

SUMMARY
Cryopyrin (CIAS1, NLRP3) and ASC are components of the inflammasome, a multiprotein complex required for caspase-1 activation and cytokine IL-1βproduction. CIAS1 mutations underlie autoinflammation characterized by excessive IL-1β secretion. Disease-associated cryopyrin also causes a program of necrosis-like cell death in macrophages, the mechanistic details of which are unknown. We find that patient monocytes carrying disease-associated CIAS1 mutations exhibit excessive necrosis-like death by a process dependent on ASC and cathepsin B, resulting in spillage of the proinflammatory mediator HMGB1. Shigella flexneri infection also causes cryopyrin-dependent macrophage necrosis with features similar to the death caused by mutant CIAS1. This necrotic death is independent of caspase-1 and IL-1β, and thus independent of the inflammasome. Furthermore, necrosis of primary macrophages requires the presence of Shigella virulence genes. While similar proteins mediate pathogen-induced cell death in plants, this report identifies cryopyrin as an important host regulator of programmed pathogen-induced necrosis in animals, a process we term pyronecrosis.
INTRODUCTION
The CATERPILLER family (Harton et al., 2002) (CLR, also known as NLR) is comprised of proteins involved in the regulation of innate immunity (Inohara and Nunez, 2003; Martinon and Tschopp, 2005). Functionally similar to the evolutionarily conserved Toll-like receptors (TLRs), increasing evidence suggests that CLRs may serve as intracellular molecules that sense pathogen-derived products (Hoffmann and Reichhart, 2002; Poltorak et al., 1998). Significant attention has been focused one CLR family member, cryopyrin, which is encoded by the gene CIAS1. CIAS1 is mutated in a trio of dominantly inherited periodic fevers: FCAS (familial cold autoinflammatory syndrome), MWS (Muckle-Wells syndrome), and CINCA/NOMID (chronic infantile neurological cutaneous and articular syndrome/neonatal onset multisystemic auto-inflammatory disease), which are proposed to represent a continuum of severity for a single condition, CIAS1-associated periodic syndrome (CAPS) (Aksentijevich et al., 2002; Feldmann et al., 2002; Hoffman et al., 2001a, 2001b).
Recent investigations have highlighted an essential role for IL-1β in the development of mutant-CIAS1-associated periodic fevers. Mutant CIAS1/NLRP3 causes elevated levels of spontaneous and induced IL-1β both in vitro and in vivo. Indeed, FCAS, MWS, and CINCA/NOMID have all been successfully treated with daily doses of the IL-1β receptor antagonist Anakinra (Kineret) (Goldbach-Mansky et al., 2006; Hawkins et al., 2004; Hoffman et al., 2004). Cryopyrin participates in the regulation of IL-1β through involvement in a multimolecular complex called the inflammasome (Agostini et al., 2004). This complex, which also includes ASC (Apoptotic Speck protein containing a CARD) and TUCAN, promotes activation of caspase-1/ICE. In turn, caspase-1 then cleaves pro-IL-1β to produce mature IL-1β, which is released from the cell. Mutations in cryopyrin result in the hyperactivation of this pathway, causing excessive IL-1β production and the severe episodes of inflammation.
The functions of cryopyrin in the immune system are not limited to autoinflammatory disorders. Several recent reports have established cryopyrin as an important adaptor capable of organizing the inflammasome to elicit IL-1β release in response to bacterial, viral, and other proinflammatory stimuli (Kanneganti et al., 2006a, 2006b; Mariathasan et al., 2006; Martinon et al., 2006; Sutterwala et al., 2006). However, it is not yet known if the protein has additional biologic functions in the containment of pathogens.
Clues regarding an additional role for cryopyrin in response to pathogen may lie within its makeup. Cryopyrin consists of an amino-terminal pyrin domain, a central NACHT (NAIP, CIITA, HET-E, TP1) domain, and seven carboxy terminal LRRs (leucine-rich repeats). This architecture is conserved in plants, where similar proteins comprise a subfamily of disease-resistant (R) proteins called NB-LRRs (Ausubel, 2005; Chisholm et al., 2006). The NB-LRR proteins respond to microbial pathogen by eliciting a hypersensitive death response in infected cells, thus resulting in elimination of the pathogen (Greenberg et al., 1994; Nimchuk et al., 2003). Similarly, one mammalian host response to microbial pathogen is macrophage/monocyte necrotic-like death, which can lead to pathogen elimination, but also to exacerbated inflammation and sepsis (Krysko et al., 2006). The participation of cryopyrin in initiating necrosis has been hinted at previously, as cryopyrin-deficient macrophages demonstrate reduced levels of cell death in response to the Gram-positive Staphylococcus bacteria (Mariathasan et al., 2006). However, the molecular players that mediate such a process and the mechanism of this form of cell death have yet been defined.
We report here that cryopyrin and ASC are required for a process of necrotic-like cell death. We furthermore expand the capabilities of cryopyrin by demonstrating that it mediates both the IL-1β and cell death response to a Gram-negative bacterium, S. flexneri, resulting in cellular necrosis and the exacerbation of inflammation. The observation that Shigella-induced cell death is independent of caspase-1 and IL-1β indicates that this process occurs independently of inflammasome formation. It further suggests that disease-associated cryopyrin represents a hyperactive form of the protein, while the function of the normal counterpart is to induce cell death only upon stimulation with bacteria or other pathogens.
RESULTS
Expression of Disease-Associated CIAS1 Mutants Induces a Necrotic-like Cell Death
Mutations in CIAS1 are associated with the periodic fever syndromes FCAS, MWS, and CINCA/NOMID. Adenoviral constructs were transduced at a moi = 1 to promote efficient exogenous expression of wild-type CIAS1 or CIAS1 containing mutations encoding the disease-associated amino acid changes A439V or R260W (Figure S1A in the Supplemental Data available with this article online). A fourth construct encoding LacZ was designed as a negative control. Expression of the disease-associated mutants dramatically decreased cell viability in the THP-1 monocytic cell line in three separate assays: the XTT assay (Figure 1A), trypan blue (Figure S1B), and Viaprobe (Figure S1C). Staurosporine was used to induce apoptosis in all of these assays. To determine the mode of cryopyrin-induced cell death, we examined the activation of caspase-3. During apoptosis, caspase-3 undergoes activating cleavage. In turn, caspase-3 cleaves PARP and other downstream substrates. Neither caspase-3 nor PARP were cleaved in cells expressing a disease-associated mutant cryopyrin, though both were cleaved in staurosporine-treated cells (Figure 1B). Further, pretreatment of cells with the pan-caspase inhibitor (zVAD-fmk) failed to abrogate cell death (Figure 1C). These results indicate that mutant-cryopyrin-induced cell death does not require or proceed via caspase activation. DNA fragmentation, another hallmark of apoptosis, was not observed in mutant-cryopyrin-expressing cells (Figure 1D), though the positive control, staurosporine, induced DNA fragmentation in a caspase-dependent manner (Figure 1D and Figure S2A). Moreover, in contrast to apoptotic cells, mutant-cryopyrin-expressing cells did not demonstrate an increase in mitochondrial membrane permeability at two time points (summarized in Figure 1E, and shown in detail in Figure S2B). Finally, electron microscopy shows that mutant-cryopyrin-expressing cells exhibit morphological features consistent with necrosis. Cells expressing mutant cryopyrin demonstrate several of these features: (1) degradation of the plasma membrane, (2) dysmorphic/swollen mitochondria, and (3) lack of chromatin condensation (Figure 1F, middle panel). Staurosporine caused a typical apoptotic morphology (Figure 1F, right panel). Taken together, our results support previous data indicating that disease-associated variants of cryopyrin induce cell death consistent with necrosis (Fujisawa et al., 2006).
Figure 1. Disease-Associated Cryopyrin Causes Necrotic-like Cell Death.
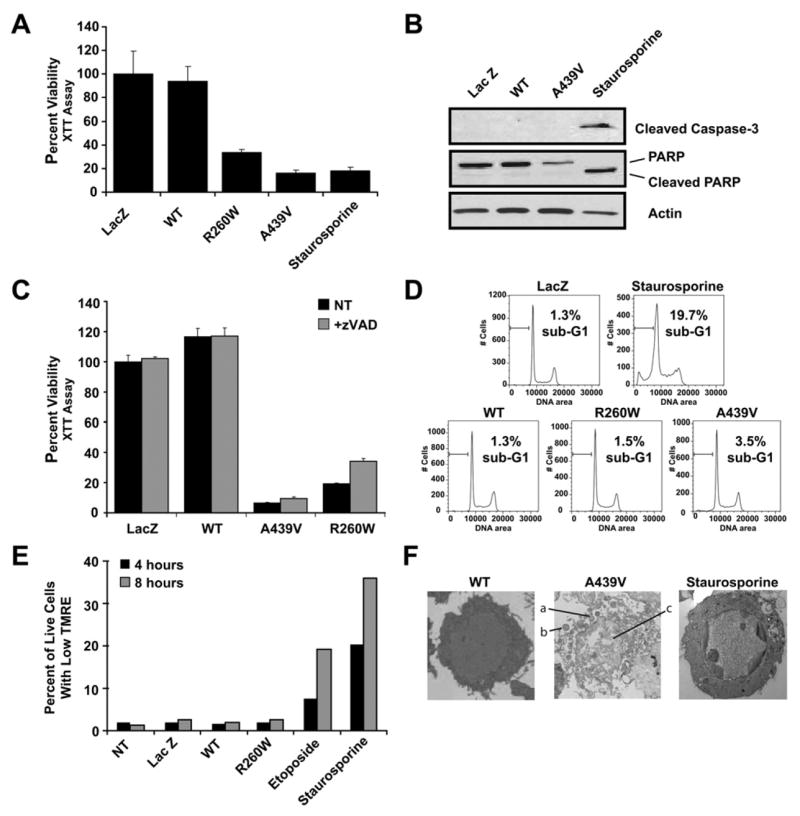
(A) Cell viability is diminished in THP-1 cells expressing disease-associated CIAS1 mutants. XTT reduction was measured 24 hr after adenoviral transduction.
