

Abstract
Secondary bacterial pneumonia frequently claimed the lives of victims during the devastating 1918 influenza A virus pandemic. Little is known about the viral factors contributing to the lethality of the 1918 pandemic. Here we show that expression of the viral accessory protein PB1-F2 enhances inflammation during primary viral infection of mice and increases both the frequency and severity of secondary bacterial pneumonia. The priming effect of PB1-F2 on bacterial pneumonia could be recapitulated in mice by intranasal delivery of a synthetic peptide derived from the C-terminal portion of the PB1-F2. Relative to its isogenic parent, an influenza virus engineered to express a PB1-F2 with coding changes matching the 1918 pandemic strain was more virulent in mice, induced more pulmonary immunopathology, and led to more severe secondary bacterial pneumonia. These findings help explain both the unparalleled virulence of the 1918 strain and the high incidence of fatal pneumonia during the pandemic.
Over the past 2 decades, the human toll from influenza has averaged 200,000 hospitalizations and 36,000 deaths per year in the United States alone (Thompson et al., 2003; Thompson et al., 2004). Few influenza viruses are sufficiently virulent to directly cause death in humans. Instead, most deaths are due to an increased physiologic load in an already compromised host, or are the outcome of the combined effects of the viral disease and a secondary bacterial infection (Mote, 1940; McCullers, 2006). Although bacterial pneumonia during or immediately following influenza is a significant contributor to morbidity and mortality (Simonsen, 1999), the pathogenic interaction between influenza viruses and bacteria is poorly understood. The 1918 influenza virus was remarkable for its lethality, accounting for more than 40 million deaths world wide (Potter, 1998). This pandemic strain was capable of causing a fatal primary pneumonia, although most fatal cases were associated with secondary bacterial pathogens (Muir and Wilson, 1919; Stone and Swift, 1919; Abrahams et al., 1919; McCullers, 2006; Morens and Fauci, 2007). The reasons for this unparalleled virulence and the strong association with secondary bacterial disease are currently unknown but are the subject of intense scientific scrutiny.
PB1-F2 is a recently described pro-apoptotic influenza A virus (IAV) protein not required for viral replication in ovo or in cultured cells. It is encoded by an alternative reading frame present in the PB1 gene of nearly all IAV isolates, including highly pathogenic avian IAVs that have infected humans (Chen et al., 2001; Obenauer et al., 2006) and the IAV whose genetic information was recovered from a victim of the 1918 pandemic (Taubenberger et al., 2005). PB1-F2 possesses a C-terminal mitochondrial targeting sequence (MTS) that is predicted to form a positively charged amphipathic helix (Gibbs et al., 2003). PB1-F2 compromises mitochondrial function and induces apoptosis, probably through its association with inner and outer mitochondrial membrane transporters ANT3 and VDAC1, respectively (Zamarin et al., 2005). Synthetic full length PB1-F2 induces cytotoxicity at concentrations of 50 nM or less when incubated with cells (Chen et al., 2001), possibly by forming pores that destabilize the plasma membrane (Chanturiya et al., 2004). PB1-F2 was recently shown to enhance viral pathogenicity in the mouse IAV infection model (Zamarin et al., 2006), raising the question of its effects on the secondary bacterial infections associated with high levels of influenza morbidity and mortality.
Results
Expression of PB1-F2 enhances secondary bacterial pneumonia
We first examined the effect of PB1-F2 expression on induction of secondary bacterial infection in a mouse model (McCullers and Bartmess, 2003; McCullers, 2004) utilizing the mouse-adapted A/Puerto Rico/8/34 (PR8) strain of influenza (wt) and an isogenic strain (mut) engineered to greatly reduce PB1-F2 expression (Zamarin et al., 2006). Groups of 6 mice were infected with wt or mut PR8 and challenged 7d later with bioluminescent Streptococcus pneumoniae. Mice infected with the wt and mut viruses had similar viral lung loads (Fig 1A) and exhibited similar weight loss through the day of pneumococcal infection (Fig. 1B; 5.4% vs. 2.2% on d0, p > 0.1). After bacterial infection (100 CFU), expression of PB1-F2 was associated with significantly enhanced weight loss (25.8% vs. 2.2% on d3, p < 0.05), greater induction of pneumonia as detected by bioluminescence, and higher mortality (5/6 dead vs. 1/6 dead following infection with mut PR8; Fig. 1A–C). When a ten-fold higher dose of bacteria (1000 CFU) was utilized, 6/6 mice died when wt PR8 was the viral prime, compared to 5/6 following infection with mut PR8 (p > 0.1). Control mice infected with either virus and then challenged with PBS instead of bacteria all survived (data not shown).
Figure 1.
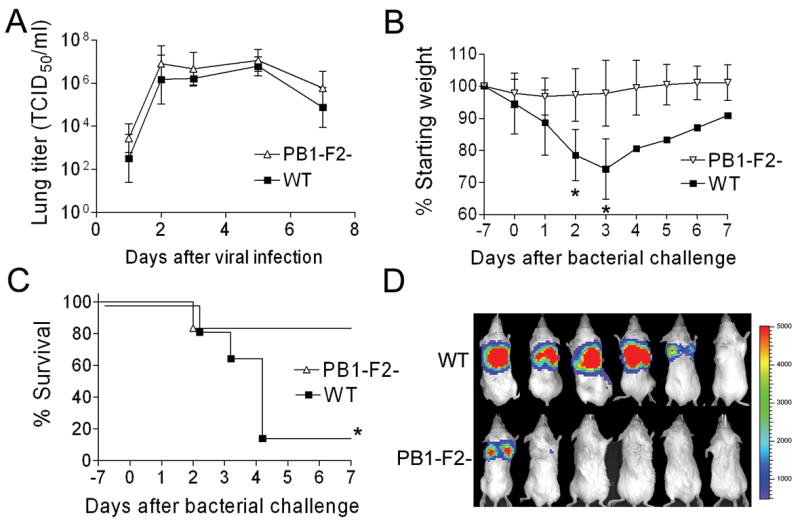
Secondary bacterial pneumonia following influenza. (A) Groups of 4–6 mice infected with either wt influenza virus PR8 (WT) or an isogenic mut virus that does not express PB1-F2 (PB1-F2-) were euthanized 1, 2, 3, 5, and 7 days after infection for determination of viral lung load. Groups of 6 mice infected with either WT or PB1-F2- were challenged with pneumococcus on d7 post-infection. (B) weight loss and (C) survival are plotted until 7 days after secondary challenge. Error bars represent standard deviation and an asterisk (*) denotes a significant difference (p < 0.05) compared to the group infected with PB1-F2- virus. (D) Pictures of anesthetized mice taken 36–48 hours after bacterial infection with luciferase-expressing pneumococci show bioluminescence indicative of pneumonia. The scale on the right indicates the number of relative light units per shaded pixel.
