enhance fusiform cell spontaneous activity. The influence from these latter sources may constitute new neuroplastic activity arising from attentional and emotional brain centers (Kaltenbach, 2006).
Two other sources of altered neural activity have been identified after peripheral ototrauma: bursting discharges and neuronal synchrony (Baguley, 2002; Eggermont, 2005). Both of these represent the emergence of a temporal pattern on what are otherwise the random discharges of SA. Bursts of activity appear in cat auditory nerve fibers after kanamycin exposure, and in the IC and AC of the rat following salicylate administration (Chen and Jastreboff, 1995; Kiang et al., 1970). It also occurs in the DCN after exposure to noise (Chen, Chang, Zhang, Kaltenbach, & Godfrey, 1999). Unfortunately, an unambiguous link between bursting and tinnitus has yet to emerge (Kaltenbach, 2000), and it is not clear if they are sustained for a long time. In addition, cross correlation of simultaneously recorded neuron activity in the primary auditory cortex (area AI), following noise exposure or quinine treatment, reveal an increase in synchronized discharges patterns (Noreña and Eggermont, 2003; Ochi and Eggermont, 1997). This emergence of synchronized activity (meaning that the SA of multiple neurons are occurring at the same time) is related to cortical reorganization and tends to increase with time. Synchronization in brainstem nuclei at present is uncertain. Moreover, how bursting and synchronization can engender the percepts of tinnitus is uncertain, but they remain additional aspects of the neuroplastic response to peripheral ear insult.
Kaltenbach et al. (2005) discussed additional aspects of plastic changes in the DCN beginning with the up and down regulation of gene expressions capable of modulating cell excitability. These modulations relate to neurotransmission and the processes that control vesicle dynamics, changes in the function of receptors on post-synaptic membranes, and the number and species of ion channels expressed in excitable membranes. Finally enzymes regulating protein production can dynamically alter the synaptic interaction of neurons by reducing or increasing the number of synapses. These aspects raise the complexity of tinnitus considerably.
6. Cortical reorganization and tinnitus
Neuroplastic events at the auditory cortex (AC) have been reported to include hyperactive SA of cortical neurons and an increase in neuronal synchronization (Noreña & Eggermont, 2003; Ochi & Eggermont, 1997; Seki & Eggermont, 2003). Recently a spiking neuron model of the AC was developed in which lateral excitation and loss of inhibition altered the strength of cortical connections causing increased SA and neuron synchrony (Dominguez, Becker, Bruce, & Read, 2006).
Loss of input to the auditory cortex from peripheral ototrauma can also lead to areas of missing frequencies in the cortical tonotopic map (Lockwood, Salvi, & Burkard, 2002; Møller, 2006; Salvi et al., 2000). The relation between cortical deafferentation, cortical tonotopic reorganization, and tinnitus has been considered in considerable detail elsewhere (Meikle, 1995; Moller, 2000; Møller, 2006; Salvi et al., 2000) and the salient points are developed here.
The model for understanding tonotopic remodeling in the auditory cortex comes from the somatosensory system, where there is a well know neuroplastic response to the elimination of peripheral input (see discussion by Salvi et al., 2000). Each neuron in the somatosensory cortex is excited by stimulation from a specific skin location, and each location is surrounded by an area, which when stimulated, results in an inhibitory input to the neuron. Light but punctate skin stimulation elicits only excitatory activity, but with greater stimulation the surrounding inhibitory region is activated, leading to suppression of the excitatory response. The cortical response is the net result of activity from the excitatory center and inhibitory surround, and the interplay of these inputs defines and enhances the neuron’s receptive field on the skin (see Salvi et al, 2000).
Amputation of a finger on the hand disrupts the equilibrium between these excitatory and inhibitory inputs and silences cortical neurons that receive input from the affected area. With the passage of time these silent neurons again become active, but now they code for the stimulation of adjacent skin areas in the palm. This reactivation of neural activity in the deafferentated area of the somatosensory cortex results from the unmasking of pre-existing excitatory inputs. These neural connections were always there, but unrecognized because their relatively weak neural inputs were masked by the presence of strong inhibitory influences. When the source of surround inhibition was removed, because of the amputation, a new excitatory region was expressed, and appeared to ‘fill in’ the deafferentated cortical area (Rauschecker, 1999). Over time this unmasked input strengthened. There are limits to this process that depend on the lateral extent of the axonal connections from one excitatory field to an adjacent field (Salvi et al., 2000).
Fig. 5 demonstrates auditory cortex reorganization after cochlear damage and there are similarities to that seen in the somatosensory cortex. The top two panels in the figure show the best frequency (frequently referred to as the characteristic frequency or CF) response for electrode penetrations along the surface of the primary auditory cortex (AI) in a normal cat and a cat with a noise-induced hearing loss above 10.0 kHz (Eggermont, 2005; Eggermont & Roberts, 2004). As can be seen in the upper left panel there is a relatively orderly progression of frequency mapped along the normal tonotopic axis. The panel to the right depicts the same mapping, but in the exposed animal. The striking difference is that the anterior region of the cortex now maps only to frequencies along the lower edge of the lesion (~10.0 kHz). This observation is quantitatively described in the panel below (AC), where frequency is mapped along the tonotopic extent of the primary auditory cortex of normal and damaged cat ears. In the normal animal, the CF of units systematically increases with more anterior locations. In the damaged animal (where the cochlear lesion destroyed the sensory surface above 18.0 kHz) the progression of the tonotopic map stops at approximately 3.2 mm with more anterior locations all showing CFs around 18.0 kHz. The loss of input in the lesion area has ‘unmasked’ otherwise silent inputs to the cortical cells arising from weaker thalamic projections arising from the lower frequency edge of the lesion (Rajan and Irvine, 1998b).