(B) Mutant CIAS1-induced cell death does not cause caspase-3 or PARP cleavage. Immunoblots for caspase-3 or its substrate PARP were performed on lysates made from cells infected with the indicated adenoviral constructs or treated with staurosporine. Cleaved caspase-3 and PARP are observed in staurosporine-treated cells but not in THP-1 cells expressing wild-type or disease-associated A439V CIAS1.
(C) Incubation with 100 μM pan-caspase (zVAD-fmk) inhibitor does not substantially block cell death.
(D) THP-1 cells expressing the R260W or A439V disease-associated mutant die without exhibiting DNA fragmentation. DNA content was measured by PI staining followed by flow cytometry. The percentage of cells with sub-G1 content, indicating the DNA fragmentation characteristic of apoptosis, is shown.
(E) Disease-associated cryopyrin expression does not promote the loss of mitochondrial membrane potential. THP-1 cells were treated as indicated. Mitochondrial membrane potential was measured with the potential-sensitive dye TMRE. Data summarized here are shown in Figure S2B.
(F) Representative EM images of wild-type cryopyrin (left), A439V-transduced (middle), and staurosporine-treated (right) THP-1 cells. A439V-transduced cells demonstrate necrotic features, including (1) degradation of the plasma membrane, (2) dysmorphic/swollen mitochondria, and (3) the lack of chromatin condensation. Staurosporine-treated cells exhibit a typical apoptotic morphology.
All values are the mean of three independent experiments. Error bars indicate standard deviation of the mean.
Disease-Associated Cryopyrin Mutants Induce Enhanced IL-1β Release, but Cell Death Is Independent of Caspase-1 and IL-1β Signaling
CIAS1-associated periodic fevers are characterized by excessive IL-1β production. To explore the mechanism by which disease-associated cryopyrin causes cell death, we first determined if this process is dependent on caspase-1 or IL-1β. In agreement with previous observations, substantially more IL-1β was released from cells expressing mutant cryopyrin than cells expressing wild-type cryopyrin (Figure 2A) (Agostini et al., 2004; Dowds et al., 2004). IL-18 is also regulated by caspase-1, and levels of IL-18 are greatly induced by disease-associated cryopyrin (Figure 2B). Treatment with YVAD-CHO, a specific peptide inhibitor of caspase-1, abrogated mutant-cryopyrin-induced release of IL-1β and IL-18 (Figures 2A and 2B.). However, while YVAD-CHO successfully blocked IL-1β and IL18, mutant-cryopyrin-induced cell death was unaffected (Figure 2C). Kineret (Anakinra), an IL-1 receptor antagonist (IL-1Ra) also failed to diminish mutant-cryopyrin-induced cell death (Figure 2D). To assure that the concentration of Kineret was adequate to block the biologic function of IL-1β, we measured its effect on IL-1β-mediated induction of IL-8 (Figure 2E). Even at a concentration a log lower than that used in the cell viability assay (Figure 2D), the induction of IL-8 by IL-1β was abolished by Kineret. Together, these results demonstrate that mutant-cryopyrin-induced cell death occurs independently of caspase-1 activity and IL-1β-mediated signaling. As previously reported, inhibition of cathepsin B with 50 μM Ca-074-Me substantially blocked cell death caused by disease-associated cryopyrin but had no effect on staurosporine-induced death (Fujisawa et al., 2006). Of note, Figure 1C shows that the pan-caspase inhibitor caused a slight reversal of disease variant cryopyrin-mediated cell death. This slight improvement in viability may be attributed to cross inhibition of cathepsin B by the zVAD peptide (Schotte et al., 1999).
Figure 2. Disease-Associated CIAS1 Induces IL-1β Release, but Cell Death Is IL-1β Independent.
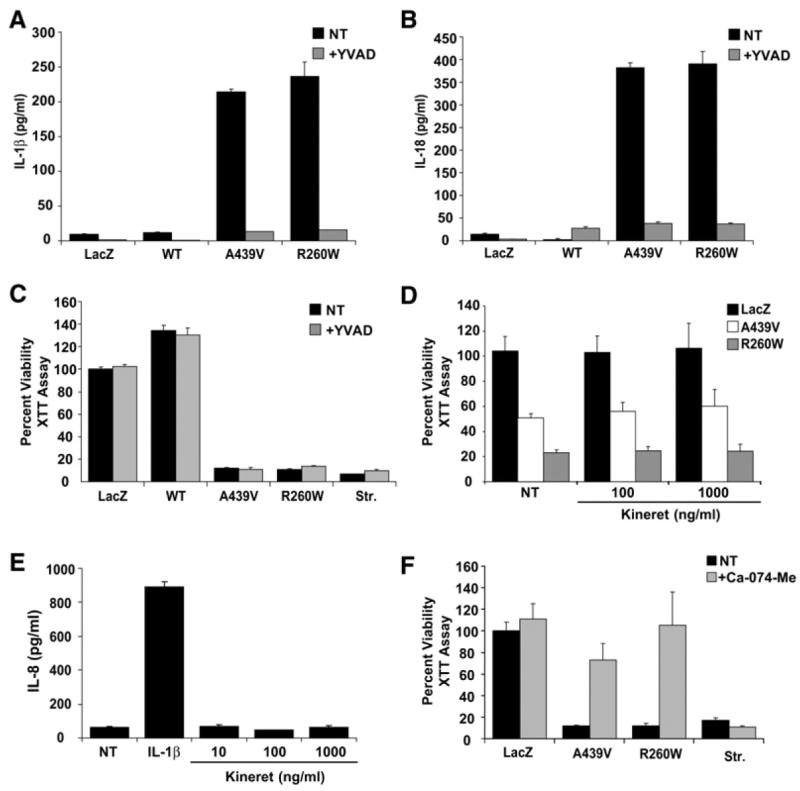
(A) IL-1β is released from THP-1 cells infected with two mutant forms of CIAS1 as measured by ELISA. IL-1β release is abrogated with 100 μM YVAD.
(B) IL-18 is released from THP-1 cells infected with two mutant forms of CIAS1 as measured by ELISA. IL-18 release is abrogated with 100 μM YVAD.
(C) Cell death induced by CIAS1 mutants is not inhibited by 100 μM YVAD. Viability was measured by XTT reduction 24 hr posttransduction.
(D) Kineret, the IL-1 receptor antagonist, does not prevent cryopyrin-induced cell death. THP-1 cells were infected with the indicated adenovirus for 24 hr in the presence or absence of Kineret. NT, not treated with Kineret.
(E) IL-8 induction in THP-1 cells by recombinant IL-1β is inhibited by Kineret. IL-1β induced a significant level of IL-8 production; this biologic effect of IL-1β was completely abrogated by Kineret.
(F) Cell death induced by cryopyrin mutants is blocked by a cathepsin B inhibitor, Ca-074-Me. THP-1 cells were infected with the indicated adenovirus for 24 hr in the presence or absence of Ca-074-Me. Viability was measured by XTT reduction after 24 hr.
All values are the mean of three independent experiments. Error bars indicate standard deviation of the mean.
Disease-Associated Mutant-Cryopyrin-Induced Cell Death Is ASC Dependent
To further explore the mechanism by which disease-associated CIAS1 causes cell death, the role of ASC was examined. Short hairpin RNA molecules (shRNAs) were designed to promote the degradation of ASC mRNA (shASC). A control shRNA with a mutated target ASC sequence was also prepared (shCntrl). These shRNAs were incorporated into retrovirus and stably transduced into THP-1 cells, resulting in stable reduction of both ASC protein and mRNA (Figures 3A and 3B). A second ASC shRNA generated the same results (data not shown). Consistent with a role for ASC in the inflammasome, shASC diminished spontaneous production of IL-1β induced by wild-type and mutant cryopyrin (Figure 3C). More importantly, shASC reverted cell death induced by the A439V disease-associated cryopyrin mutant (Figure 3D). These results demonstrate that ASC is required for mutant-cryopyrin-induced cell death as well as IL-1β production in monocytic/macrophage cell types.