Expression of PB1-F2 enhances immunopathology of secondary bacterial pneumonia
Having established that expression of PB1-F2 in this model increases the incidence and exacerbates bacterial pneumonia, we next addressed whether expression of PB1-F2 influences the pathogenesis of bacterial pneumonia once it has developed. Mice were challenged with pneumococcus (1000 CFU) 7 days after infection with either wt or mut PR8, and bioluminescent imaging was used to choose pneumonic mice for detailed characterization as described (McCullers and Bartmess, 2003; McCullers, 2004). The higher dose of bacteria was used in this experiment to insure that mice in both groups had pneumonia in order that the comparison of the lungs be relevant. Under these conditions, expression of PB1-F2 was not associated with significant differences in thoracic bacterial bioluminescence or bacterial lung titers at either early (first detection of pneumonia) or late (after 24 hours of pneumonia) timepoints during the course of pneumonia (Fig. 2). We did, however detect significant PB1-F2-associated differences in immunopathology. PB1-F2 expression significantly (p < 0.05) increased numbers of white blood cells present in broncho-alveolar lavage fluids (BALFs). This difference was due to increases in the absolute numbers of neutrophils (Fig. 3A), macrophages (Fig. 3B), and T-cells (Fig. 3C), but not B-cells (Fig. 3D). Based on the lack of differences in viral (Fig 1A) and bacterial (Fig 2) parameters in this comparison of mice infected with the wt and mut PR8, it is unlikely that enhanced viral virulence from expression of the PR8 PB1-F2 is the sole or most important factor engendering the increased inflammation. However, Zamarin et al. showed differences in viral lung load in mice at day 6 (but not day 3) using a different pair of viruses based on the strain WSN (Zamarin et al., 2006) suggesting that this conclusion may not hold for all viruses.
Figure 2.
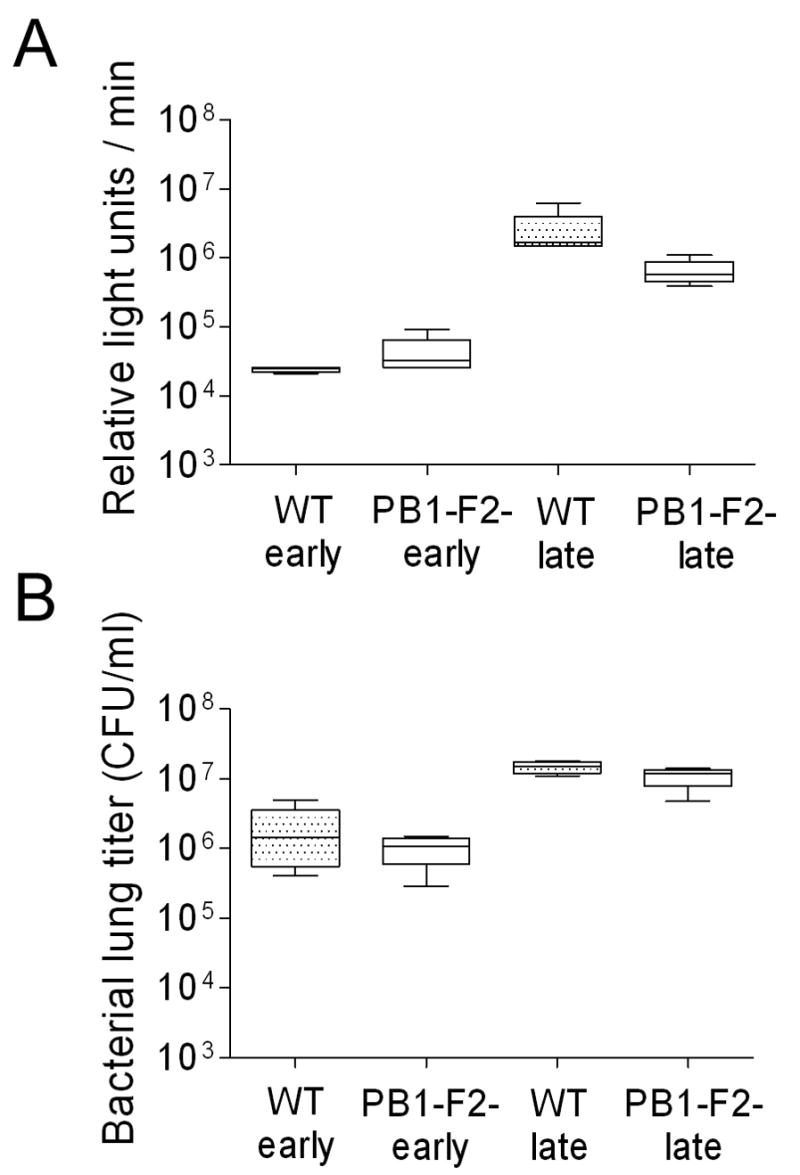
Titers from mice with pneumonia. Groups of mice were infected with either PR8 wt (WT) or an isogenic mut strain of PR8 unable to express PB1-F2 (PB1-F2-) and challenged 7 days later with pneumococcus. Four mice per group were assayed when they developed early pneumonia (> 10,000 relative light units (RLU)/min from the thorax) and late pneumonia (24 h of visual bioluminescence and > 1,000,000 RLU/min from the thorax) for A) mean flux of RLU/min and B) bacterial lung titers (CFU/ml lung homogenate). There were no significant differences between mice infected with PR8 wt and PR8 mut at any time point for any measurement (p > 0.1).
Figure 3.
