

SUMMARY
Staphylococcus aureus, a bacterium responsible for tremendous morbidity and mortality worldwide, exists as a harmless commensal organism in approximately 25% of the human population. Identifying the molecular machinery that is activated upon infection is central to understanding staphylococcal pathogenesis. We describe here the Heme-Sensor System (HssRS) that responds to heme exposure and activates expression of the Heme Regulated Transporter (HrtAB). The coordinated activities of HssRS and HrtAB maintain intracellular heme homeostasis and modulate S. aureus virulence. Inactivation of the Hss or Hrt systems leads to increased virulence in a vertebrate infection model, a phenotype that is associated with an inhibited innate immune response. Genomic analyses have identified orthologous Hss and Hrt systems in Bacillus anthracis, Listeria monocytogenes, and Enterococcus faecalis, suggesting a conserved regulatory system by which Gram positive pathogens sense heme as a molecular marker of internal host tissue and modulate virulence.
INTRODUCTION
Staphylococcus aureus is one of the most significant infectious threats to global public health (Fridkin et al., 2005). Infections with S. aureus result in diverse human diseases ranging from skin and soft tissue infections to endocarditis, septicemia and toxic shock syndrome (Brook, 2002; Fowler et al., 2005). Paradoxically, 25% of the human population is harmlessly colonized by S. aureus residing as normal flora of the skin and anterior nares (Wertheim et al., 2004). In order for S. aureus to initiate invasive infection, it must gain access to internal tissues or vasculature of its host. Once inside the host, S. aureus likely undergoes a shift in gene expression resulting in the controlled production of virulence determinants that facilitate infection. Although virulence gene regulation is one of the most well studied aspects of staphylococcal pathogenesis (Bronner et al., 2004), the environmental cues and corresponding staphylococcal regulatory systems that are active during invasive infection have not been defined.
S. aureus pathogenesis is dependent on the secretion of an array of virulence factors and the surface exposure of multiple cell wall anchored proteins (Foster, 2005). The expression of these effectors in vivo is presumably coordinated by a network of two-component systems (TCS) and transcriptional regulators. Although the contributions of a subset of TCS (agr, saeRS, srrAB, arlSR and lytRS) to virulence gene expression have been studied extensively (Bronner et al., 2004; Cheung et al., 2004; Novick, 2003), genomic analyses reveal that the majority of staphylococcal TCS remain unassigned. In this regard, environmental cues such as high salt, cell density, glucose, energy availability, pH, and subinhibitory antibiotics have been found to affect S. aureus virulence gene expression in laboratory conditions (Bronner et al., 2004; Cheung et al., 2004; Novick, 2003). However, the specific host molecules recognized by staphylococcal regulatory systems remain elusive. Decoding the signals sensed by S. aureus inside the host will set the stage to identify the molecular machinery that is activated during invasive infection.
One of the most significant obstacles that bacterial pathogens encounter when infecting vertebrates is iron limitation. Iron is an essential cofactor for many biochemical processes and thus required by virtually all pathogenic bacteria to establish infection (Bullen, 1999). The majority of vertebrate iron is in the form of the metalloporphyrin heme, the functional cofactor of hemoglobin and myoglobin, the oxygen transport and storage proteins of blood and muscle, respectively (Deiss, 1983). S. aureus acquire heme through the elaboration of transport systems which rapidly transport host-derived heme into the staphylococcal cytoplasm for use as a nutrient source (Mazmanian et al., 2003; Skaar et al., 2004; Torres et al., 2006; Vermeiren et al., 2006). Staphylococci likely facilitate this process through the hemolysin-mediated rupture of erythrocytes upon entry into the blood stream (Bernheimer et al., 1968; Skaar and Schneewind, 2004).