Figure 3. Mutant-CIAS1-Induced THP-1 Cell Death Is ASC Dependent.
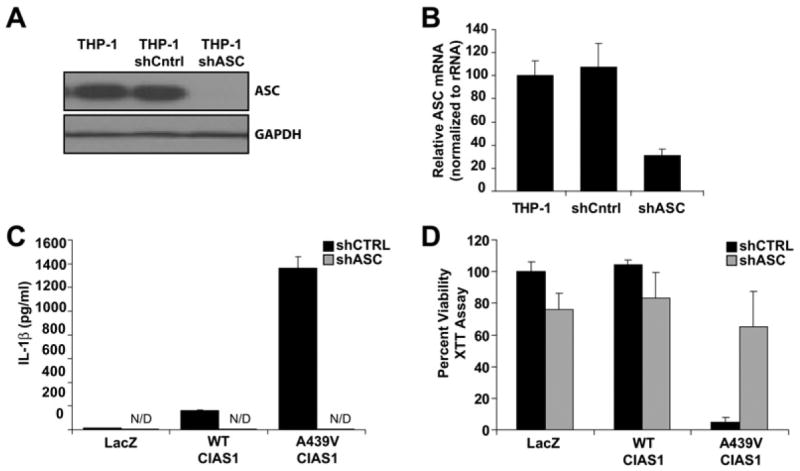
(A and B) Expression of ASC is markedly decreased in cells stably transduced with shRNA designed to promote the degradation of ASC mRNA (shASC). Immunoblot analysis and real-time PCR of the indicated stable cell lines verify a decrease of ASC protein (A) and mRNA (B).
(C) IL-1β release following infection with CIAS1-containing adenovirus is abrogated in cells with shASC. IL-1β was determined by ELISA. Values < 10 pg/ml are considered not detectable (“N/D”).
(D) Mutant CIAS1-induced cell death is abrogated in cells with decreased expression of ASC caused by shASC. XTT reduction was measured 24 hr after adenoviral infection.
All values are the mean of three independent experiments. Error bars indicate standard deviation of the mean.
Disease-Associated Cryopyrin Mutants Induce HMGB1 Release
HMGB1 is emerging as an important therapeutic target for sepsis, cancer, and other conditions. Normally maintained as a nuclear factor within the healthy cell, HMGB1 takes on the role of a strong proinflammatory factor when released from cells undergoing necrosis (Scaffidi et al., 2002). This prompted us to examine the release of HMGB1 in the presence of disease-associated cryopyrin. As measured by western analysis, HMGB1 release from cells transduced with the wild-type CIAS1-adenovirus is barely detectable, but a high level is released by cells expressing either of two disease-associated forms of CIAS1 (Figure 4A). Though ASC is essential for HMGB1 release (Figure 4B), caspase-1 activity is not (Figure 4C). HMGB1 release following the induction of apoptosis with staurosporine is not observed at the 6 hr time point but is only observed 24 hr posttreatment. This is likely the consequence of secondary necrosis caused by longer treatment time (Figure S2C). Collectively, the results presented in Figures 3 and 4 suggest that mutant cryopyrin causes necrotic-like cell death and subsequent HMGB1 release in an ASC-dependent but caspase-1-independent fashion.
Figure 4. HMGB1 Is Released from THP-1 Cells Following Expression of Disease-Associated CIAS1 Mutants.
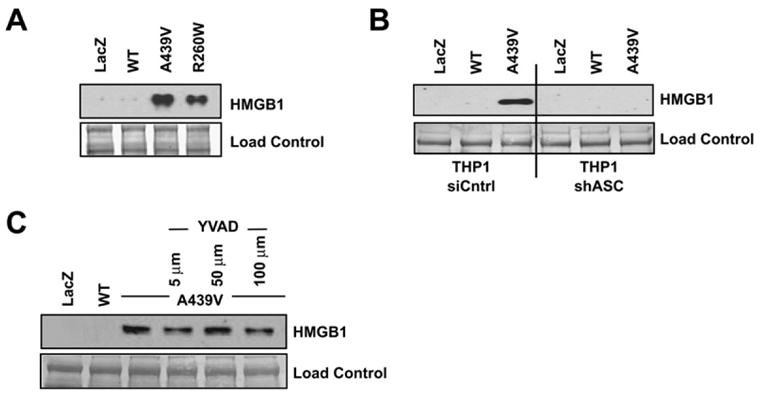
(A) Two disease-associated CIAS1 mutants induce substantially more HMGB1 release than the wild-type gene.
(B) HMGB1 release is abrogated in cells with shASC, which reduced ASC expression.
(C) Inhibition of caspase-1 with YVAD-CHO does not substantially affect HMGB1 release. Nitrocellulose membranes stained with amido black are provided as loading controls.
LPS Induces Death of FCAS Patient Cells
We next sought to determine the effects of CIAS1 mutation on cell viability in peripheral blood mononuclear cells (PBMCs) from FCAS patients with confirmed CIAS1 mutations. Samples were obtained from patients with CIAS1 mutations who have not undergone anti-inflammatory treatment. The endotoxin lipopolysaccharide (LPS) has been used by others to induce monocytic cell death (Karahashi and Amano, 1998). LPS is also known to induce CIAS1 mRNA and protein expression, both of which are very low in resting mononuclear cells (O’Connor et al., 2003). LPS challenge resulted in a dose-dependent decrease in cell viability in patient PBMCs but not healthy controls, supporting the conclusions made from exogenous expression of disease-associated cryopyrin (Figure 5A). As disease-associated cryopyrin variants are generally accepted as gain-of-function mutants, properties observed with disease-associated cryopyrin are expected to be observed with wild-type cryopyrin, either at a reduced level or under stimulated conditions. While adenovirus with disease-associated CIAS1 caused substantial cell death at moi = 1, transduction of adenovirus containing wild-type cryopyrin into THP-1 cells also resulted in cell death when a higher moi was used (Figure 5B). These findings prompted us to determine whether wild-type cryopyrin plays a role in cell necrosis associated with bacterial pathogenesis.
Figure 5. Cell Death in Cells Isolated from FCAS Patients and in Cells Expressing Wild-Type CIAS1.
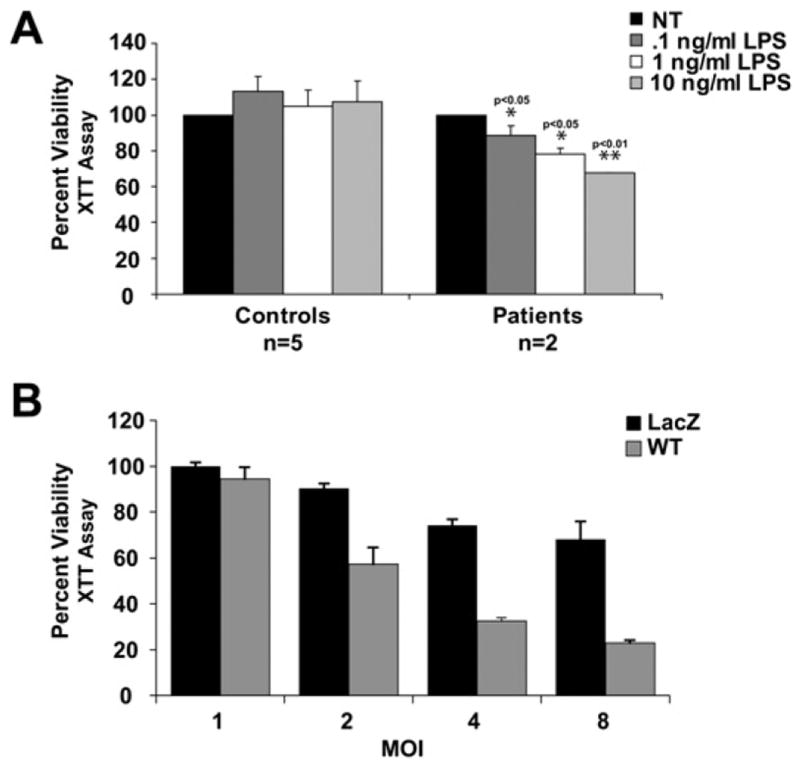
(A) Cell viability is decreased in PBMCs from FCAS patients in response to LPS. XTT reduction was used to assay cell viability and was measured 72 hr after stimulation. *p < 0.05, **p < 0.01 when compared to controls.
(B) Cell viability is diminished in THP-1 cells expressing wild-type CIAS1 at higher multiplicities of infection. XTT reduction was measured 48 hr after adenoviral infection.
All values are the mean of three independent experiments. Error bars indicate standard deviation of the mean.