Fig. 5.

Top two panels show the surface of the cat auditory cortex (AI). Superimposed on the cortex are the characteristic frequencies detected at different locations. The left panel shows a control animal and the right from a sound damaged animal with cochlear damage above 10.0 kHz. The tonotopic map of the damaged animals is modified with tonotopic representation absent for frequencies above 10.0 kHz (top panels permission from Eggermont, 2005). The bottom left panel (IC) shows tonotopic organization in the IC of a chinchilla with a hearing loss above 1.0 kHz. The black bar shows the frequency extent of the lesion and hearing loss. The solid line and the 95% confidence intervals depict the tonotopic progression with IC depth in normal animals. The black dots indicate tonotopic progression with depth in the exposed animals. The frequency increases as depth increases to about 2.4 mm. Deeper penetrations reveal that the CF of units remains fixed at around 1.0 kHz. This represents tonotopic reorganization of the IC (redrawn from Salvi et al., 2000). The right lower panel shows the change in CF with progressive distance along the auditory cortex in control animals and in cats with inner ear damage above 19.0 kHz (the black bar again shows the extent of the lesion). The control cats show a normal increase in CF as more distal locations are sampled. In the exposed animals the area above 18.0 kHz is only responsive to frequencies at the edge of the lesion (redrawn from Rajan and Irvine, 1998b; Rauschecker, 1999).
In the lower left panel another tonotopic map is seen for the chinchilla IC (Salvi et al., 2000; Salvi, Wang, & Powers, 1996). The solid line is the normal tonotopic progression of CFs plotted against penetration depth in the nucleus (the dashed lines show the 95% confidence intervals). The black data points represent results from a single animal with a severe high frequency hearing loss above 1.0 kHz. The CF increases in an orderly manner with increasing electrode depth until about 2.3 mm. At progressively deeper locations the CF remains around 1.0 kHz. The reduced input from the periphery and the unmasking of pre-existing neuronal connections could also explain the reorganization in the IC. This, of course, raises the question of whether the cortical reorganization in the other panels of Fig. 5 are simply a passive reflection of events in the IC. Interestingly, tonotopic maps of the DCN in control and noise damaged cats were the same, indicating the absence of reorganization at this level of the auditory pathway (Rajan and Irvine, 1998a).
Fig. 6 helps to clarify the process of unmasking. An array of hair cells is depicted along the cochlear partition from low to high frequency. The central projections from these hair cells remain tonotopically organized to the medial geniculate body (MGB) of the thalamus. Cells in the AC are also tonotopically organized, but the axons from the MGB send collateral projections, in a progressively weaker manner, to adjacent cortical cells. This is why tuning curves at the cortex often appear to have multiple CFs. For the most part, activity in these collateral fibers is masked by the primary input from the MGB and by inhibition present on the cortical cells. To the left there is a lesion on the organ of Corti and this causes diminished input to the respective cortical cells. The colatteral inputs from the adjacent cells are now ‘unmasked’ and the neurons in the deafferentated region of the cortex come to exhibit CFs derived from the unmasked input from the low frequency edge of the lesion. The consequence of this is an extension of the edge frequencies into the deafferentated region of the map. This may have importance for tinnitus since the perception of the phantom sound is often associated with the frequencies at the edge of the hearing loss.
Fig. 6.

A Cartoon of how cochlear damage unmasks lateral axonal connections from MGB cells lying at the low frequency edge of the lesion. Neuroplastic changes in spontaneous activity and neuron synchronization in the auditory cortex may arise because of deafferentation and disinhibition of the fiber tracks coming out of the peripheral ear. See text for details (redrawn and modified from Eggermont, 2005).
On the right hand side of Fig. 6 is seen an inhibitory interneuron (ii), indicated in black. This provides a source of feed-forward inhibition to the cortical cell. A number of these inhibitory interneurons are shown in the frequency region of the lesion but omitted (to avoid cluttering the figure) in the normal projection area. When inhibitory input to the cortical cell in the deafferentated area is removed, the disinhibited cortical cell will exhibit increased SA (Komiya & Eggermont, 2000; Noreña & Eggermont, 2003; Seki & Eggermont, 2003). This is a process not unlike that described at the DCN and attributable to the same sort of neuroplastic cellular mechanisms described above. The increment in cortical SA might represent another sustained source of hyperactivity signaling tinnitus.
Also important is the occurrence of synchronized output from many cortical cells. If the unmasked SA input from a given MGB cell at the lesion edge activates multiple cortical cells simultaneously, then their spontaneous output will have a high degree of coincidence. The increase in synchronization raises the possibility of coincident inputs occurring on cells in cortical associational areas (AII or various structures in the limbic system) that would otherwise never experience coincident inputs. There may also be coincidence in corticothalamic fibers feeding back on common MGB cells. Coincident neural inputs are known to enhance synaptic efficiency through NMDA receptors and the rules of Hebbian conditioning (Cotman et al., 1989; Guitton et al., 2003). Such conditioning would represent a long-term neuroplastic change in the auditory connections of the CNS, and could result in self-sustained positive feedback between the primary auditory cortex and the MGB, and with the cortex and limbic system structures such as the amygdala (Mahlke & Walläusser-Franke, 2004; Walläusser-Franke et al., 2003).