Although heme is a valuable nutrient source to invading pathogens, the intracellular accumulation of heme is toxic due to the molecule's reactivity. Therefore, organisms that acquire exogenous heme to satisfy nutrient iron needs must have adaptable mechanisms to avoid surplus heme accumulation. In this regard, we have recently reported the identification of a subset of staphylococcal proteins that are affected by changes in environmental hemin (the oxidized form of heme) concentration (Friedman et al., 2006). In particular, exposure to exogenous hemin results in the 45-fold up-regulation of the Heme Regulated Transporter, HrtAB (Friedman et al., 2006). The dramatic up-regulation of HrtAB upon exposure to hemin suggests that S. aureus possess systems capable of sensing heme and subsequently altering protein expression. The association of heme with the major protein constituents of blood and muscle establishes heme as a molecular marker that can potentially be exploited by infecting bacteria to distinguish internal host tissue from surface colonization sites. Hence, heme sensing systems may represent a mechanism by which bacterial pathogens sense when the surface tissues of the host have been breached.
In this study, we identified a S. aureus TCS which we have called the Heme-Sensor System (HssRS). HssRS responds to heme exposure and activates the expression of HrtAB, an efflux pump that plays a pivotal role in intracellular heme homeostasis. Inactivation of the Hss or Hrt systems results in enhanced liver-specific S. aureus virulence which correlates with a reduced innate immune response to infection. Staphylococcal strains unable to sense and excrete surplus heme exhibit increased virulence factor expression and secretion, providing a mechanistic explanation for the observed immunomodulation. Importantly, Hss and Hrt systems are present in Bacillus anthracis, Listeria monocytogenes, Staphylococcus epidermidis, and Enterococcus faecalis, suggesting a conserved mechanism by which these important human pathogens sense their vertebrate hosts to modulate virulence.
RESULTS
Staphylococcus aureus adapt to avoid heme toxicity
Heme-iron acquisition is vital to staphylococcal pathogenesis (Skaar et al., 2004; Torres et al., 2006). However, the value of heme as an iron source must be balanced against its toxicity at high concentrations (Everse and Hsia, 1997). In this regard, we found that S. aureus growth is slightly inhibited when bacteria are cultured in iron-replete medium supplemented with 5 μM hemin (Figure 1A). Exposure to concentrations of hemin at or above 10 μM severely inhibited staphylococcal growth in these same culture conditions (Figure 1A), highlighting the acute sensitivity of S. aureus to excess hemin.