Shigella flexneri-Induced Cell Death Requires Cryopyrin and ASC, but Not Caspase-1
Necrosis of monocytes and macrophages is a documented response to pathogenic bacteria, although proteins that control this process are not well defined (Golstein and Kroemer, 2007; Zong and Thompson, 2006). Significant evidence indicates that Shigella causes a necrotic-like cell death, although apoptosis has also been reported (Koterski et al., 2005; Nonaka et al., 2003; Suzuki et al., 2005; Zychlinsky and Sansonetti, 1997). This led us to examine the potential of wild-type cryopyrin to mediate necrosis in response to Shigella flexneri. Stable reduction of cryopyrin protein in THP-1 cells was achieved utilizing retroviral-transduced shRNAs specific for CIAS1, which caused a near-ablation of targeted gene expression (Figure 6A). The induction of IL-1β by S. flexneri was nearly abolished in cryopyrin-deficient cells as measured by ELISA, providing a biologic assay to assure that the CIAS1 shRNA caused the intended biologic effect (Figure 6B). More importantly, S. flexneri-induced death was substantially abrogated in shCIAS1 cells (Figure 6C). The presence of shCIAS1 did not affect staurosporine-induced cell death, indicating specificity of cryopyrin for S. flexneri-induced death. To confirm that cryopryin-mediated cell death requires ASC, we tested the ability of Shigella to elicit cell death and IL-1β in ASC-deficient THP1 cells. As expected, both cell death and IL-1β release were substantially abrogated in the shASC cells (Figures 6D and 6E).
Figure 6. Shigella flexneri-Induced Cell Death and IL-1β Require CIAS1 and ASC but Not Caspase-1.
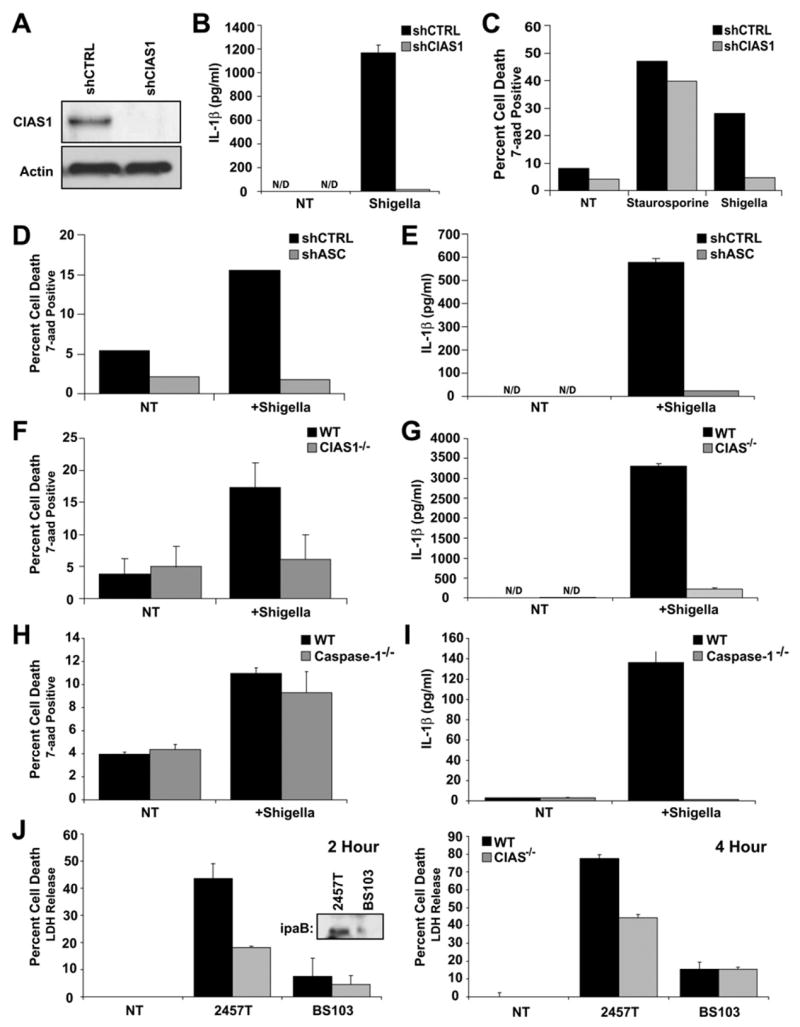
(A) Cryopyrin protein expression is decreased in THP-1 cells transduced with cryopyrin-specific shRNA (shCIAS1).
(B) S. flexneri-induced IL-1β release is diminished in CIAS1-deficient THP1 cells.
(C) THP-1 cells with shCIAS1 resist S. flexneri-induced death but not staurosporine-induced death.
(D and E) ASC is required for S. flexneri-induced cell death (D) and IL-1β release (E) in THP-1 cells.
(F) CIAS1−/− bone marrow-derived macrophages exhibit decreased levels of cell death in response to S. flexneri.
(G) S. flexneri-induced IL-1β release from thioglycolate-elicited peritoneal macrophages is reduced in CIAS1−/− macrophages.
(H) S. flexneri does not require Caspase-1 to initiate cell death.
(I) S. flexneri requires caspase-1 to induce IL-1β production in bone marrow-derived macrophages.
(J) Cryopyrin initiates cell death in response to virulent Shigella. Bone marrow-derived macrophages were infected with either 2457T (virulent) or BS103 (avirulent) S. flexneri at a moi of 50 for 2 or 4 hr. Absence of virulence plasmid in BS103 was verified by ipaB immunoblot (inset). In all cases, cell death was measured by 7-aad uptake or LDH release and IL-1β determined by ELISA.
All values are the mean of three independent experiments. Error bars indicate standard deviation of the mean.
To examine the physiologic importance of these results, we utilized macrophages isolated from wild-type and CIAS1 gene-ablated mice. In wild-type bone marrow-derived macrophages, Shigella caused a 2.5- to 3-fold increase in cell death compared to uninfected macrophages. However, in CIAS1−/− macrophages, Shigella failed to initiate cell death above the level of uninfected cells (Figure 6F). Moreover, the level of IL-1β secretion was reduced approximately 15-fold in peritoneal macrophages from CIAS1−/− mice (Figure 6G). The requirement of CIAS1 for both cell death and IL-1β release in response to S. flexneri establishes wild-type cryopyrin as a critical host adaptor capable of responding to a Gram-negative bacterial pathogen. In contrast, bone marrow-derived macrophages isolated from mice lacking caspase-1 demonstrated no differential cell death response to S. flexneri (Figure 6H), though caspase-1 remained essential for IL-1β activation (Figure 6I). These results indicate that cryopyrin and ASC, but not caspase-1, are required for S. flexneri-induced macrophage necrosis.
Cryopyrin-dependent cell death is not observed in cells infected with another intracellular bacteria, Salmonella typhi, thus indicating that the role of cryopyrin cannot be generalized to all intracellular bacteria (Figure S3) (Mariathasan et al., 2006). To assess if cryopyrin-induced cell death is caused by virulence factors expressed by S. flexneri, we compared avirulent, plasmid-cured S. flexneri (BS103) and the virulent parental strain (2457T). The former lacks a 230 kb virulence plasmid that encodes the invasion plasmid antigens IpaB, IpaC, and IpaD. These antigens are essential for S. flexneri virulence and entrance into the host cell (Menard et al., 1993). Immunoblot analysis was used to confirm the plasmid-cured Shigella strain lacks the IpaB protein, while this protein is detected in the parental strain (Figure 6J, left panel, inset). Cell death was assayed at two (2 and 4 hr) different time points. At both time points, virulent Shigella (2457T) caused cell death, and this process was reduced in macrophages lacking the CIAS1 gene (Figure 6J, right and left panels). As expected, when macrophages were infected with the plasmid-cured BS103 strain, cell death is reduced by >80%. This residual level of cell death was not affected by the absence of CIAS1. These results indicate that virulence factors expressed by S. flexneri cause CIAS1-dependent cell death.
Cryopyrin Mediates Shigella-Induced Necrotic-like Cell Death with Properties Similar to Mutant-Cryopyrin-Induced Death
Having established that cryopyrin was essential for Shigella-mediated cell death, we sought to determine the nature of cell death and whether it was consistent with the necrotic-like death induced by mutant cryopyrin. Infection of shCTRL THP-1 cells with S. flexneri for 6 hr resulted in cell death with features morphologically consistent with necrosis (Figure 7A, compare 7Ai and 7Aii). To verify that the necrotic cells contain bacteria, we performed electron microscopy at an earlier time point (2 hr after infection) when the cell morphology is less fragmented. Cells with a negative control shRNA that are infected with bacteria demonstrate the initial loss of cytoplasm, indicating necrosis (Figure 7Aiii). In contrast, cellular morphology consistent with cell death was not observed in shCIAS1 cells, despite the presence of several intracellular Shigella bacteria (Figure 7Aiv). Next, we examined biochemical properties of cryopyrin-initiated IL-1β and cell death response to Shigella. In contrast to the apoptotic control staurosporine, Shigella-induced cell death did not result in the cleavage of PARP after 6 hr (Figure 7B). Earlier reports have suggested that S. flexneri induces apoptosis in macrophages at early time points, while necrosis is observed at later points (Zychlinsky et al., 1992; Koterski et al., 2005). We observed that treatment with the pan-caspase inhibitor zVAD slightly diminished cell death after 2 hr of infection, but not after 6 hr of infection. The caspase-1-specific inhibitor YVAD had no effect. Attempts to inhibit cell death and IL-1β release with glycine pretreatment, previously shown to reduce Shigella-induced apoptosis, were also ineffective (Figure S4) (Edgeworth et al., 2002). However, the cathep-sin B inhibitor Ca-074-Me substantially blocked cell death, further validating that mutant cryopyrin and Shigella-induced necrosis occurred via the same pathway (Figure 7C). Although the caspase-1 inhibitor YVAD failed to block cell death, it substantially abrogated both IL-1β and IL-18 in response to Shigella in shCTRL cells, indicating that cell death is predominantly caspase-1, IL-1β, and IL-18 independent (Figures 7D and 7E). As expected, shCIAS1 nearly abolished both IL-1β and IL-18. Finally, Shigella-mediated cell death was associated with the release of HMGB1 (Figure 7F). In contrast, treatment with apoptosis-inducing staurosporine for 2 and 6 hr did not cause HMGB1 release.