The techniques of brain imaging, including positron emission tomography (PET) and magnetic resonance imaging (MRI)), as well recordings of magnetic-evoked responses, are noninvasive methods for assessing brain function. The technical aspects of these were discussed in detail elsewhere (Salvi et al, 2000). These methods have been applied with success in human tinnitus patients to identify various brain areas active during phantom perceptions, and to examine the effects of some treatments (see for example Giraud et al., 1999; Mirz et al., 1999; 2000; Reyes et al., 2002). There is now good evidence from PET and magnetic encephalography that a reorganization of the frequency axis in the human auditory cortex occurs much as described above (Lockwood et al., 1998; Mühlnickel et al., 1998). Nevertheless, a more recent study using neuromagnetic recordings (this permitted identification of highly specific brain locations) suggested that equivalent dipole activity at the edge of hearing loss was no more active than control locations (Weisz, Wienburch, Dohrmann, & Elbert, 2005). This observation questions whether map reorganization by itself is responsible for the emergence of tinnitus.
High resolution MRI and voxel-based morphometry recently identified specific regions of gray matter change in the brain. The resulting images can be combined to represent group data, and comparisons were made among healthy control and tinnitus patients. Grey matter increased at the level of the auditory thalamus, but decreased in the subcallosal area (Mühlau et al., 2006). The increase in posterior MGB grey matter was thought to be the result of reorganization in this nucleus perhaps from sustained activity in corticothalamic feedback loops. The authors suggested that sustained activity in the MGB was then relayed to the limbic system, contributing to the formation of emotional associations that exacerbated tinnitus percepts.
An interesting clinical report recently described the loss of tinnitus following a cerebral vascular accident (CVA) (Lowery, Eisenman, & Saunders 2004). The patient was a physician and upon realizing that his tinnitus of 40 years disappeared, he initiated a thorough work-up, including audiometric assessment of hearing and MRI imaging. The disappearance of the tinnitus was not related to an additional hearing loss from the CVA since pre and post audiograms showed no change in hearing. Fig. 7 shows the MRI scans of the patient. Panel A is a coronal section showing the CVA in the left hemisphere. The lesion is located in the white matter of the dorsal corona radiata projecting to the frontal parietal cortex. Panels B and C are horizontal sections at different dorsal levels (as indicated in panel A). These sections show the anterior-posterior extent of the lesion and indicate that it reached into the neostriatum, involving parts of the caudate nucleus and the putamen. This CVA clearly impacted on the thalamocortical radiations and the descending corticothalamic projections. The lesion was further interpreted to impact on corticocortico fibers in the superior longitudinal fasciculus. The loss of tinnitus in this patient might be attributable to the interruption of communication between the cortex and thalamus (and vice versa), and the projections from the auditory cortex to other non-sensory areas of the brain. This reversal is not so surprising given the cortical involvement in neuroplastic events associated with tinnitus noted above.
Fig. 7.
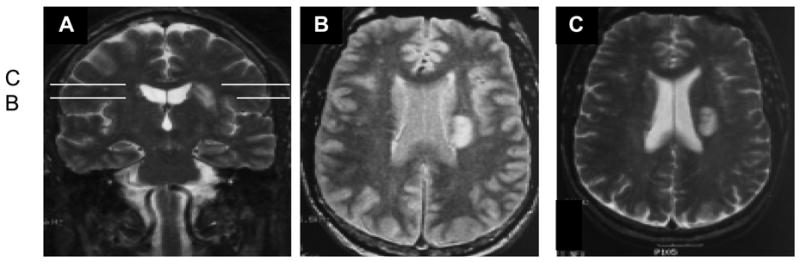
PET images from a patient with a CVA that abolished tinnitus. A. A coronal section. B. and C. Horizontal sections at different depths. The lesion affected white matter in the cortical radiations between the thalamus and auditory cortex, and in more extensive non-sensory tracks (modified from Lowery et al., 2004).
7. Conclusions
In the introduction it was noted that tinnitus is a mysterious and perplexing disorder of hearing. The number of variables associated with the symptoms and the plethora of underlying biologic factors are difficult to sift through and coherently organize. Nevertheless, advances in understanding the neurobiology of tinnitus have been exciting during the past 15 years. Although not considered here, there are now convincing behavioral paradigms for use with animals that clearly indicate that they experience the sensations of a phantom sound. These have been effectively used to compare both brain activity with tinnitus perceptions (e.g. Brozoski, Bauer, & Caspary, 2002), and constitute a powerful tool for exploring brain function and tinnitus, as well as interventional procedures.
The neurophysiological model remains much as proposed by Jastreboff (1990) in that damage to the peripheral organ serves as a trigger for tinnitus. It is then sustained by events occurring in the central auditory pathway. These changes most likely result from neuroplastic processes that in and of themselves are maladaptive. They may also extend to non-sensory areas of the brain giving rise to the attentional and emotional aspects that often accompany the disorder. Hyperactivity of spontaneous activity is a current target phenomenon, and it has been demonstrated in the brainstem and auditory cortex and appears to correlate with tinnitus. Similarly, reorganization of the tonotopic representation of frequency in brainstem and cortical areas is another neuroplastic response to inner ear damage, and it is argued that this may also contribute to tinnitus. Tonotopic reorganization appears to pave the way for additional neural connectivity in non-sensory brain areas, ascribing additional and perhaps negative attributes to the tinnitus percepts. The identification of increments in spontaneous activity, enhanced synchronization, remodeling of tonotopic organization, or the extension of tinnitus activity into non-sensory areas remain phenomena that are correlated with various aspects of tinnitus. They hint at underlying cause-and effect mechanisms but do not yet prove it.
It is useful to conclude with the observation that tinnitus must be now thought of as a ‘constellation’ of neural changes that are becoming increasingly complex with advancing research. This complexity, and the growing array of contributing and associated brain factors further suggest why tinnitus has not been amenable to a single treatment. Indeed, future treatment strategies may have to target multiple factors at the same time (Kaltenbach et al., 2005).