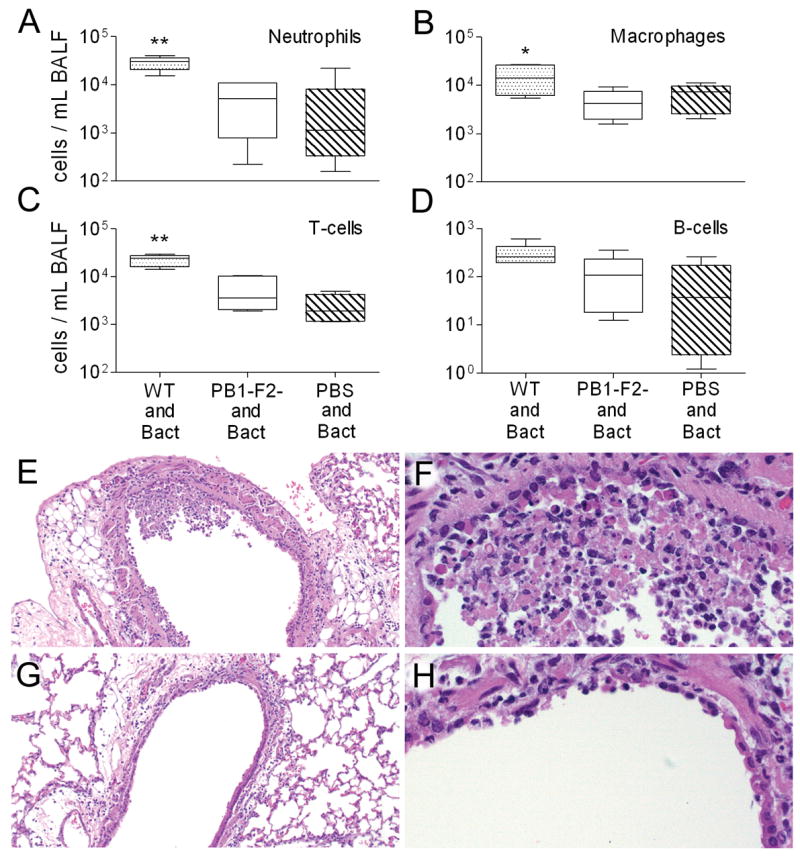
Cell counts from broncho-alveolar fluid (BALF) of secondarily infected mice with pneumonia. Mice were infected with PR8 wt (WT), an isogenic mut strain of PR8 unable to express PB1-F2 (PB1-F2-), or PBS, followed 7 days later by pneumococcus. Selected mice that developed early pneumonia (6–10 per group) were assayed for number of (A) neutrophils, (B) macrophages, (C) T-cells, and (D) B-cells from BALF samples. The 25th – 75th percentiles are represented by the shaded box-plots with the horizontal bar indicating the mean value. Error bars indicate the standard deviation of the measurements. A double asterisk (**) indicates a significant difference (p < 0.05) compared to all other groups, an asterisk (*) compared to the mut PR8 group. In a separate experiment, lungs were taken from mice 3 days after infection with either PR8 wt (E, F) or PR8 mut (G, H). Pictured are representative sections at either 10X (E, G) or 40X (F, H) magnification. Necrosis of the airway epithelium, manifest as killed cells with disruptions in the epithelial layer and deposition of debris, and inflammatory infiltration with neutrophils are more prominent in the mice infected with the wt virus.
This increased influx of leukocytes into the pulmonary compartment was correlated with histological changes apparent in the lungs of infected mice. As reported previously (McCullers and Rehg, 2002), infection with wt PR8 resulted in multiple parenchymal foci of infection characterized by alveolar inflammation, alveolar epithelial cell hypertrophy and hyperplasia, and occasional alveolar necrosis and fibrin deposition. Inflammatory cell infiltrates with lymphocytes, neutrophils, and macrophages were seen, particularly in perivascular regions. Notably, the degree of parenchymal inflammation was diminished in the mut PR8-infected mice infected (mean pathological score of 0.8 ± 0.5 on d3 compared to 2.3 ± 0.5 in wt-infected mice) (Fig. 3E–H). Airway involvement in PR8 infected mice was characterized by inflammatory cells in the lumen and/or mucosa as well as epithelial necrosis and epithelial hyperplasia. While wt and mut both induced foci of necrosis/apoptosis and sloughing of airway epithelium, expression of PB1-F2 was associated with increased inflammation in the airways (mean pathological scores of 2.8 ± 0.5 (wt) vs. 1.0 ± 0.0 (mut) on d3). The inflammatory infiltrate induced by PB1-F2 was dominated by macrophages. Bacterial superinfection of both groups increased the pathological alterations; viz. inflammatory infiltrates, epithelial cell hypertrophy and hyperplasia, necrosis, and fibrin deposition. These changes were exacerbated by PB1-F2 expression.
C-terminal PB1-F2 peptide primes for bacterial infection
In the context of viral infection, expression of the PB1-F2 protein enhanced secondary bacterial infections. To determine whether this property was mediated by other viral proteins or was intrinsic to the PB1-F2 itself, we administered peptides derived from the amino acid sequence of PR8 PB1-F2 to mice and then challenged them with bacteria 24 hours later. Two peptides were utilized, one from the C-terminal region of PB1-F2 including the mitochondrial targeting sequence and one from the N-terminal region as a control. Mice became ill and lost significant weight when given the C-terminal peptide, but were unaffected by the N-terminal peptide or PBS (Fig 4A). Following bacterial challenge, all mice primed with the C-terminal peptide succumbed within 8 days, while no mice died in either control group (Fig 4B). Because the C-terminal peptide is bacteriocidal for pneumococcus in vitro at concentrations ranging between 16 and 256 μM (data not shown), this finding could have been mediated by peptide induced bacterial death and the subsequent inflammatory response. Thus, to determine whether the inflammatory changes seen in the BAL were a direct effect of PB1-F2 or were mediated through an interaction with the bacteria, mice were given peptides or PBS, and then sacrificed for collection of BALF 72 hours later. A 2–3 log increase in neutrophil and macrophage numbers were seen in the BALF of mice which were given the C-terminal peptide (Fig 4C, D). T-cells and B-cells were not increased relative to control groups. Administration of bacteria (100 CFU) in the absence of peptide did not increase the cellularity of the BAL (data not shown). Therefore, the PB1-F2 can elicit inflammation and promote severe bacterial pneumonia outside the context of the virus, a function which maps to the C-terminal region of the protein.
Figure 4.

Peptide from the C-terminal portion of PB1-F2 elicits an inflammatory response and primes for secondary bacterial pneumonia. Groups of 5 mice were given peptides or PBS as a control intranasally on day -1 then challenged with 100 CFU of bacteria 24 hours later. (A) Weight loss and (B) mortality are compared. The peptide from the C-terminal region of the PB1-F2 protein caused more weight loss prior to and after bacterial challenge (* p < 0.05 by Student’s t-test) and less survival (* p < 0.01 by log-rank test on the Kaplan-Meier survival data) than did a peptide from the N-terminal region or PBS. Groups of 5 mice were assayed for number of (C) neutrophils and (D) macrophages from broncho-alveolar lavage fluid samples 3 days after exposure to peptide alone. The 25th – 75th percentiles are represented by the shaded box-plots with the horizontal bar indicating the mean value. Error bars indicate the standard deviation of the measurements. An asterisk (*) indicates a significant difference (p < 0.05) by ANOVA compared to both controls at that timepoint.
The 1918 PB1-F2 contributes to virulence
Could the 1918 PB1-F2 have contributed to the pandemic strain’s unparalleled virulence? To examine this question we modified the PR8 PB1 gene segment at eight positions within the PB1-F2 ORF where the PR8 and 1918 pandemic strain sequences differ. Thus, a new virus was rescued which differs from the PR8 version by 7 amino acid substitutions and a single residue extension to the COOH-terminus of the PB1-F2 protein, recapitulating differences between the wt and the 1918 PB1-F2. None of the non-synonymous mutations in the PB1-F2 reading frame result in non-synonymous mutations in the overlapping region of the PB1. Expression of the 1918-like PB1-F2 protein enhanced the growth of the PR8-PB1-F21918 virus in tissue culture compared to wt (Fig. 5A), and significantly increased the mean plaque size (1.33 mm ± 0.36) compared to either wt (0.79 mm ± 0.35) or mut PR8 (0.79 mm ± 0.20) (Fig 5B; p < 0.001). PR8-PB1-F21918 was unable to grow in the absence of trypsin (data not shown), a characteristic of the reconstituted 1918 strain (Tumpey et al., 2005). PR8-PB1-F21918 was more virulent than wt in mice (Fig. 6A) and reached significantly higher mean titers 2 days after infection (Fig. 6B; p < 0.01). However, this accelerated growth at early timepoints in mice did not result in a higher peak titer, and no differences in viral lung load were present at either 3 or 7 days post infection. We next examined the cellularity of the BALF of mice infected with wt PR8, mut PR8, or PR8-PB1-F21918 1, 3, and 7 days after infection. Expression of 1918 PB1-F2 increased pulmonary inflammation on d7 (but not d1 or d3) with increases in absolute numbers of pulmonary neutrophils, macrophages, and T-cells compared to wt and mut PR8 (Fig. 6C–F).