Figure 7. Shigella flexneri Induces Cryopyrin-Dependent Necrosis.
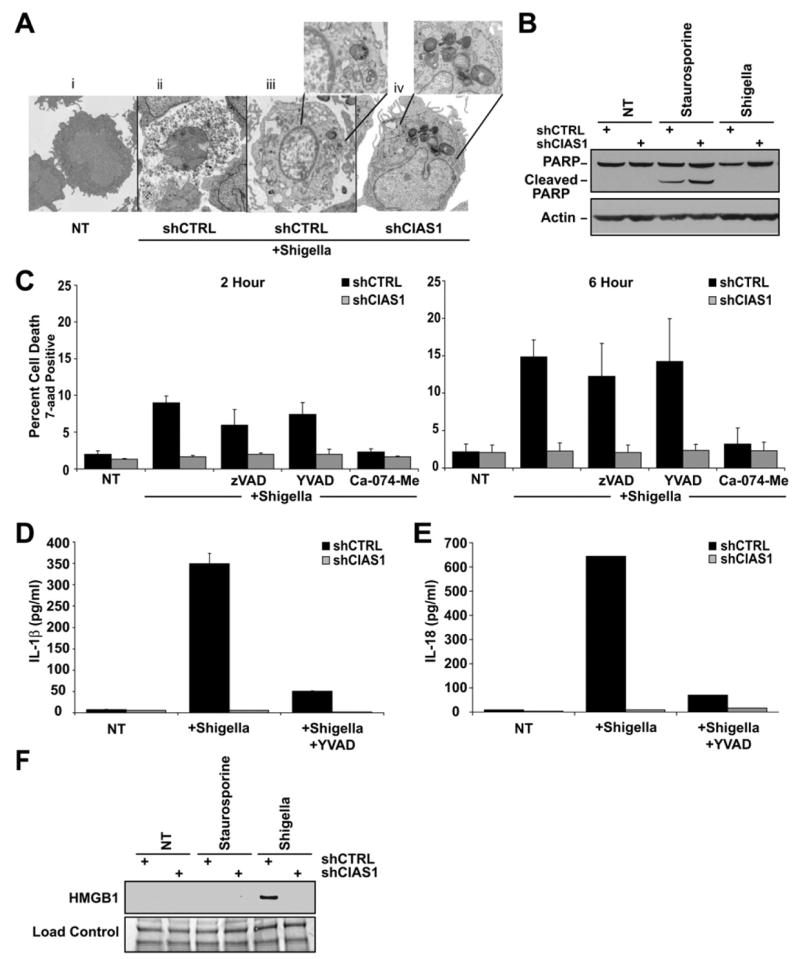
(A) Infection with S. flexneri for 6 hr induced cell death that is morphologically consistent with necrosis (see Ai and Aii). To detect intracellular bacteria, a shorter infection time (2 hr) was used so that the cells are just entering the initial phase of cell death. Cells with shCTRL, but not shCIAS1, exhibited a lost of cytoplasmic content as determined by EM imaging. Insets show the presence of bacteria (opaque round or oblong structures).
(B) PARP is not cleaved following S. flexneri infection.
(C) Cathepsin B inhibitor (50 μM) (Ca-074-Me) substantially abrogates S. flexneri cell death. In contrast, 100 μM pan-caspase (zVAD-fmk) and 100 μM caspase-1 specific (YVAD-CHO) inhibitors fail to block S. flexneri-induced cell death in shCTRL and shCIAS1 cells at 6 hr.
(D and E) S. flexneri-induced IL-1β (D) and IL-18 (E) release is reduced in shCIAS1 THP-1 cells and in cells treated with 100 μM YVAD-CHO.
(F) S. flexneri-induced HMGB1 release is abrogated by shCIAS1, and thus is cryopyrin dependent. In all cases, cell death was measured by 7-aad uptake. IL-18 and IL-1β release were determined by ELISA.
All values are the mean of three independent experiments. Error bars indicate standard deviation of the mean.
Thus, S. flexneri-induced death shared multiple characteristics associated with the necrotic-like cell death observed with disease-associated cryopyrin, suggesting that this process is mediated by cryopyrin and ASC and proceeds through cathepsin B independent of either caspase-1 or IL-1β, resulting in HMGB1 release.
DISCUSSION
We report a necrotic-like cell death caused by disease-associated mutants of CIAS1, which is dependent on ASC but not on caspase-1 or IL-1β. This process results in release of the proinflammatory mediator HMGB1, which likely propagates the inflammatory response. Additionally, we show that ASC and native, wild-type cryopyrin are required for S. flexneri-induced cell death, which proceeds in a manner identical to that induced by disease-associated cryopyrin. These results implicate cryopyrin as a crucial regulator of pathogen-induced necrotic-like death, a process proposed to play an important role in the pathogenesis of a myriad of infectious diseases, which we propose to call pyronecrosis.
From the perspective of cryopyrin-associated periodic fever, these results and the accumulated data in the literature suggest that cryopyrin mutants directly regulate disease progression through at least two distinct signaling pathways. It is known that cryopyrin participates in the activation of caspase-1, the maturation of IL-1β, and subsequent hyperactivation of the inflammatory process via an ASC-dependent process (Agostini et al., 2004). Previous reports have indicated that disease-associated mutants demonstrate a gain-of-function phenotype with respect to these properties leading to enhanced IL-1β release (Dowds et al., 2004). We report a second gain-of-function, the induction of necrotic-like cell death. The manner of death is particularly important. While it is generally believed that apoptotic cells die an orderly death with minimal impact on inflammation, necrosis involves the spilling of cellular contents into the environment, intensifying local inflammation and damaging neighboring cells (Krysko et al., 2006). It is important to keep in mind that the IL-1 receptor antagonist Anakinra has been successfully used to treat patients suffering from CAPS, indicating that excessive IL-1β production underlies disease. This would seem to suggest that the inflammasome function of cryopyrin is solely responsible for periodic fever in these patients. However, given that necrosis allows for the release of proinflammatory factors such as HMGB1, which in turn promote further release of IL-1β, it is likely that cryopyrin-mediated cell death also contributes to disease state in patients.
Both disease-associated cryopyrin expression and S. flexneri infection triggered the release of HMGB1, a chromatin-associated protein released by necrotic cells. Once HMGB1 is released, it acts as a potent danger indicator, inducing several proinflammatory cytokines by signaling through the RAGE, TLR2, and TLR4 receptors to elicit a severe inflammatory response (Andersson et al., 2000; Hori et al., 1995; Park et al., 2004). Serum HMGB1 is increased both during endotoxin exposure in mice and in septic patients who succumbed to infection (Wang et al., 1999). HMGB1 neutralization has been shown to significantly reduce inflammation and improve survival in animal models of established sepsis (Yang et al., 2004). The release of HMGB1 elicited by bacterial-induced cell death further supports the use of HMGB1 antagonists to reduce inflammation during sepsis.
The results shown here demonstrate a requirement for ASC in cryopyrin-induced cell death. ASC was initially identified as a cytosolic protein aggregated into specks in myeloid cells undergoing apoptosis and has since been implicated in several cell death pathways in nonmyeloid cells (Masumoto et al., 1999). Functionally ASC is a bipartite adaptor protein comprised of N-terminal pyrin domain (PD) and C-terminal CARD. It is proposed that these domains each engage in homotypic interactions, thereby linking the PD of cryopyrin to the CARD region of caspases. Constitutive interactions between several transfected disease-associated cryopyrin mutants and ASC have been reported when overexpressed in a HEK293T cells (Dowds et al., 2004). Overexpressed ASC has also been shown to interact with caspases-8 and-9, and several studies have implicated ASC in the progression of apoptosis. The concurrent overexpression of a related protein, Ipaf, with ASC in HEK293T cells results in caspase-8-dependent apoptosis, while overexpression of ASC alone induces caspase-9-mediated cell death in HEK293 cells (Masumoto et al., 2003; McConnell and Vertino, 2000). These observations suggest the involvement of apoptotic caspases in ASC-induced cell death in nonmonocytic cells (Dowds et al., 2004; Masumoto et al., 2003; Wang et al., 2004). However, coimmunoprecipitation experiments in monocytic THP-1 cells indicate that ASC/caspase interaction is limited to caspase-1, suggesting that ASC is not involved in the initiation of apoptotic caspases in these cells (Stehlik et al., 2003). Our results in a monocytic cell type demonstrate that ASC is important in a necrotic-like cell death pathway that is caspase independent.