Acknowledgments
The preparation of this paper was partially supported by an award from the NIDCD to JCS (DC-000710). The author appreciates the critical comments and suggestions of Dr. Michael Anne Gratton, Mr. Michael Avissar, and Ms. Shachee Doshi.
Appendix A. Continuing education
-
Ototrauma to the cochlea has what consequence on activity in fibers of the auditory nerve?
There is enhanced neural synchronization in nerve fibers tuned to the same frequency
There is enhanced spontaneous activity for all damaging conditions except after
Treatment with high doses of salicylate
There are no changes or only a general depression in spontaneous activity following exposure to intense sound
There is mild enhancement of sound-driven activity
Sharpened neural tuning curves result from diminished efferent feedback in the cochlea.
-
Neuroplastic changes in the brain serve what major function?
They eliminate non-homeostatic brain chemistry
They all result in positive brain adaptations to a changing environment
They filter maladaptive experience from causing non-adaptive brain changes
They permit the brain to reorganize its neural networks on the basis of new experiences
They organize brain functions during development into fixed and unchanging patterns of activity
-
How can hyperactive brain activity contribute to tinnitus?
Hyperactive spontaneous activity arising from the dorsal cochlear nucleus represents a sustained input to the brain that might be interpreted as a tinnitus percept
Hyperactive sound-driven activity can produce tinnitus sensations directly
Transient hyperactive neural activity activates attentional and emotional centers of the brain which directly leads to tinnitus
It will increase inhibitory events in the central nervous system thus enhancing neural responses that activate tinnitus
It plays no role in maintaining the percepts of tinnitus
-
Why may hyperactive spontaneous discharges in the dorsal cochlear nucleus not be the cause for tinnitus?
Because it causes hyperactive spontaneous activity in all auditory nuclei
Because ablating the dorsal cochlear nucleus in animals does not eliminate behaviors indicative of tinnitus perceptions
Because it arises from excessive input from the auditory nerve
Because of the general brain increase in the inhibitory neurotransmitter GABA
Because it can be modulated by descending cortiofugal fibers from deeper brain centers
-
Reorganization of the auditory cortex tonotopic map, following peripheral ear damage arises from what conditions?
Increased spontaneous activity from peripheral brain centers that cause an excitotoxic response leading to massive reorganization of the entire tonotopic axis at the cortex
The enhancement of excitatory input and increased inhibitory activity influencing cortical neurons
It arises exclusively from the activity of inhibitory interneurons which causes suppression of spontaneous activity in cortical neurons
General atrophy and loss of unused cortical neurons
Reduced input from the periphery due to cochlear damage and the unmasking of weaker collateral inputs from MGB cells lying on the low frequency side of the lesion.
Appendix A. Continuing education (correct answers*)
-
Ototrauma to the cochlea has what consequence on activity in fibers of the auditory nerve?
There is enhanced neural synchronization in nerve fibers tuned to the same frequency
There is enhanced spontaneous activity for all damaging conditions except after
Treatment with high doses of salicylate*
There are no changes or only a general depression in spontaneous activity following exposure to intense sound
There is mild enhancement of sound-driven activity
Sharpened neural tuning curves result from diminished efferent feedback in the cochlea.
-
Neuroplastic changes in the brain serve what major function?
They eliminate non-homeostatic brain chemistry
They all result in positive brain adaptations to a changing environment
They filter maladaptive experience from causing non-adaptive brain changes
They permit the brain to reorganize its neural networks on the basis of new experiences*
They organize brain functions during development into fixed and unchanging patterns of activity
-
How can hyperactive brain activity contribute to tinnitus?
Hyperactive spontaneous activity arising from the dorsal cochlear nucleus represents a sustained input to the brain that might be interpreted as a tinnitus percept*
Hyperactive sound-driven activity can produce tinnitus sensations directly
Transient hyperactive neural activity activates attentional and emotional centers of the brain which directly leads to tinnitus
It will increase inhibitory events in the central nervous system thus enhancing neural responses that activate tinnitus
It plays no role in maintaining the percepts of tinnitus
-
Why may hyperactive spontaneous discharges in the dorsal cochlear nucleus not be the cause for tinnitus?
Because it causes hyperactive spontaneous activity in all auditory nuclei
Because ablating the dorsal cochlear nucleus in animals does not eliminate behaviors indicative of tinnitus perceptions*
Because it arises from excessive input from the auditory nerve
Because of the general brain increase in the inhibitory neurotransmitter GABA
Because it can be modulated by descending cortiofugal fibers from deeper brain centers
-
Reorganization of the auditory cortex tonotopic map, following peripheral ear damage arises from what conditions?