Figure 5.

Growth and plaque size of a virus expressing the 1918 PB1-F2. An otherwise isogenic virus engineered to express the PB1-F2 protein from the 1918 pandemic strain (1918) was compared to the wt PR8 strain (WT). (A) HA titer is compared for a virus expressing the 1918 PB1-F2 to wt PR8. Error bars indicate standard deviation of the measurements and an asterisk (*) indicates a significant difference at that timepoint by the paired Student’s t-test (p < 0.05). (B) Plaque size was measured for a virus expressing the 1918 PB1-F2 (1918) compared to wt PR8 (WT) and a virus that does not express the PB1-F2 protein (PB1-F2-). An asterisk (*) indicates a significant difference (P < 0.00001) compared to both other groups.
Figure 6.

Virulence of and inflammatory response to a virus expressing the 1918 PB1-F2. An otherwise isogenic virus engineered to express the PB1-F2 protein from the 1918 pandemic strain (1918) was compared to the wt PR8 strain (WT). (A) Survival of groups of 4 mice infected with 500 or 1000 TCID50 of wt PR8 or PR8-PB1-F21918. (B) Viral titers from lung homogenates taken from groups of 6–12 mice at multiple timepoints after infection with wt PR8 or PR8-PB1-F21918. Error bars indicate standard deviation of the measurements and an asterisk (*) indicates a significant difference at that timepoint by ANOVA compared to the wt PR8. In a separate experiment, mice were infected with PR8 wt or PR8-PB1-F21918 and were assayed for number of (C) neutrophils, (D) macrophages, (E) T-cells, and (F) B-cells from broncho-alveolar lavage fluid samples 1, 3, and 7 days after infection. The 25th – 75th percentiles are represented by the shaded box-plots with the horizontal bar indicating the mean value. Error bars indicate the standard deviation of the measurements. An asterisk (*) indicates a significant difference (p < 0.05) by ANOVA compared to wt PR8 at that timepoint.
The 1918 PB1-F2 primes for secondary bacterial pneumonia more efficiently than wt PR8
We next determined the effect of PB1-F21918 expression on bacterial superinfection. Groups of 10 mice infected with wt PR8 or PR8-PB1-F21918 were challenged with bacteria 7 days later. Expression of PB1-F21918 significantly enhanced mortality (p < 0.01) (Fig. 7A) and increased bacterial growth (p < 0.01) 1, 2, and 3 days following bacterial challenge (Fig. 7B). Control mice infected with either virus at this dose and then challenged with PBS instead of bacteria all survived (data not shown). Histopathologic examination of pneumonic lungs taken from wt PR8 infected mice revealed changes similar to those observed in earlier experiments described above. Infection with PR8-PB1-F21918 resulted in a severe, purulonecrotic, lobar pneumonia characterized by diffuse coagulative necrosis of the interstitium, marked pleuritis, and massive neutrophilic bronchitis and bronchiolitis (Fig. 7C, D). Extravasation of lymphocytes and plasma cells was evident perivascularly and along the airways. Compared to the patchy distribution of consolidated foci seen in the mice pre-infected with wt PR8, entire lung lobes were diffusely involved in mice infected with PR8-PB1-F21918. The finding of necrotic changes in the airways and the prominence of neutrophils and macrophages resemble the pathology reported for mice infected with the fully reconstructed 1918 strain (Tumpey et al., 2005), although in the present instance it is greatly exacerbated by secondary bacterial infection.
Figure 7.
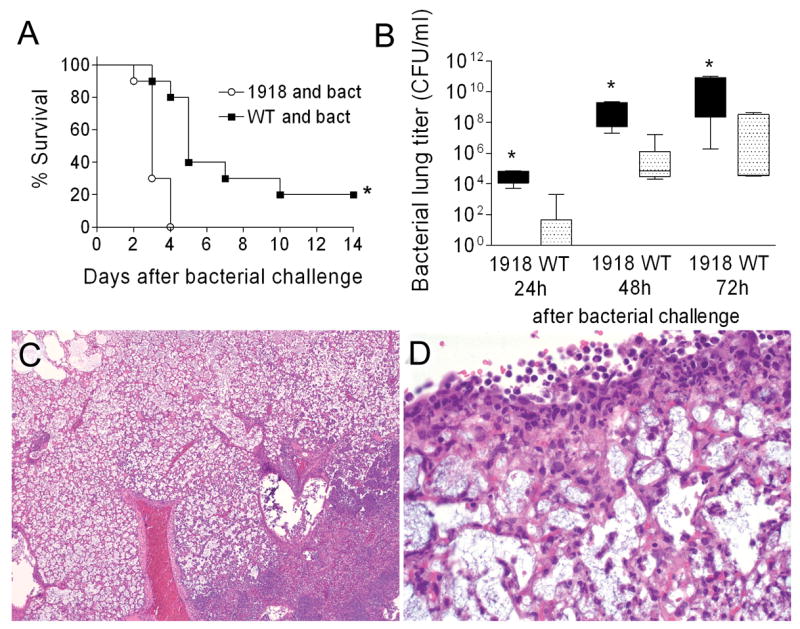
1918 PB1-F2 and secondary bacterial pneumonia. Groups of 10 mice were infected with 50 TCID50 of wt PR8 or PR8-PB1-F21918 followed 7 days later by bacterial challenge. (A) Survival following bacterial challenge is plotted, an asterisk (*) indicates a significant difference in survival by the log-rank test on the Kaplan-Meier data. (B) Bacterial lung loads from 5 mice per group and timepoint were determined 24, 48, and 72 hours after bacterial challenge. The 25th – 75th percentiles are represented by the shaded box-plots with the horizontal bar indicating the mean value. Error bars indicate the standard deviation of the measurements. An asterisk (*) indicates a significant difference (p < 0.05) by ANOVA compared to wt PR8 at that timepoint. Lungs were taken from 5 mice per group with evidence of pneumonia by bioluminescence 3 days after infection with either wt PR8 (data not shown) or PB8-PB1-F21918 (C, D). Pictured are representative sections at either 10X (C) or 40X (D) magnification. Massive, lobar, coagulative necrosis with pleuritis and leukocytic infiltration are evident.