The results here delineate an endogenous program of necrosis initiated by cryopyrin and ASC. This pathway proceeds through cathepsin B, yet occurs independent of caspase-1, IL-1β, and the inflammasome. Recently, ASC has also been implicated in the initiation of another rapid form of inflammatory cell death called pyroptosis. In contrast to the necrosis initiated by cryopyrin and ASC, pyroptosis requires caspase-1 activation by ASC following the dimerization of ASC into speck-like pyroptosomes (Fernandes-Alnemri et al., 2007). Pyroptosomes per se do not contain detectable cryopyrin; however, the role of cryopyrin in the formation of this structure was not tested. This form of cell death has features of both apoptosis and oncosis. Unlike the cryopyrin/ASC necrosis pathway described here, pyroptosis can be blocked by exogenous caspase inhibitors; however, the requirement for cathepsin B has not been investigated (Fernandes-Alnemri et al., 2007). The involvement of ASC in the induction of both pyronecrosis and pyroptosis establish ASC as a key component in the host defense arsenal to invading pathogens.
Several recent reports establish cryopyrin as a pivotal regulator of IL-1 and IL-18 release to both bacterial and viral challenges. Utilizing macrophages isolated from CIAS1−/− mice, components of the immune response to Gram-positive bacteria such as Staphylococcus aureus and Listeria monocytogenes, as well as Sendai and influenza viruses, have been shown to require cryopyrin (Kanneganti et al., 2006a; Mariathasan et al., 2006). However, infection with Gram-negative Salmonella typhimurium or Francisella tularensis elicits caspase-1 activation and IL-1β release in a cryopyrin-independent fashion (Mariathasan et al., 2005; Sutterwala et al., 2006). Macrophage cell death induced by Salmonella and Francisella are similarly unaffected by CIAS1 deficiency (Mariathasan et al., 2005). Alternate proteins mediate the response to these pathogens. Ipaf, which participates in its own inflammasome, is responsible for initiating inflammation in response to Salmonella typhimurium (Mariathasan et al., 2004). It has been suggested that cryopyrin does not mediate the recognition of Gram-negative bacteria. However, our results indicate that cryopyrin mediates both IL-1β release and cell death in response to the Gram-negative bacteria S. flexneri in THP-1 cells and mouse macrophages (Figure 7). Thus, cryopyrin mediates IL-1β processing and secretion in response to specific Gram-positive and -negative bacteria.
Despite substantial progress made in understanding pathogen-induced host cell death, the mechanisms governing Shigella-induced host cell death have not been conclusively delineated (Haimovich and Venkatesan, 2006). This study illuminates a necrotic host cell-death pathway detonated by Shigella requiring both cryopyrin and ASC. While the strain M90T (Serogroup 5) has been reported to induce both necrosis and caspase-1-dependent apoptosis (Francois et al., 2000; Raqib et al., 2002; Zychlinsky et al., 1992), many others have observed caspase-1-independent necrosis initiated by strains 2457T (Serogroup 2A), YSH6000 (Serotype 2A), and now 12022 (Serogroup 2B) (Fernandez-Prada et al., 1997, 2000; Koterski et al., 2005; Nonaka et al., 2003). These differences have been attributed to differences in species and infected cell types, Shigella strains, duration and multiplicity of infection, and conclusions regarding the nature of cell death drawn from incomplete methodology (Nonaka et al., 2003). The results presented here verify and define cryopyrin and ASC as key components of a caspase-1-independent mechanism by which Shigella induces necrosis and should do much to advance the understanding of Shigella pathogenesis.
Further resolved is the putative involvement of caspase-1 in mediating Shigella-induced cell death. Previous reports have suggested that the virulence factor IpaB is secreted by Shigella via a type III secretion system upon contact with host cells and subsequently binds directly to caspase-1 to induce cell death (Blocker et al., 1999; Chen et al., 1996; Menard et al., 1994). Our results clearly demonstrate a caspase-1-independent pathway, as both caspase-1-specific inhibitors and macrophages lacking caspase-1 were equally susceptible to Shigella-induced cell death as wild-type controls, a result consistent with other reports (Suzuki et al., 2005). It should be noted that Suzuki et al. observed an early apoptotic event requiring caspase-1, and IpaB preceded a later necrotic event that occurs independent of caspase-1 or IpaB, and thus both outcomes are possible. While we show here that cryopyrin and ASC are required for Shigella-initiated necrosis, the activation of NF-κB and JNK in epithelial cell line following Shigella infection has been reported to require the overexpression of NOD1/CARD4 (Girardin et al., 2001). Although verification of this finding using primary epithelial cells from mice lacking NOD1/CARD4 has not been performed to confirm the physiologic relevance of this data, taken together, these data suggest that several CLR proteins might mediate different aspects of the immune response to Shigella in a cell-type specific fashion.
In summary, we report that disease-associated cryopyrin mediates a form of necrotic-like cell death that is replicated when normal monocytes encounter the bacterial pathogen S. flexneri. These findings suggest that the gain-of-function mutant cryopyrin in patients might propagate an inflammatory response without the normal stimulation caused by pathogens. These findings also show a parallel with pathogen-induced, NB-LRR-mediated cell death found in plants. A long history of elegant studies in plants has shown that cell death is a major mechanism by which plant R proteins mediate host response to multiple microbial pathogens (Belkhadir et al., 2004). It will be important to determine if other NBD-LRR proteins also cause the induction of cell death in myeloid and nonmyeloid cells, and if these proteins mediate both apoptotic and necrotic death in a pathogen-specific fashion.
EXPERIMENTAL PROCEDURES
Cell Lines and Reagents
THP-1 cells were purchased from American Type Culture Collection (ATCC) and cultured as described previously (Williams et al., 2005). Anti-caspase-3 antibody was purchased from Cell Signaling; anti-PARP, anti-ipaB, anti-Actin, and HRP-conjugated secondary antibodies from Santa Cruz Biotechnology; anti-HMGB1 antibody from Abcam; anti-ASC antibody from Immuno Diagnostic Oy; anti-Cryopyrin antibody from Alexis Biochemicals; Super Signal ECL reagent from Bio-Rad; E. coli LPS from Chemicon. Detailed methods for preparation of retroviral shuttle vectors, transduction, and sorting to generate THP-1 cell lines stably expressing shRNA have been described (Taxman et al., 2006). The shRNA target sequences are as follows: shASC-GCTCTTCAGTTTCACACCA, shCtrl-GCTCTTCctggcCACAC CA, shCIAS-GGATGAACCTGTTCCAAAA. Stable expression of shRNA did not induce interferon response as assessed by OAS1 expression (data not shown).
Generation of Recombinant Adenoviruses
Recombinant adenovirus expressing CIAS1 or LacZ was generated using Adeno-X Expression System (Clontech). Briefly, genes were subcloned into pShuttle2 intermediate vector and ligated into the modified type 5 human adenoviral genome vector Adeno-X following excision with PI-Sce and I-Ceu enzymes. Recombinant adenovirus was then amplified in HEK293 cells and purified using Adeno-X Virus Purification Kit (Clontech). Viral titers were determined by UNC Viral Vector Core Facility (UNC-Chapel Hill).
Adenovirus Transduction of THP-1 Cells
THP-1 cells were aliquoted into Falcon 2059 polypropylene tubes at a density of 106/ml in RPMI 1640 containing 10% FBS. After addition of adenovirus (moi = 1 unless otherwise noted), cells were centrifuged at 2000 × g for 2 hr at 37°C. Immediately after centrifugation, cells were resuspended and incubated at 5 × 105/ml following the addition of fresh RPMI 1640 containing 10% FBS.
XTT Assay
Cells were plated into 96-well plates at 20,000 cells per well 24 hr after adenovirus transduction. Fifty microliters of serum-free media containing 25 μM phenazine methosulfate and 1 mg/ml XTT was added to each well. Plates were read at 450 nM after 4 hr of incubation.
Mitochondrial Membrane Potential Staining
Cells were stained with tetramethylrhodamine ethyl ester, perchlorate (TMRE) for 25 min at 37°C at a final concentration of 5 nM. After staining, cells were rinsed in PBS, resuspended in 0.5 ml PBS, and analyzed using a FACScan (Becton Dickinson) in FL2.