Increased spontaneous activity from peripheral brain centers that cause an excitotoxic response leading to massive reorganization of the entire tonotopic axis at the cortex
The enhancement of excitatory input and increased inhibitory activity influencing cortical neurons
It arises exclusively from the activity of inhibitory interneurons which causes suppression of spontaneous activity in cortical neurons
General atrophy and loss of unused cortical neurons
Reduced input from the periphery due to cochlear damage and the unmasking of weaker collateral inputs from MGB cells lying on the low frequency side of the lesion*
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott SD, Hughes LF, Bauer CA, Salvi R, Caspary DM. Detection of glutamate decarboxylase isoforms in rat inferior colliculus following acoustic exposure. Neuroscience. 1999;93:1375–1381. doi: 10.1016/s0306-4522(99)00300-0. [DOI] [PubMed] [Google Scholar]
- Baguley DM. Mechanisms of tinnitus. British Medical Bulletin. 2002;63:195–212. doi: 10.1093/bmb/63.1.195. [DOI] [PubMed] [Google Scholar]
- Bashtanov ME, Goodyear RJ, Richardson GP, Russel IJ. The mechanical properties of the chick (Gallus domesticus) sensory hair bundles: Relative contributions of structures sensitive to calcium chelation and subtilisin treatment. Journal of Physiology. 2004;559:287–299. doi: 10.1113/jphysiol.2004.065565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CA. Effects of chronic salicylate on GABAergic activity in the rat inferior colliculus. Hearing Research. 2000;147:175–182. doi: 10.1016/s0378-5955(00)00130-1. [DOI] [PubMed] [Google Scholar]
- Bauer CA. Mechanisms of tinnitus generation. Current Opinions in Otolaryngology: Head and Neck Surgery. 2004;12:413–417. doi: 10.1097/01.moo.0000134443.29853.09. [DOI] [PubMed] [Google Scholar]
- Brozoski TJ, Bauer CA. The effects of dorsal cochlear nucleus ablation on tinnitus in rats. Hearing Research. 2005;206:227–236. doi: 10.1016/j.heares.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Brozoski TJ, Bauer CA, Caspary DM. Elevated fusiform cell activity in the dorsal cochlear nucleus of the chinchilla with psychophysical evidence of tinnitus. Journal of Neuroscience. 2002;22:2383–2390. doi: 10.1523/JNEUROSCI.22-06-02383.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Jastreboff PJ. Salicylate induced abnormal activity in the inferior colliculus of rats. Hearing Research. 1995;82:158–178. doi: 10.1016/0378-5955(94)00174-o. [DOI] [PubMed] [Google Scholar]
- Chen K, Chang H, Zhang J, Kaltenbach JA, Godfrey DA. Altered spontaneous activity in rat dorsal cochlear nucleus following loud tone exposure. In: Hazell JWP, editor. Proceedings of the sixth international tinnitus seminar. London: Tinnitus and Hyperacusis Center; 1999. pp. 212–217. [Google Scholar]
- Chery-Croze S, Truy E, Morgon A. Contralateral suppression of transiently evoked otoacoustic emissions and tinnitus. British Journal of Audiology. 1994;28:255–266. doi: 10.3109/03005369409086575. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Bridges RJ, Taube JS, Clark AS, Gedded JW, Monaghan DT. The role of the NMDA receptor in central nervous system plasticity and pathology. Journal of NIH Research. 1989;1:65–74. [Google Scholar]
- Dobie RA. Depression and tinnitus. Otolaryngology Clinics of North America. 2003;36:383–388. doi: 10.1016/s0030-6665(02)00168-8. [DOI] [PubMed] [Google Scholar]
- Dominguez M, Becker S, Bruce I, Read H. A spiking neuron model of cortical correlates of sensorineural hearing loss: Spontaneous firing, synchrony, and tinnitus. Neural Computation. 2006;18:2942–2958. doi: 10.1162/neco.2006.18.12.2942. [DOI] [PubMed] [Google Scholar]
- Drenckhahn D, Engle K, Hofer D, Merte C, Tilney L, Tilney M. Three different actin filament assemblies occur in every hair cell: Each contains a specific actin crosslinking protein. Journal of Cell Biology. 1991;112:641–651. doi: 10.1083/jcb.112.4.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan RK, Saunders JC. Stereocilia injury mediates hair bundle stiffness loss and recovery following intense water-jet stimulation. Journal of Comparative Physiology A. 2000;186:1095–1106. doi: 10.1007/s003590000164. [DOI] [PubMed] [Google Scholar]
- Eggermont JJ. Tinnitus: Neurobiological substrates. Drug Discovery Today. 2005;10:1283–1290. doi: 10.1016/S1359-6446(05)03542-7. [DOI] [PubMed] [Google Scholar]
- Eggermont JJ, Roberts LE. The neuroscience of tinnitus. Trends in Neuroscience. 2004;27:676–682. doi: 10.1016/j.tins.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Giraud AL, Chery CS, Fischer G, Fischer C, Vighetto A, Gregoire MC, et al. A selective imaging of tinnitus. Neuroreports. 1999;10:1–5. doi: 10.1097/00001756-199901180-00001. [DOI] [PubMed] [Google Scholar]
- Grati M, Schneider ME, Lipkow K, Strehler EE, Wenthold RJ, Kachar B. Rapid turnover of stereocilia membrane proteins: Evidence from trafficking and mobility of plasma membrane CA(2+)-ATPase 2. Journal of Neuroscience. 2006;26:6386–6395. doi: 10.1523/JNEUROSCI.1215-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guitton MJ, Caston R, Ruel J, Johnson RM, Pujol R, Puel JL. Salicylate induced tinnitus through activation of NMDA receptors. Journal of Neuroscience. 2003;23:3944–3952. doi: 10.1523/JNEUROSCI.23-09-03944.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller AJ. Classification and epidemiology of tinnitus. Otolaryngology Clinics of North America. 2003;36:239–248. doi: 10.1016/s0030-6665(02)00160-3. [DOI] [PubMed] [Google Scholar]