The 1918 PB1-F2 promotes cytokine and chemokine release during secondary bacterial pneumonia
We further characterized the effect of PB1-F21918 on the pathological process by measuring a set of six cytokines and chemokines (TNF-α, IL-1-α, IL-6, IL-10, KC, MIP-1α) previously implicated in the pathogenesis of secondary bacterial infections in the mouse model (Smith et al., 2007). No significant differences were seen in this panel on d3 and d7 after primary infection with wt vs. PR8-PB1-F21918 (Fig. 8A–F). Three days after bacterial infection (10 days after viral infection), however, the entire panel except IL-10 demonstrate significant elevations in mice infected with PR8 PB1-F21918 vs. wt PR8 (Fig. 8A–F).
Figure 8.

Cytokines and chemokines in secondary bacterial pneumonia. Groups of 10 mice were infected with wt PR8 or PR8-PB1-F21918 followed 7 days later by bacterial challenge. Levels of (A) TNF-α, (B) IL-1α, (C) IL-6, (D), IL-10, (KC), and (F) MIP-1α from lung homogenates taken from mice 3 and 7 days after viral infection (prior to bacterial challenge) and 72 hours after bacterial challenge (10 days after viral infection) are presented. The 25th – 75th percentiles are represented by the shaded box-plots with the horizontal bar indicating the mean value. Error bars indicate the standard deviation of the measurements. An asterisk (*) indicates a significant difference by ANOVA compared to the wt PR8 group (p < 0.05).
Discussion
Taken together, our findings indicate that PB1-F2 plays an important role in promoting lung pathology in both primary viral infection and secondary bacterial infection. The numbers of IAV-associated deaths vary from season to season, and secondary bacterial pneumonia is a significant contributor to this yearly toll (Thompson et al., 2003). Differences in viral virulence factors likely contribute to fluctuations in secondary bacterial pneumonia and annual mortality (Peltola et al., 2005; McCullers, 2006). IAVs are subtyped by differences in their hemagglutinin (HA) and neuraminidase (NA) glycoproteins. Over the last 2 decades infections with H1N1 strains have caused less morbidity and mortality than H3N2 strains (Thompson et al., 2003; Thompson et al., 2004; Simonsen et al., 2000). Post-1956 H1N1 viruses possess PB1 gene segments that encode truncated PB1-F2 proteins of 67 amino acids due to the introduction of a stop codon. Such PB1-F2s lack the MTS required for induction of apoptosis, which is in the region important for induction of inflammation in our experiments with synthesized peptides (Gibbs et al., 2003). This defect might contribute to the decreased pathogenicity of contemporary H1N1 strains and their reduced capacity to promote secondary bacterial infections (Peltola et al., 2006). The predicted amino acid sequence of the PB1-F2 from the 1918 strain differs at eight positions from the sequence of PR8, five of them in the C-terminal region. It is of obvious interest and importance to correlate genetic variation in this and other PB1-F2 proteins with alterations in PB1-F2 function. The enhancing effect of these substitutions on viral replication, completely unexpected from previous findings that PB1-F2 knockdown does not effect replication in vitro (Chen et al., 2001; Zamarin et al., 2006), suggests that PB1-F2 can modify the function of one or more IAV gene products that are required for viral replication.
The mechanism by which PB1-F2 facilitates inflammation and secondary bacterial pneumonia is of prime interest. A synthetic version of PB1-F2 has been shown to be a potent inducer of cell death (Chen et al., 2001) and a synthetic peptide from the C-terminal region provided a strong pro-inflammatory stimulus in mice (Fig. 4). In vivo, the protein may be released from moribund or dead cells and promote cell death in areas surrounding foci of infection or in first responder infected mononuclear cells. Release of cell wall components from pneumococci, such as lipoteichoic acids and peptidoglycan, along with the cytotoxin pneumolysin, activates the innate immune system through Toll-like receptors 2 and 4 leading to production of pro-inflammatory cytokines (Yoshimura et al., 1999; Malley et al., 2003; McCullers and Tuomanen, 2001). PB1-F2 mediated cell death may trigger a positive feedback cytokine loop, amplified by bacterial superinfection, that enhances the pulmonary inflammatory response to IAV leading to the immunopathological death of the host. This hypothesis builds on earlier work demonstrating that influenza infection prior to bacterial superinfection causes a synergistic increase in the cytokine response with a resultant adverse outcome (Smith et al., 2007), and treatment of secondary pneumonia by antibiotic-mediated lysis of pneumococcus does not reduce mortality (McCullers, 2004). Recent work from Kash et al. supports this view, as expression of inflammatory and death receptor genes linked to mitochondrial apoptosis is increased by infection with the reconstructed 1918 virus (Kash et al., 2006). Although these effects could not be tied directly to a specific gene product through analyses using the full virus, our data suggest that the PB1-F2 is a leading candidate. However, differences in lung cytokines were not seen in our experiments until after bacterial challenge, suggesting that expression of PB1-F2 in the context of the entire 1918 genome may be necessary for the full impact on inflammation.
While this model is compelling, the pro-inflammatory effect of PB1-F2 may be completely unrelated to its pro-apoptotic functions. PB1-F2 may be directly recognized by pattern recognition receptors of the innate immune system, or serve as a chemoattractant similar to human beta-defensins released from dead cells (Yang et al., 1999). The predicted homology of the C-terminal cationic helical domain of PB1-F2 to similar regions of the human beta-defensins lends some credence to this theory. The immune effector cells recruited through this mechanism may contribute to immunopathology while accomplishing their primary function of clearing the infection. Alternatively, expression of PB1-F2 may alter the expression or function of other viral proteins that impact viral pathogenesis. Further studies are necessary to investigate these hypotheses in detail.