Immunoblotting
Immunoblots were performed as described previously (Williams et al., 2005). Cryopyrin was immunoprecipitated with rabbit anti-CIAS-1 peptide IgG. Expression was confirmed by probing immunoblots with anti-CIAS1. HMGB1 blots were performed directly on culture supernatants and developed as indicated.
Quantitative PCR
Total RNA was isolated, cDNA was reversed transcribed, and quantitative PCR was performed using Absolute SYBR green mix (ABgene, UK) to assess ASC mRNA expression as described (Taxman et al., 2006). Real-time values were standardized to the expression of 18 s rRNA and normalized to 100 in control (untransduced) cells. Primers used for real-time PCR are as follows: ASC-[AACCCAAGCAAGA TGCGGAAG, TTAGGGCCTGGAGGAGCAAG], 18 s-[CGGCTACCAC ATCCAAGG, GCTGCTGGCACCAGACTT].
Viaprobe and 7-AAD Cell Staining
Cells were collected and rinsed twice in cold PBS. Pellets were resuspended in 0.5 ml PBS with 3 μl Viaprobe (Becton Dickinson) or 1 μl 7-AAD (BD PharMingen). Cells were incubated in the dark for 15 min before analysis on a FACScan (BD).
Propidium Iodide Staining
Following treatment, cells were collected and pelleted via centrifugation. Pelleted cells were fixed in 70% ethanol for a minimum of 2 hr, rinsed once in PBS, then resuspended in PBS containing 1% Triton, 20 μg/ml propidium iodide (Sigma Chemical Co.), and 200 μg/ml RN-ase A (QIAGEN). The cells were allowed to stain for 15 min or longer and then analyzed using a FACScan (BD).
ELISA
THP1 samples were harvested 24 hr posttransduction and assayed with BD OptEIA Human IL-1β ELISA Set (BD Biosciences) and Human IL-18 ELISA (MBL International). Mouse samples were taken at indicated times postinfection and assayed with BD OptEIA Mouse IL-1β ELISA Set (BD Biosciences).
Electron Microscopy
THP-1 cells were infected with adenoviruses at moi = 1 and fixed in 2% paraformaldehyde, 2.5% glutaraldehyde, in 0.15 M sodium phosphate (pH 7.4) 2 or 6 hr postinfection. Electron microscopy was performed at the UNC Microscopy Services Laboratory.
Patient Cells
Two female patients (ages 62 and 71) with FCAS were included in the study. Both FCAS patients had classic clinical presentation and met diagnostic criteria, as described previously (Hoffman et al., 2001b). Neither FCAS subject was experiencing significant inflammatory symptoms at the time of study, nor on regular anti-inflammatory medications. Two female controls (ages 39 and 41) were studied simultaneously with the FCAS patients. However, one control was later found to have significantly abnormal inflammatory responses and therefore was not included. An additional four male controls aged 27–34 were subsequently studied. PBMCs were isolated and prepared as previously described (Stack et al., 2005).
CIAS1 and Caspase-1-Deficient Mice
CIAS1−/− and Caspase-1−/− mice were described previously and were respectively produced by Millenium Inc. and Dr. Richard Flavell, Yale University (Sutterwala et al., 2006). They were backcrossed for a minimum of six generations to C57BL/6. Macrophages were obtained by peritoneal lavage 5 days after intraperitoneal injection with 4% thioglycollate and cultured in DMEM supplemented with 10% fetal calf serum and 50 μg/ml penicillin and streptomycin. Bone marrow macrophages were harvested from 6- to 8-week-old mice and cultured for 7 days in 30% M-CSF conditioned media.
Bacterial Infections
Shigella flexneri strain 12022 was obtained from ATCC. 2457T and BS103 have been described previously (Fernandez-Prada et al., 1997). THP-1 cells were cultured at 106/ml in antibiotic-free RPMI. All samples were infected with S. flexneri or S. typhi bacteria at a moi of 50 at 37°C for the indicated amount of time. Samples were centrifuged at 650 × g for 10 min immediately following addition of bacteria. Gentamicin (50 μg/ml) was added to cultures 2 hr postinfection.
Supplementary Material
Supplemental Data
The Supplemental Data include four supplemental figures and can be found with this article online at http://www.cellhostandmicrobe.com/cgi/content/full/2/3/147/DC1/.
Acknowledgments
This work is supported by National Institutes of Health Grants AI63031, AI57175, AI057175, and DE16326. J.P.-Y.T. is a member of SERCEB and a SPAR awardee. J.A.D. was supported by the Pfizer Fellowship in Infectious Diseases and NIH K12RR023248. We thank all members of the Hoffman Lab (La Jolla, California) for their help and advice. We thank Millenium Pharmaceuticals for the CIAS1−/− mice. We are grateful to everyone in the UNC Microscopy Services Laboratory, especially Victoria Madden, for experimental direction and outstanding technical support. We also thank the UNC Gene Therapy Center, specifically Xinghua Zhang, for assistance in preparing adenoviral constructs.
References
- Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–325. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, Stein L, Russo R, Goldsmith D, Dent P, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): A new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46:3340–3348. doi: 10.1002/art.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–570. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel FM. Are innate immune signaling pathways in plants and animals conserved? Nat Immunol. 2005;6:973–979. doi: 10.1038/ni1253. [DOI] [PubMed] [Google Scholar]
- Belkhadir Y, Subramaniam R, Dangl JL. Plant disease resistance protein signaling: NBS-LRR proteins and their partners. Curr Opin Plant Biol. 2004;7:391–399. doi: 10.1016/j.pbi.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Blocker A, Gounon P, Larquet E, Niebuhr K, Cabiaux V, Parsot C, Sansonetti P. The Tripartite type III secreton of Shigella flexneri inserts IpaB and IpaC into host membranes. J Cell Biol. 1999;147:683–693. doi: 10.1083/jcb.147.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Smith MR, Thirumalai K, Zychlinsky A. A bacterial invasin induces macrophage apoptosis by binding directly to ICE. EMBO J. 1996;15:3853–3860. [PMC free article] [PubMed] [Google Scholar]
- Chisholm ST, Coaker G, Day B, Staskawicz BJ. Host-microbe interactions: Shaping the evolution of the plant immune response. Cell. 2006;124:803–814. doi: 10.1016/j.cell.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Dowds TA, Masumoto J, Zhu L, Inohara N, Nunez G. Cryopyrin-induced Interleukin 1β secretion in monocytic cells: Enhanced activity of disease-associated mutants and requirement for ASC. J Biol Chem. 2004;279:21924–21928. doi: 10.1074/jbc.M401178200. [DOI] [PubMed] [Google Scholar]
- Edgeworth JD, Spencer J, Phalipon A, Griffin GE, Sansonetti PJ. Cytotoxicity and interleukin-1 processing following Shigella flexneri infection of human monocyte-derived dendritic cells. Eur J Immunol. 2002;32:1464–1471. doi: 10.1002/1521-4141(200205)32:5<1464::AID-IMMU1464>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, Teillac-Hamel D, Fischer A, de Saint Basile G. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71:198–203. doi: 10.1086/341357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Wu J, Yu J-W, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, Alnemri ES. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590–1604. doi: 10.1038/sj.cdd.4402194. Published online June 29, 2007. 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Prada C, Hoover D, Tall B, Venkatesan M. Human monocyte-derived macrophages infected with virulent Shigella flexneri in vitro undergo a rapid cytolytic event similar to oncosis but not apoptosis. Infect Immun. 1997;65:1486–1496. doi: 10.1128/iai.65.4.1486-1496.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Prada CM, Hoover DL, Tall BD, Hartman AB, Kopelowitz J, Venkatesan MM. Shigella flexneri IpaH7.8 facilitates escape of virulent bacteria from the endocytic vacuoles of mouse and human macrophages. Infect Immun. 2000;68:3608–3619. doi: 10.1128/iai.68.6.3608-3619.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois M, Le Cabec V, Dupont MA, Sansonetti PJ, Maridonneau-Parini I. Induction of necrosis in human neutrophils by Shigella flexneri requires type III secretion, IpaB and IpaC invasins, and actin polymerization. Infect Immun. 2000;68:1289–1296. doi: 10.1128/iai.68.3.1289-1296.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa A, Kambe N, Saito M, Nishikomori R, Tanizaki H, Kanazawa N, Adachi S, Heike T, Sagara J, Suda T, et al. Disease-associated mutations in CIAS1 induce cathepsin B-dependent rapid cell death of human THP-1 monocytic cells. Blood. 2006;7:2903–2911. doi: 10.1182/blood-2006-07-033597. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Tournebize R, Mavris M, Page AL, Li X, Stark GR, Bertin J, DiStefano PS, Yaniv M, Sansonetti PJ, Philpott DJ. CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2001;2:736–742. doi: 10.1093/embo-reports/kve155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, Kim HJ, Brewer C, Zalewski C, Wiggs E, et al. Neonatal-onset multisystem inflammatory disease responsive to Interleukin-1β inhibition. N Engl J Med. 2006;355:581–592. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golstein P, Kroemer G. Cell death by necrosis: Towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Greenberg JT, Guo A, Klessig DF, Ausubel FM. Programmed cell death in plants: A pathogen-triggered response activated coordinately with multiple defense functions. Cell. 1994;77:551–563. doi: 10.1016/0092-8674(94)90217-8. [DOI] [PubMed] [Google Scholar]