Figure 1. S. aureus adapt to avoid the toxic effects of hemin.
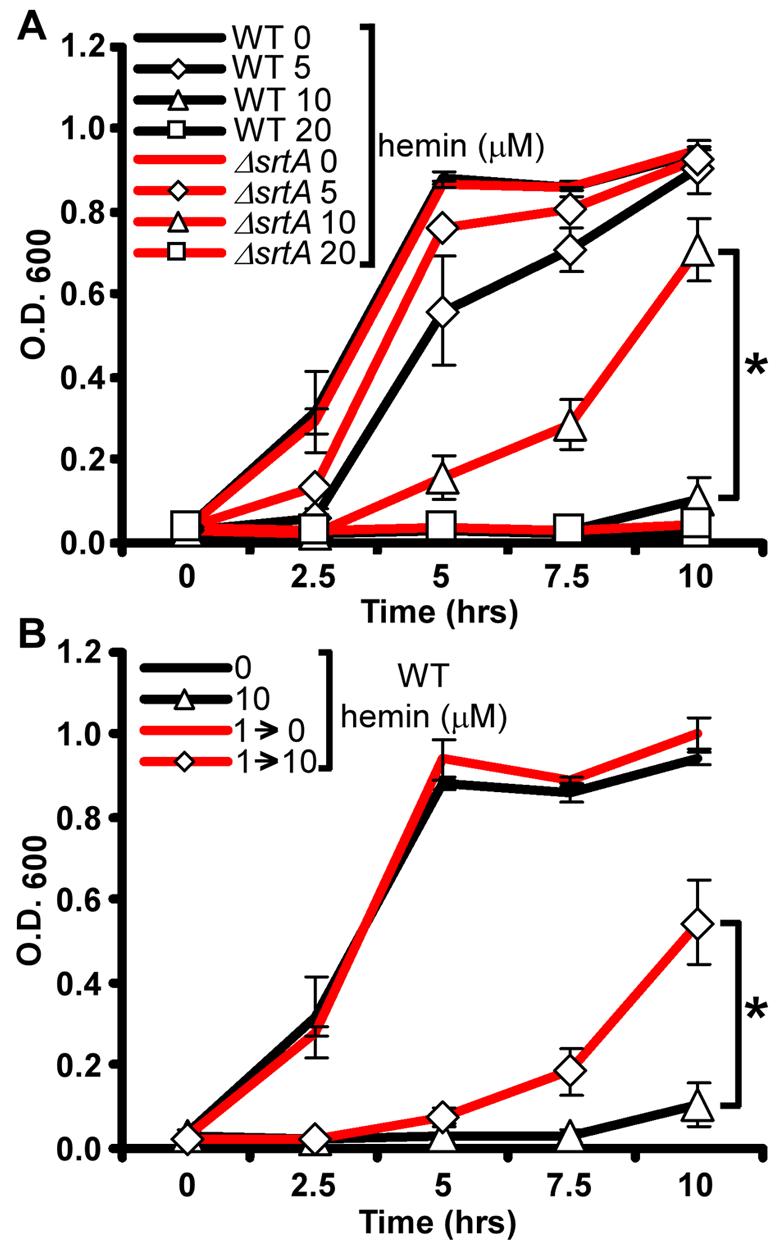
A) S. aureus wildtype and ΔsrtA were grown O/N in TSB and then subcultured into TSB supplemented with 0, 5, 10, or 20 μM hemin. Growth rates were determined by measuring the absorbance (O.D. 600) at the indicated time points. B) S. aureus wildtype was grown O/N in TSB supplemented with 0 or 1 μM hemin (1→) and then subcultured into TSB supplemented with 0 or 10 μM hemin (→0 or →10). The results represent the mean ± S.D. from triplicate experiments. Asterisks denote statistically significant differences as determined by Student's t test (p≤0.05).
The initial interaction between staphylococci and host heme sources is mediated by bacterial receptors that are covalently anchored to the cell wall by the action of the transpeptidase sortase A (SrtA) (Mazmanian et al., 2003; Torres et al., 2006; Vermeiren et al., 2006). To investigate the contribution of staphylococcal heme and hemoglobin receptors to heme toxicity, we compared wildtype S. aureus and a mutant lacking srtA (ΔsrtA) for their sensitivity to hemin toxicity. Growth curve analyses demonstrate that ΔsrtA proliferates in medium containing up to 10 μM hemin (Figure 1A), presumably due to the absence of surface linked heme receptors leading to a decreased ability to internalize free hemin. These data show that hemin-mediated growth inhibition is reliant on the same SrtA-dependent pathways required for the utilization of heme as a nutrient source.
S. aureus heme acquisition is a highly efficient process that results in the rapid cytoplasmic accumulation of heme (Mazmanian et al., 2000; Skaar et al., 2004). The strict requirement for heme uptake systems in staphylococcal virulence (Skaar et al., 2004; Torres et al., 2006) implies that S. aureus contain adaptable mechanisms that exploit heme as a nutrient iron source while avoiding heme-mediated toxicity. To explore this adaptation mechanism in more detail, we investigated whether pre-exposing S. aureus to sub-inhibitory concentrations of hemin increases hemin tolerance. Staphylococcal cultures grown in sub-inhibitory hemin concentrations (1 μM) exhibited a pronounced resistance to hemin toxicity when subcultured at concentrations up to 10 μM (Figure 1B). These findings demonstrate that S. aureus undergo an adaptive response to exogenous hemin resulting in increased resistance to hemin toxicity.