One intriguing result was the shortened time to peak viral titer observed both in vitro (Fig 5A) and in vivo in the virus expressing the 1918-like PB1-F2 (Fig 6B). This appears to be a unique feature of the 1918-like PB1-F2, since deletion of PB1-F2 from wt PR8 (data not shown) or WSN (Zamarin et al., 2006) did not alter viral replication in vitro. In mice, virulence is not diminished by deletion of PB1-F2 from wt PR8 (data not shown) or WSN (Zamarin et al., 2006), and the kinetics of viral infection do not differ in a PR8 background (Fig 1A). Transfer of the PR8 PB1 gene to a WSN background did reveal differences in virulence and clearance of virus from the lungs of mice, although viral titers did not differ at an early timepoint (day 3) and in vitro replication was again not affected (Zamarin et al., 2006). Thus, it could be postulated that the accelerated tempo of early lung infection of the virus expressing the 1918-like PB1-F2 allows the virus to outpace innate immune mechanisms of control. This could contribute to virulence as has been suggested for PR8 (Grimm et al., 2007), highly pathogenic H5N1 viruses (Gao et al., 1999) and the 1957 H2N2 pandemic strain (Legge and Braciale, 2003). While this is an interesting hypothesis to explain the increased virulence of the 1918 strain, it fails to explain the enhancement of bacterial superinfections because this enhancement is seen in both the wt PR8 knockout model (Fig 1B–D), where there are no differences in viral kinetics (Fig 1A), as well as in the 1918 PB1-F2 model (Fig. 6). Several factors suggest that other mechanisms must be considered for the exacerbation of secondary bacterial infections with the 1918 PB1-F2, or at the least that there are both direct and indirect contributions of this protein to the pathogenesis. First, the differences in cellularity of the BALF (Fig. 6C–E) were initially detected on d7, a time when viral titers (Fig. 6B) and lung pathology (data not shown) did not significantly differ. Second, the enhanced secondary bacterial pneumonia and elevated cytokines (Fig. 8A–F) also occurred after viral lung loads had equalized. Had the increased tempo of early infection been responsible these differences would have been expected to be detectable earlier as was seen in experiments with H5N1 viruses (Gao et al., 1999). Third, even when wt PR8 was given in much higher doses, or with much higher doses of bacteria, it never induced pathological alterations of similar character and magnitude as PB8-PB1-F21918 (data not shown). And finally, topical administration of a peptide derived from the C-terminal end of the PB1-F2 protein recapitulates the effects on bacterial superinfection independent of viral infection (Fig. 4).
Clearly, much remains to be learned about PB1-F2 and its contribution to viral virulence. We have demonstrated here that the protein is pro-inflammatory, can contribute to virulence, and facilitates secondary bacterial infections. The ability of 1918 PB1-F2 to enhance the pathogenicity of PR8 may only represent the tip of the iceberg of the full pathogenic potential of this protein when expressed in its natural context of the complete 1918 strain genome. Given the importance of IAV as a leading cause of virus-induced morbidity and mortality year in and year out, and its potential to kill tens of millions in the inevitable pandemic that may have its genesis in the viruses currently circulating in southeast Asia, it is imperative to understand the role of PB1-F2 in IAV pathogenicity and transmission in humans and animals. Our data help explain why the 1918 pandemic strain was so efficient at supporting bacterial pneumonia and reinforce the recent suggestion of the American Society of Microbiology that nations should stockpile antibiotics in anticipation for the next pandemic (American Society for Microbiology, 2006).
Materials and Methods
Influenza viruses
The Mount Sinai strain of mouse adapted influenza virus A/Puerto Rico/8/34 (H1N1), hereafter referred to as PR8, was genetically engineered to interfere with PB1-F2 expression by substituting in the PB1 gene T ⇒ C at position 120 and C ⇒ G at position 153 (altering the initiating Met to Ser and inserting a stop codon after 11 residues). Both of the substitutions result in synonymous codons in the PB1 reading frame. This virus and its wild-type parent (provided by Dr. Peter Palese, Mt. Sinai Medical School, NY, NY) were used in experiments described in the text referring to Figures 1, 2, and 3.
For subsequent experiments, a second set of isogenic viruses were generated at St. Jude Children’s Research Hospital using PR8 plasmids derived from the St. Jude influenza virus repository. Full length A/Brevig Mission/1/1918 (H1N1) and PR8 PB1 sequences were aligned and differences within the PB1-F2 encoding region used for primer design. Generation of a plasmid coding for the 1918 PB1-F2 within the PR8-PB1 backbone was achieved using Quickchange site directed mutagenesis (Qiagen) at nucleotides coding for the 8 predicted amino acid positions where the sequences differ, changing these predicted amino acids as follows: R33H, G40D, R60Q, N66S, I68T, L69P, F71S, and END88W (primer sequences available on request). The resulting plasmid coded for a PB1 protein identical in predicted amino acid sequence to the wt PR8 parent. This mutated plasmid and the wildtype plasmid were utilized in a 7+1 reverse genetics system to generate recombinant viruses as described (Hoffmann et al., 2002). The sequence of the PB1 gene of the parental PR8 strain generated by reverse genetics and used in these studies differs from the published sequence (NCBI NC-002021) at two positions in the PB1-F2 ORF, resulting in conservative changes R29K and K59R. These changes were retained in the PR8-PB1-F21918 virus so the viruses would be completely isogenic other than at positions within the PB1-F2 where the sequence of PR8 differs from A/Brevig Mission/1/1918. Mutation at these positions to match the NCBI published sequence did not change the phenotype of the virus in vitro or in mice (data not shown).
All viruses were passaged once through MDCK cells, stocks were grown by a single passage through eggs, and allantoic fluid stored at −80°C. The resultant viral stocks were sequenced to ensure the desired mutations and no additional inadvertent mutations were present, were characterized via TCID50 assay in MDCK cells and EID50 assay in embryonated hen’s eggs, as well as HA titer and determination of the number of plaque forming units (PFU/mL) in MDCK cells. Growth was assessed by serial HA titer on 4–8 wells every 4–8 hours for 72 hours in comparison to the wt PR8 parent. The sequences of all eight gene segments of these viruses were identical other than the mutations described above in the PB1-F2 ORF.
Bacterial strains
Streptococcus pneumoniae A66.1, a type 3 encapsulated strain, was obtained from Dr. Elaine Tuomanen at SJCRH and engineered to express luciferase (Kevin Francis and Jun Yu, Xenogen Corporation, Alameda, CA). Pneumococci were grown in Todd Hewitt broth (Difco Laboratories, Detroit, MI, USA) to an OD620 of approximately 0.4 then frozen at −80ºC mixed 2:1 with 5% sterile glycerol. The titer of the frozen stocks were quantitated on tryptic soy agar (Difco Laboratories, Detroit, MI, USA) supplemented with 3% v/v sheep erythrocytes.
Mice
Six to eight week old female Balb/c mice (Jackson Laboratory, Bar Harbor, ME) were maintained in a Biosafety Level 2 facility in the Animal Resource Center at SJCRH. All experimental procedures were approved by the Animal Care and Use Committee at SJCRH and were done under general anesthesia with inhaled isoflurane 2.5% (Baxter Healthcare Corporation, Deerfield, IL).
Infectious model
Infectious agents were diluted in sterile PBS and administered intranasally in a volume of 100 μl (50 μl per nostril) to anesthetized mice held in an upright position. Groups of 6 to 10 mice were weighed and followed at least daily for illness and mortality. Mice found to be moribund were euthanized and considered to have died on that day. In experiments comparing wt and mut PR8, influenza virus was given at a dose of 100 TCID50 per mouse followed 7 days later by pneumococcal challenge with 100 or 1000 CFU per mouse and mice were followed for 21 days. For experiments comparing wt PR8 to PR8-PB1-F21918, influenza virus was given to groups of 4 mice at a dose of 50, 100, 500, or 1000 TCID50 per mouse (to determine the dose lethal for 50% of mice (MLD50)), or to groups of 6–10 mice at a dose of 50 TCID50 per mouse either as a sole infection (collection of BALF for cell counts or lungs for viral titers, histopathology, or cytokines) or followed 7 days later by challenge with 100 CFU of pneumococcus (all assays following bacterial challenge). This lower dose of 50 TCID50 was to ensure a sub-lethal dose was being used, since some mice died after infection with 100 TCID50 of the PR8-PB1-F21918 virus alone. Survival from secondary bacterial pneumonia comparing the wt PR8 to the PR8-PB1-F21918 virus using 100 TCID50 (and excluding mice that died from viral infection alone) yielded results similar to those depicted in Fig 6A (data not shown).