- House JW, Brackman DE. Tinnitus: Surgical treatment. In: Evered D, Lawrenson G, editors. CIBA symposium 85: Tinnitus. London: Pitman Press; 1981. pp. 204–2120. [DOI] [PubMed] [Google Scholar]
- Husbands JM, Steinberg SA, Kurian R, Saunders JC. Tip-link integrity on chick tall hair cell stereocilia following intense sound exposure. Hearing Research. 1999;135:135–145. doi: 10.1016/s0378-5955(99)00101-x. [DOI] [PubMed] [Google Scholar]
- Jastreboff PJ. Phantom auditory perception (tinnitus): Mechanisms of generation and perception. Neuroscience Research. 1990;8:221–254. doi: 10.1016/0168-0102(90)90031-9. [DOI] [PubMed] [Google Scholar]
- Kaltenbach JA. Neurophysiologic mechanisms of tinnitus. Journal of the American Academy of Audiology. 2000;11:125–137. [PubMed] [Google Scholar]
- Kaltenbach JA. The dorsal cochlear nucleus as a participant in the auditory, attentional and emotional components of tinnitus. Hearing Research. 2006;217:224–234. doi: 10.1016/j.heares.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Kaltenbach JA, Zhang J, Finlayson P. Tinnitus as a plastic phenomenon and its possible neural underpinnings in the dorsal cochlear nucleus. Hearing Research. 2005;206:200–226. doi: 10.1016/j.heares.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Kaltenbach JA, Zhang J, Afman CE. Plasticity of spontaneous neural activity in the dorsal cochlear nucleus after intense sound exposure. Hearing Research. 2000;147:282–292. doi: 10.1016/s0378-5955(00)00138-6. [DOI] [PubMed] [Google Scholar]
- Kiang NY, Moxon EC, Levine RA. Auditory-nerve activity in cats with normal and abnormal cochleas. In: Wolstenholm GEW, Knight J, editors. Sensorineural hearing loss. London: Churchill Livingston; 1970. pp. 241–276. [DOI] [PubMed] [Google Scholar]
- Komiya H, Eggermont JJ. Spontaneous firing activity of cortical neurons in adult cats with reorganized tonotopic map following pure-tone trauma. Acta Otolaryngologica. 2000;120:750–756. doi: 10.1080/000164800750000298. [DOI] [PubMed] [Google Scholar]
- König O, Schaette R, Kempter R, Gross M. Course of hearing loss and occurrence of tinnitus. Hearing Research. 2006;221:59–64. doi: 10.1016/j.heares.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Kurian R, Krupp NL, Saunders JC. Tip link loss and recovery on short hair cells following intense exposure to sound. Hearing Research. 2003;181:40–50. doi: 10.1016/s0378-5955(03)00165-5. [DOI] [PubMed] [Google Scholar]
- LePage EL. A model for cochlear origin of subjective tinnitus: Excitatory drift in the operating point of inner hair cells. In: Vernon JA, Moller AR, editors. Mechanisms of tinnitus. London: Allyn and Bacon; 1995. pp. 115–148. [Google Scholar]
- Liberman MC. Chronic ultrastructural changes in acoustic trauma: Serial-section reconstruction of stereocilia and cuticular plates. Hearing Research. 1987;26:65–88. doi: 10.1016/0378-5955(87)90036-0. [DOI] [PubMed] [Google Scholar]
- Liberman MC, Kiang NY. Acoustic trauma in cats. Cochlear pathology and auditory-nerve activity. Acta Otolaryngologica. 1978;(Suppl 358):1–63. [PubMed] [Google Scholar]
- Lockwood AH. Tinnitus. Neurologic Clinics. 2005;23:893–900. doi: 10.1016/j.ncl.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Lockwood AH, Salvi RJ, Burkard RF. Tinnitus. New England Journal of Medicine. 2002;347:904–910. doi: 10.1056/NEJMra013395. [DOI] [PubMed] [Google Scholar]
- Lockwood AH, Salvi RJ, Coad ML, Towsley ML, Wack DS, Murphy BW. The functional neuroanatomy of tinnitus: Evidence for limbic system links and neural plasticity. Neurology. 1998;50:114–120. doi: 10.1212/wnl.50.1.114. [DOI] [PubMed] [Google Scholar]
- Long GR, Tubis A. Modification of spontaneous and evoked emissions and associated psychoacoustic microstructure by asprin consumption. Journal of the Acoustic Society of America. 1988;84:1343–1353. doi: 10.1121/1.396633. [DOI] [PubMed] [Google Scholar]
- Lowery LD, Eisenman LM, Saunders JC. An absence of tinnitus. Otology and Neurotology. 2004;25:474–478. doi: 10.1097/00129492-200407000-00013. [DOI] [PubMed] [Google Scholar]
- Ma WLD, Hidaka H, May BJ. Spontaneous activity in the inferior colliculus of CBA/J mice after manipulations that induce tinnitus. Hearing Research. 2006;212:9–21. doi: 10.1016/j.heares.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Mahlke C, Wallhäusser-Franke E. Evidence for tinnitus-related plasticity in the auditory and limbic system, demonstrated by arg3.1 and c-fos immunocytochemistry. Hearing Research. 2004;195:17–34. doi: 10.1016/j.heares.2004.03.005. [DOI] [PubMed] [Google Scholar]
- McFadden SL, Kasper C, Ostrowski J, Ding D, Salvi RJ. Effects of inner ear hair cell loss on inferior colliculus evoked potential thresholds, amplitudes, and forward masking functions in chinchillas. Hearing Research. 1998;120:121–132. doi: 10.1016/s0378-5955(98)00052-5. [DOI] [PubMed] [Google Scholar]