- Haimovich B, Venkatesan MM. Shigella and Salmonella: Death as a means of survival. Microbes Infect. 2006;8:568–577. doi: 10.1016/j.micinf.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Harton JA, Linhoff MW, Zhang J, Ting JPY. Cutting edge: CATERPILLER: A large family of mammalian genes containing CARD, Pyrin, nucleotide-binding, and leucine-rich repeat domains. J Immunol. 2002;169:4088–4093. doi: 10.4049/jimmunol.169.8.4088. [DOI] [PubMed] [Google Scholar]
- Hawkins PN, Lachmann HJ, Aganna E, McDermott MF. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50:607–612. doi: 10.1002/art.20033. [DOI] [PubMed] [Google Scholar]
- Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001a;29:301–305. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman HM, Wanderer AA, Broide DH. Familial cold autoinflammatory syndrome: Phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol. 2001b;108:615–620. doi: 10.1067/mai.2001.118790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann JA, Reichhart JM. Drosophila innate immunity: An evolutionary perspective. Nat Immunol. 2002;3:121–126. doi: 10.1038/ni0202-121. [DOI] [PubMed] [Google Scholar]
- Hoffman HM, Rosengren S, Boyle D, Cho J, Nayar J, Mueller J, Anderson J, Wanderer A, Firestein G. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364:1779–1785. doi: 10.1016/S0140-6736(04)17401-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. J Biol Chem. 1995;270:25752–25761. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- Inohara N, Nunez G. NODS: Intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3:371–382. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- Kanneganti TD, Body-Malapel M, Am A, Park JH, Whitfield J, Taraporewala ZF, Miller D, Patton JT, Inohara N, Nunez G. Critical role for cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006a;281:36560–36568. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- Kanneganti TD, Ozoren N, Body-Malapel M, Am A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006b;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- Karahashi H, Amano F. Apoptotic changes preceding necrosis in lipopolysaccharide-treated macrophages in the presence of cycloheximide. Exp Cell Res. 1998;241:373–383. doi: 10.1006/excr.1998.4062. [DOI] [PubMed] [Google Scholar]
- Koterski JF, Nahvi M, Venkatesan MM, Haimovich B. Virulent Shigella flexneri causes damage to mitochondria and triggers necrosis in infected human monocyte-derived macrophages. Infect Immun. 2005;73:504–513. doi: 10.1128/IAI.73.1.504-513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko DV, D’Herde K, Vandenabeele P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis. 2006;11:1709–1726. doi: 10.1007/s10495-006-9527-8. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med. 2005;202:1043–1049. doi: 10.1084/jem.20050977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Martinon F, Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol. 2005;26:447–454. doi: 10.1016/j.it.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, Hidaka E, Katsuyama T, Higuchi T, Sagara J. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem. 1999;274:33835–33838. doi: 10.1074/jbc.274.48.33835. [DOI] [PubMed] [Google Scholar]
- Masumoto J, Dowds TA, Schaner P, Chen FF, Ogura Y, Li M, Zhu L, Katsuyama T, Sagara J, Taniguchi S, et al. ASC is an activating adaptor for NF-κB and caspase-8-dependent apoptosis. Biochem Biophys Res Commun. 2003;303:69–73. doi: 10.1016/s0006-291x(03)00309-7. [DOI] [PubMed] [Google Scholar]
- McConnell BB, Vertino PM. Activation of a Caspase-9-mediated apoptotic pathway by subcellular redistribution of the novel caspase recruitment domain protein TMS1. Cancer Res. 2000;60:6243–6247. [PubMed] [Google Scholar]
- Menard R, Sansonetti PJ, Parsot C. Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J Bacteriol. 1993;175:5899–5906. doi: 10.1128/jb.175.18.5899-5906.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard R, Sansonetti P, Parsot C. The secretion of the Shigella flexneri Ipa invasins is activated by epithelial cells and controlled by IpaB and IpaD. EMBO J. 1994;13:5293–5302. doi: 10.1002/j.1460-2075.1994.tb06863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchuk Z, Eulgem T, Holt BF, III, Dangl JL. Recognition and response in the plant immune system. Annu Rev Genet. 2003;37:579–609. doi: 10.1146/annurev.genet.37.110801.142628. [DOI] [PubMed] [Google Scholar]
- Nonaka T, Kuwabara T, Mimuro H, Kuwae A, Imajoh-Ohmi S. Shigella-induced necrosis and apoptosis of U937 cells and J774 macrophages. Microbiology. 2003;149:2513–2527. doi: 10.1099/mic.0.26341-0. [DOI] [PubMed] [Google Scholar]
- O’Connor W, Jr, Harton JA, Zhu X, Linhoff MW, Ting JPY. Cutting edge: CIAS1/Cryopyrin/PYPAF1/NALP3/CAT-ERPILLER 1.1 is an inducible inflammatory mediator with NF-κB suppressive properties. J Immunol. 2003;171:6329–6333. doi: 10.4049/jimmunol.171.12.6329. [DOI] [PubMed] [Google Scholar]
- Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of Toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCrmMice: Mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Raqib R, Ekberg C, Sharkar P, Bardhan PK, Zychlinsky A, Sansonetti PJ, Andersson J. Apoptosis in acute shigellosis is associated with increased production of Fas/Fas ligand, Perforin, Caspase-1, and Caspase-3 but reduced production of Bcl-2 and Interleukin-2. Infect Immun. 2002;70:3199–3207. doi: 10.1128/IAI.70.6.3199-3207.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Schotte P, Declercq W, Van Huffel S, Vandenabeele P, Beyaert R. Non-specific effects of methyl ketone peptide inhibitors of caspases. FEBS Lett. 1999;442:117–121. doi: 10.1016/s0014-5793(98)01640-8. [DOI] [PubMed] [Google Scholar]
- Stack JH, Beaumont K, Larsen PD, Straley KS, Henkel GW, Randle JCR, Hoffman HM. IL-converting enzyme/caspase-1 inhibitor VX-765 blocks the hypersensitive response to an inflammatory stimulus in monocytes from familial cold autoinflammatory syndrome patients. J Immunol. 2005;175:2630–2634. doi: 10.4049/jimmunol.175.4.2630. [DOI] [PubMed] [Google Scholar]
- Stehlik C, Lee SH, Dorfleutner A, Stassinopoulos A, Sagara J, Reed JC. Apoptosis-associated Speck-like protein containing a caspase recruitment domain is a regulator of Procaspase-1 activation. J Immunol. 2003;171:6154–6163. doi: 10.4049/jimmunol.171.11.6154. [DOI] [PubMed] [Google Scholar]
- Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichten-berger GS, Grant EP, Bertin J, Coyle AJ, Galan JE, Askenase PW, Flavell RA. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Nakanishi K, Tsutsui H, Iwai H, Akira S, Inohara N, Chamaillard M, Nunez G, Sasakawa C. A novel caspase-1/toll-like receptor 4-independent pathway of cell death induced by cytosolic shigella in infected macrophages. J Biol Chem. 2005;280:14042–14050. doi: 10.1074/jbc.M414671200. [DOI] [PubMed] [Google Scholar]
- Taxman DJ, Livingstone LR, Zhang J, Conti BJ, Iocca HA, Williams KL, Lich JD, Ting JP, Reed W. Criteria for effective design, construction, and gene knockdown by shRNA vectors. BMC Biotechnol. 2006;6:7. doi: 10.1186/1472-6750-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hasegawa M, Imamura R, Kinoshita T, Kondo C, Konaka K, Suda T. PYNOD, a novel Apaf-1/CED4-like protein is an inhibitor of ASC and caspase-1. Int Immunol. 2004;16:777–786. doi: 10.1093/intimm/dxh081. [DOI] [PubMed] [Google Scholar]
- Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, Moore C, Kurtz S, Coffield VM, Accavitti-Loper MA, Su L, et al. The CATERPILLER protein Monarch-1 Is an antagonist of Toll-like receptor-, tumor necrosis factor α-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem. 2005;280:39914–39924. doi: 10.1074/jbc.M502820200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev. 2006;20:1–15. doi: 10.1101/gad.1376506. [DOI] [PubMed] [Google Scholar]
- Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358:167–169. doi: 10.1038/358167a0. [DOI] [PubMed] [Google Scholar]
- Zychlinsky A, Sansonetti PJ. Apoptosis as a proinflammatory event: What can we learn from bacteria-induced cell death? Trends Microbiol. 1997;5:201–204. doi: 10.1016/S0966-842X(97)01044-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data
The Supplemental Data include four supplemental figures and can be found with this article online at http://www.cellhostandmicrobe.com/cgi/content/full/2/3/147/DC1/.