Peptides
Using the predicted amino acid sequence of the PR8 PB1-F2 protein, peptides from the C-terminal (WLSLRNPILVFLKTRVLKRWRLFSKHE) and N-terminal (MGQEQDTPWILSTGHISTQK) ends were synthesized on an Apex 396 multiple organic synthesizer (Aaaptec, Louisville, KY) and suspended in PBS at a concentration of 1 mM. Mice were given peptides in a volume of 100 μl intranasally while under light isoflurane anesthesia.
Imaging of live mice
Mice were infected with a strain of pneumococcus A66.1 engineered to express luciferase (Kevin Francis and Jun Yu, Xenogen Corporation, Alameda, CA). They were then imaged for 60 seconds using an IVIS™ CCD camera (Xenogen Corporation, Alameda, CA) 24, 48, and 72 h after pneumococcal challenge. Total photon emission from selected and defined areas within the images of each mouse was quantified using the LivingImage™ software package (Xenogen Corporation, Alameda, CA) as described (Francis et al., 2001), and expressed as the flux of relative light units per minute. Pneumonia was defined as visible bioluminescence within the thorax and detection of a flux of > 20,000 relative light units (RLU) per minute. Pneumonia was designated “early” when less than 1×105 RLU/min were present on the initial measurement. Pneumonia was designated “late” when it had been present for at least 24 hours and more than 1×106 RLU/min were present on the final measurement. These definitions were developed and validated so relevant comparisons could be made between mice at similar stages of the pneumonic process, avoiding the problem of comparing mice with large bacterial loads to those with small bacterial loads that is common to many models of bacterial pneumonia (Smith et al., 2007).
Histo-pathologic examination
Lungs were removed immediately following euthanasia, inflated, and fixed in 10% neutral buffered formalin overnight. The lungs were processed routinely, embedded in paraffin, sectioned at five microns, stained with hematoxylin and eosin, and examined microscopically for histopathologic alterations. The pulmonary system was divided into the following segments, bronchi, bronchioles, interstitium and pleura. Each segment was assigned a grade 0–3 based on the histologic character of the lesions. A score of 1 was given to mild findings including minimal infiltrates of lymphocytes and plasma cells around airways and vessels, minimal epithelial hyperplasia, minimal leukocyte infiltration of alveolar spaces, and < 10% of the lung affected. A score of 2 was given for moderate findings including moderate infiltrates of lymphocytes and plasma cells around airways and vessels, moderate epithelial hyperplasia with focal necrosis, focally extensive infiltration of the alveolar spaces by leukocytes with some consolidation, focal pleuritis, and >10% but <30% of the lung affected. A score of 3 was given for more severe findings including extensive necrosis of airway epithelium and the interstitium, extensive leukocyte infiltration and consolidation, severe pleuritis, and lobar involvement. Grading and description of pathology were performed by an experienced veterinary pathologist blinded to the composition of the groups (KLB).
Broncho-alveolar lavage (BAL) for cell counts
Following euthanasia by CO2 inhalation, the trachea was exposed and cannulated with a 18 gauge plastic catheter (BD Insyte, Becton Dickinson, Sandy, UT). Lungs were lavaged thrice with 1 ml of cold, sterile PBS. The number of WBC per ml of the resulting suspension was then determined on a Hemavet 3700 (Drew Scientific, Dallas, TX) using a 100 μL aliquot. Flow cytometry (LSRII, Becton Dickinson, San Jose, California) was performed on the BALF suspension via staining (1μL/106cells) with MHCII(FITC)/CD11b(PE)/B220-CD3e(PeCy-7)/Gr1(APC)/CD11c(APC Cy-7)/SA-APC-Cy7 (Becton Dickinson, San Jose, California). Viable cells were gated (DAPI−) and the proportion of T cells (MHCII−, within lymphocyte (FSClow, B220+, CD3+) region), B cells (MHCII+, within lymphocyte region), Neutrophils (Gr1high, MHCII− within non-lymphocyte region), Macrophages (CD11c+, CD11b+ MHCII−, within the non-lymphocyte region) and Dendritic cells (CD11c+, CD11b+, MHCII+, within non-lymphocyte region) were assessed. Analysis of BALF cell composition was based upon the proportion of viable events analyzed by Flow Cytometry as related to the number of WBC/mL.
Measurement of cytokines and chemokines
Lung homogenates were centrifuged at 10,000 × g for 5 minutes and the supernatants frozen. The production of interleukins (IL)-1α, IL-6, IL-10, and chemokines KC, macrophage inflammatory (MIP)-1α and tumor necrosis factor (TNF)-α was measured in lungs by using the “mouse 18-plex” cytokine assay (Bio-Rad Laboratories, Hercules, CA) read on a Luminex 100 reader (Luminex Corp., Austin, TX) according to the manufacturer’s instructions. Samples were diluted 1:4 and run in duplicate in all assays with appropriate internal controls.
Statistical Analysis
Comparison of survival between groups of mice was done with the Log Rank chi-squared test on the Kaplan-Meier survival data. Comparison of viral or bacterial lung titers, cell counts in BALF or cytokine and chemokine levels between groups was done using analysis of variance (ANOVA). Comparison of weight loss between groups of mice was done using the Student’s t-test for pair-wise comparisons and ANOVA for multiple comparisons. A p-value of < 0.05 was considered significant for these comparisons. SigmaStat for Windows (SysStat Software, Inc., V 3.11) was utilized for all statistical analyses.
Acknowledgments
We are grateful to Dr. Peter Palese for providing viruses for use in this study. This work was supported by NIH (grants AI-66349 and AI-54802), the NIAID intramural research program, and by the American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Abrahams A, Hallows N, French H. A further investigation into influenzo-pneumococcal and influenzo-streptococcal septicaemia. Lancet. 1919:1–11. [Google Scholar]
- American Society for Microbiology. December 5th, 2005 - ASM Statement on Pandemic Influenza Plan. 2006 http://www.asm.org/Policy/index.asp?bid=39508.