- Milbrandt JC, Holder TM, Wilson MC, Salvi RJ, Caspary DM. GAD levels and muscimol binding in rat inferior colliculus following acoustic trauma. Hearing Research. 2000;147:251–260. doi: 10.1016/s0378-5955(00)00135-0. [DOI] [PubMed] [Google Scholar]
- Mirz F, Gjedde A, Ishizu K, Pedersen CB. Cortical networks subserving the perception of tinnitus- A PET study. Acta Otolaryngologica. 2000;(Suppl 543):241–243. doi: 10.1080/000164800454503. [DOI] [PubMed] [Google Scholar]
- Mirz F, Pederson B, Ishizu K, et al. Positron emission tomography of cortical centers of tinnitus. Hearing Research. 1999;134:133–144. doi: 10.1016/s0378-5955(99)00075-1. [DOI] [PubMed] [Google Scholar]
- Moller AR. Similarities between severe tinnitus and chronic pain. Journal of the American Academy of Audiology. 2000;11:115–124. [PubMed] [Google Scholar]
- Møller AR. Neural plasticity and tinnitus. In: Moller AR, editor. Progress in brain research. Vol. 157. New York: Elsevier; 2006. pp. 365–372. [DOI] [PubMed] [Google Scholar]
- Morgenstern L. The bells are ringing. Perspectives in Biology and Medicine. 2005;48:396–407. doi: 10.1353/pbm.2005.0075. [DOI] [PubMed] [Google Scholar]
- Mühlau M, Rauschecker JP, Oestreicher E, Gaser C, Röttinger M, Wohlschläger AM, et al. Structural brain changes in tinnitus. Cerebral Cortex. 2006;16:1283–1288. doi: 10.1093/cercor/bhj070. [DOI] [PubMed] [Google Scholar]
- Mühlnickel W, Elbert T, Taub E, Flor H. Reorganization of auditory cortex in tinnitus. Proceedings of the National Academy of Science, USA. 1998;95:10340–10343. doi: 10.1073/pnas.95.17.10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulheran M. The effects of quinine on cochlear nerve fiber activity in the guinea pig. Hearing Research. 1999;134:145–152. doi: 10.1016/s0378-5955(99)00076-3. [DOI] [PubMed] [Google Scholar]
- Müller M, Klinke R, Arnold W, Oestreicher E. Auditory nerve fiber responses to salicylate revisited. Hearing Research. 2003;183:37–43. doi: 10.1016/s0378-5955(03)00217-x. [DOI] [PubMed] [Google Scholar]
- Noreña AJ, Eggermont JJ. Changes in spontaneous neural activity immediately after an acoustic trauma: Implications for neural correlates of tinnitus. Hearing Research. 2003;183:137–153. doi: 10.1016/s0378-5955(03)00225-9. [DOI] [PubMed] [Google Scholar]
- Ochi K, Eggermont JJ. Effects of quinine on neural activity in the cat primary auditory cortex. Hearing Research. 1997;105:105–118. doi: 10.1016/s0378-5955(96)00201-8. [DOI] [PubMed] [Google Scholar]
- Patuzzi R. In: Patuzzi R, editor. Outer hair cells, EP regulation and tinnitus; Proceedings of the VIIth international tinnitus seminar; Perth: University of Western Australia; 2002. pp. 16–24. [Google Scholar]
- Penner MJ, Burns EM. The dissociation of SOAEs and tinnitus. Journal of Speech and Hearing Research. 1987;30:396–403. doi: 10.1044/jshr.3003.396. [DOI] [PubMed] [Google Scholar]
- Penner MJ, Coles RRA. Indications for asprin as a palliative for tinnitus caused by SOAEs: A case study. British Journal of Audiology. 1992;26:91–96. doi: 10.3109/03005369209077876. [DOI] [PubMed] [Google Scholar]
- Pickles JO, Osborne MP, Comis SD. Vulnerability of tip links between stereocilia to acoustic trauma in the guinea pig. Hearing Research. 1987;25:173–183. doi: 10.1016/0378-5955(87)90089-x. [DOI] [PubMed] [Google Scholar]
- Rajan R, Irvine DR. Absence of plasticity of the frequency map in dorsal cochlear nucleus of adult cats after unilateral partial cochlear lesions. Journal of Comparative Neurology. 1998a;399:35–46. [PubMed] [Google Scholar]
- Rajan R, Irvine DR. Neural responses across cortical field AI in plasticity induced by peripheral field auditory organ damage. Audiology and Neurootology. 1998b;3:123–144. doi: 10.1159/000013786. [DOI] [PubMed] [Google Scholar]
- Rauschecker JP. Auditory cortical plasticity: A comparison with other sensory systems. Trends in Neuroscience. 1999;22:74–80. doi: 10.1016/s0166-2236(98)01303-4. [DOI] [PubMed] [Google Scholar]
- Reyes SA, Salvi RJ, Burkard RF, Coad ML, Wack DS, Galantowicz PJ, et al. Brain imaging of the effects of lidocaine on tinnitus. Hearing Research. 2002;171:43–50. doi: 10.1016/s0378-5955(02)00346-5. [DOI] [PubMed] [Google Scholar]
- Rzadzinska AK, Schneider ME, Davies C, Riordan GO, Kachar B. An actin molecular treadmill and myosins maintain stereocilia functional architecture and self-renewal. Journal of Cell Biology. 2004;164:887–897. doi: 10.1083/jcb.200310055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahey TL, Nodar RH. A biochemical model of peripheral tinnitus. Hearing Research. 2001;151:43–54. doi: 10.1016/s0378-5955(00)00235-5. [DOI] [PubMed] [Google Scholar]
- Salvi RJ, Lockwood AH, Burkard R. Neural plasticity and tinnitus. Ch. 5. In: Tyler R, editor. Tinnitus handbook. San Diego: Singular; 2000. pp. 123–148. [Google Scholar]
- Salvi RJ, Powers NL, Saunders SS, Boettcher FA, Clock AE. Enhancement of evoked response amplitude and single unit activity after noise exposure. In: Dancer A, Henderson D, Salvi RJ, Henderson RJ, editors. Noise induced hearing loss. St. Louis: Mosby Year Book; 1992. pp. 156–171. [Google Scholar]
- Salvi RJ, Saunders SS, Gratton MA, Arehole S, Powers N. Enhanced evoked response amplitudes in the inferior colliculus of the chinchjilla following acoustic trauma. Hearing Research. 1990;50:245–258. doi: 10.1016/0378-5955(90)90049-u. [DOI] [PubMed] [Google Scholar]
- Salvi RJ, Wang J, Powers NL. Plasticity and reorganization in the auditory brainstem: Implications for tinnitus. In: ReichJE GE, Vernon JA, editors. Proceedings of the fifth international tinnitus seminar. Portland, OR: American Tinnitus Association; 1996. pp. 457–466. [Google Scholar]
- Saunders JC, Bock GR, James R, Chen CS. Effects of priming for audiogenic seizures on auditory evoked responses in the cochlear nucleus and inferior colliculus of BALB/c mice. Experimental Neurology. 1972;37:388–394. doi: 10.1016/0014-4886(72)90082-9. [DOI] [PubMed] [Google Scholar]
- Saunders JC, Cohen YE, Szymko YM. The structural and functional consequences of acoustic injury in the cochlea and peripheral auditory system: A five year up year update. Journal of the Acoustical Society of America. 1991;90:136–146. doi: 10.1121/1.401307. [DOI] [PubMed] [Google Scholar]
- Saunders JC, Flock A. Changes in cochlear hair-cell stereocilia stiffness following overstimulation. Hearing Research. 1986;22:323. doi: 10.1016/0378-5955(86)90113-9. [DOI] [PubMed] [Google Scholar]
- Saunders JC, Schneider ME, Dear SP. The anatomical consequences of acoustic trauma. A review and tutorial. Journal of the Acoustical Society of America. 1985;78:833–860. doi: 10.1121/1.392915. [DOI] [PubMed] [Google Scholar]
- Schleuning AJ. Management of the patient with tinnitus. Medical Clinics of North America. 1991;75:1225–1237. doi: 10.1016/s0025-7125(16)30383-2. [DOI] [PubMed] [Google Scholar]
- Schneider ME, Belyantseva IA, Azevedo RB, Kachar B. Rapid renewal of auditory hair bundles. Nature. 2002;418:837–838. doi: 10.1038/418837a. [DOI] [PubMed] [Google Scholar]
- Seki S, Eggermont JJ. Changes in spontaneous firing rate and neural synchrony in cat primary auditory cortex after localized tone-induced hearing loss. Hearing Research. 2003;180:28–38. doi: 10.1016/s0378-5955(03)00074-1. [DOI] [PubMed] [Google Scholar]
- Sindhusake D, Golding M, Wigney D, Newell P, Jakobsen K, Mitchell O. Factors predicting severity of tinnitus: A Population-based assessment. Journal of the American Academy of Audiology. 2004;15:269–280. doi: 10.3766/jaaa.15.4.2. [DOI] [PubMed] [Google Scholar]
- Stouffer JL, Tyler RS. Characterization of tinnitus by tinnitus patients. Journal of Speech and Hearing Disorders. 1990;55:439–453. doi: 10.1044/jshd.5503.439. [DOI] [PubMed] [Google Scholar]
- Stypulkowski PH. Mechanisms of salicylate ototoxicity. Hearing Research. 1990;46:113–146. doi: 10.1016/0378-5955(90)90144-e. [DOI] [PubMed] [Google Scholar]
- Szymko YM, Nelson PM, Saunders JC. Stiffness changes in chick hair bundles following in vitro overstimulation. Journal of Comparative Physiology A. 1995;176:727–735. doi: 10.1007/BF00192621. [DOI] [PubMed] [Google Scholar]
- Tilney JG, Saunders JC, Egelman E, DeRosier DJ. Changes in the organization of actin filaments in the stereocilia of noise-damaged lizard cochleae. Hearing Research. 1982;7:181–197. doi: 10.1016/0378-5955(82)90013-2. [DOI] [PubMed] [Google Scholar]
- Wallhäusser-Franke E, Mahlke C, Oliva R, Braun S, Wenz G, Langner G. Expression of c-fos in auditory and non-auditory brain regions of the gerbil after manipulations that induce tinnitus. Experimental Brain Research. 2003;153:649–654. doi: 10.1007/s00221-003-1614-2. [DOI] [PubMed] [Google Scholar]
- Wang J, Salvi RJ, Powers N. Plasticity of response properties of inferior colliculus neurons following acute cochlear damage. Journal of Neurophysiology. 1996;75:171–183. doi: 10.1152/jn.1996.75.1.171. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hirose K, Liberman MC. Dynamics of noise-induced cellular injury and repair in the mouse cochlea. Journal of the Association for Research in Otolaryngology. 2002;3:248–268. doi: 10.1007/s101620020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz N, Wienbruch C, Dohrmann K, Elbert T. Neuromagnetic indicators of auditory cortical reorganization of tinnitus. Brain. 2005;128:2722–2731. doi: 10.1093/brain/awh588. [DOI] [PubMed] [Google Scholar]
- Willott JF, Lu SM. Noise induced hearing loss can alter neural coding and increase excitability in the central nervous system. Science. 1982;216:1331–1334. doi: 10.1126/science.7079767. [DOI] [PubMed] [Google Scholar]
- Yang G, Lobarinas EL, Zang L, Turner J, Stolzberg D, Salvi R, et al. Salicylate induced tinnitus: Behavioral measures and neural activity in auditory cortex of awake rats. Hearing Research, Web Publication. doi: 10.1016/j.heares.2006.06.013. in press. Retrieved from www.elsevier.com/locate/heares. [DOI] [PubMed]
- Zheng L, Sekerkova G, Vranich K, Tilney LG, Mugnaini E, Bartles JR. The deaf jerker mouse has a mutation in the gene encoding the espin actin-bundling proteins of hair cell stereocilia and lacks espins. Cell. 2000;102:377–385. doi: 10.1016/s0092-8674(00)00042-8. [DOI] [PMC free article] [PubMed] [Google Scholar]