- Chanturiya AN, Basanez G, Schubert U, Henklein P, Yewdell JW, Zimmerberg J. PB1-F2, an influenza A virus-encoded proapoptotic mitochondrial protein, creates variably sized pores in planar lipid membranes. J Virol. 2004;78:6304–6312. doi: 10.1128/JVI.78.12.6304-6312.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O’Neill R, Schickli J, Palese P, Henklein P, Bennink JR, Yewdell JW. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med. 2001;7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- Francis KP, Yu J, Bellinger-Kawahara C, Joh D, Hawkinson MJ, Xiao G, Purchio TF, Caparon MG, Lipsitch M, Contag PR. Visualizing pneumococcal infections in the lungs of live mice using bioluminescent Streptococcus pneumoniae transformed with a novel gram- positive lux transposon. Infect Immun. 2001;69:3350–3358. doi: 10.1128/IAI.69.5.3350-3358.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Watanabe S, Ito T, Goto H, Wells K, McGregor M, Cooley AJ, Kawaoka Y. Biological heterogeneity, including systemic replication in mice, of H5N1 influenza A virus isolates from humans in Hong Kong. J Virol. 1999;73:3184–3189. doi: 10.1128/jvi.73.4.3184-3189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs JS, Malide D, Hornung F, Bennink JR, Yewdell JW. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J Virol. 2003;77:7214–7224. doi: 10.1128/JVI.77.13.7214-7224.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Staeheli P, Hufbauer M, Koerner I, Martinez-Sobrido L, Solorzano A, Garcia-Sastre A, Haller O, Kochs G. Replication fitness determines high virulence of influenza A virus in mice carrying functional Mx1 resistance gene. Proc Natl Acad Sci U S A. 2007;104:6806–6811. doi: 10.1073/pnas.0701849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann E, Krauss S, Perez D, Webby R, Webster RG. Eight-plasmid system for rapid generation of influenza virus vaccines. Vaccine. 2002;20:3165–3170. doi: 10.1016/s0264-410x(02)00268-2. [DOI] [PubMed] [Google Scholar]
- Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O, Thomas MJ, Basler CF, Palese P, Taubenberger JK, Garcia-Sastre A, Swayne DE, Katze MG. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;443:578–581. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legge KL, Braciale TJ. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 2003;18:265–277. doi: 10.1016/s1074-7613(03)00023-2. [DOI] [PubMed] [Google Scholar]
- Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A. 2003;100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers JA. Effect of antiviral treatment on the outcome of secondary bacterial pneumonia after influenza. J Infect Dis. 2004;190:519–526. doi: 10.1086/421525. [DOI] [PubMed] [Google Scholar]
- McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev. 2006;19:571–582. doi: 10.1128/CMR.00058-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers JA, Bartmess KC. Role of neuraminidase in lethal synergism between influenza virus and Streptococcus pneumoniae. J Infect Dis. 2003;187:1000–1009. doi: 10.1086/368163. [DOI] [PubMed] [Google Scholar]
- McCullers JA, Rehg JE. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis. 2002;186:341–350. doi: 10.1086/341462. [DOI] [PubMed] [Google Scholar]
- McCullers JA, Tuomanen EI. Molecular pathogenesis of pneumococcal pneumonia. Front Biosci. 2001;6:D877–D889. doi: 10.2741/mccullers. [DOI] [PubMed] [Google Scholar]
- Morens DM, Fauci AS. The 1918 influenza pandemic: insights for the 21st century. J Infect Dis. 2007;195:1018–1028. doi: 10.1086/511989. [DOI] [PubMed] [Google Scholar]
- Mote JR. Virus and rickettsial diseases. Cambridge, Massachusetts: Harvard University Press; 1940. Human and swine influenzas; pp. 429–516. [Google Scholar]
- Muir R, Wilson GH. Influenza and its complications. Br Med J. 1919;1:3–5. doi: 10.1136/bmj.1.3027.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer JC, Denson J, Mehta PK, Su X, Mukatira S, Finkelstein DB, Xu X, Wang J, Ma J, Fan Y, Rakestraw KM, Webster RG, Hoffmann E, Krauss S, Zheng J, Zhang Z, Naeve CW. Large-Scale Sequence Analysis of Avian Influenza Isolates. Science. 2006;311:1576–1580. doi: 10.1126/science.1121586. [DOI] [PubMed] [Google Scholar]
- Peltola VT, Boyd KL, McAuley JL, Rehg JE, McCullers JA. Bacterial sinusitis and otitis media following influenza virus infection in ferrets. Infect Immun. 2006;74:2562–2567. doi: 10.1128/IAI.74.5.2562-2567.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltola VT, Murti KG, McCullers JA. Influenza virus neuraminidase contributes to secondary bacterial pneumonia. J Infect Dis. 2005;192:249–257. doi: 10.1086/430954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter CW. Chronicle of influenza pandemics. In: Nicholson KG, Webster RG, Hay AJ, editors. Textbook of Influenza. London: Blackwell Scientific Publications; 1998. pp. 3–18. [Google Scholar]
- Simonsen L. The global impact of influenza on morbidity and mortality. Vaccine. 1999;17(Suppl 1):S3–10. doi: 10.1016/s0264-410x(99)00099-7. [DOI] [PubMed] [Google Scholar]
- Simonsen L, Fukuda K, Schonberger LB, Cox NJ. The impact of influenza epidemics on hospitalizations. J Infect Dis. 2000;181:831–837. doi: 10.1086/315320. [DOI] [PubMed] [Google Scholar]
- Smith MW, Schmidt JE, Rehg JE, Orihuela C, McCullers JA. Induction of pro- and anti- inflammatory molecules in a mouse model of pneumococcal pneumonia following influenza. Comp Med. 2007;57:12–18. [PMC free article] [PubMed] [Google Scholar]
- Stone WJ, Swift GW. Influenza and influenzal pneumonia at Fort Riley, Kansas. JAMA. 1919;72:487–493. [Google Scholar]
- Taubenberger JK, Reid AH, Lourens RM, Wang R, Jin G, Fanning TG. Characterization of the 1918 influenza virus polymerase genes. Nature. 2005;437:889–893. doi: 10.1038/nature04230. [DOI] [PubMed] [Google Scholar]
- Thompson WW, Shay DK, Weintraub E, Brammer L, Bridges CB, Cox NJ, Fukuda K. Influenza-associated hospitalizations in the United States. JAMA. 2004;292:1333–1340. doi: 10.1001/jama.292.11.1333. [DOI] [PubMed] [Google Scholar]
- Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA. 2003;289:179–186. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solorzano A, Swayne DE, Cox NJ, Katz JM, Taubenberger JK, Palese P, Garcia-Sastre A. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. 2005;310:77–80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schroder JM, Wang JM, Howard OM, Oppenheim JJ. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science. 1999;286:525–528. doi: 10.1126/science.286.5439.525. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163:1–5. [PubMed] [Google Scholar]
- Zamarin D, Garcia-Sastre A, Xiao X, Wang R, Palese P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005;1:e4. doi: 10.1371/journal.ppat.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamarin D, Ortigoza MB, Palese P. Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J Virol. 2006;80:7976–7983. doi: 10.1128/JVI.00415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]