S. aureus adaptation to hemin toxicity is dependent on HrtAB
The ability of S. aureus to adapt to hemin toxicity is likely the result of coordinated changes in protein expression that occur upon hemin exposure. In this regard, we have recently identified the heme regulated transport system (HrtAB) which increases expression by approximately 45-fold upon exposure to hemin (Friedman et al., 2006). HrtAB is composed of an ATP-binding protein (HrtA) and permease (HrtB) making up a canonical ABC-type transporter system. Genomic analyses suggest that HrtAB is a member of the MacAB family of ABC-type efflux carriers, which have been implicated in the export of small molecules (Kobayashi et al., 2001). Based on these facts, we predicted that HrtAB contributes to S. aureus avoidance of hemin toxicity by exporting surplus hemin from the bacterial cytoplasm. To facilitate experiments aimed at testing this prediction, we created a S. aureus strain in which hrtA was deleted (ΔhrtA). S. aureus wildtype and ΔhrtA proliferated at comparable rates in hemin-free medium; however, the ΔhrtA strain was significantly more susceptible to hemin toxicity (Figure 2A and 2B). The ability of staphylococci to adapt and avoid hemin toxicity was fully dependent on the presence of a functional HrtAB system (Figure 2C). The increased sensitivity to hemin toxicity exhibited by ΔhrtA was restored by providing a wildtype copy of the hrtA gene in trans (Figure 2D). These findings are in accordance with a model whereby S. aureus sense heme, resulting in the elaboration of a transport system (HrtAB) dedicated to the excretion of surplus heme to protect against toxicity.
Figure 2. The HrtAB system is required for staphylococcal heme adaptation.
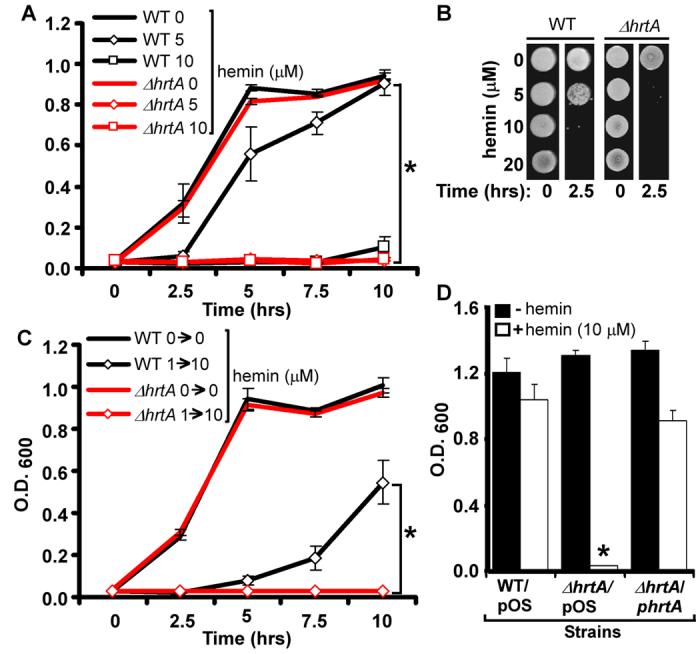
A) S. aureus wildtype and ΔhrtA were grown O/N in TSB and then subcultured into TSB supplemented with 0, 5, or 10 μM hemin. Growth rates were determined by measuring the absorbance (O.D. 600) at the indicated time points. B) S. aureus wildtype and ΔhrtA were grown O/N in TSB and then subcultured for 2.5 hrs into TSB supplemented with 0, 5, 10 or 20 μM hemin. The cultures were then serially diluted and plated on TSA plates. C) S. aureus wildtype and ΔhrtA were grown O/N in TSB and TSB supplemented with 1 μM hemin (1→) and then subcultured into TSB supplemented with 0 or 10 μM hemin (→0 or →10). Growth rates were determined by measuring the absorbance (O.D. 600) at the indicated time points (hrs). D) S. aureus wildtype and ΔhrtA transformed with vector alone (WT/pOS and ΔhrtA/pOS) and ΔhrtA transformed with vector containing a wildtype copy of hrtA (ΔhrtA/phrtA) were grown in TSB supplemented with 1 μM hemin and then subcultured into TSB supplemented with +/− 10 μM hemin and grown for 15 hrs. The results represent the mean ± S.D. from triplicate experiments. Asterisks denote statistically significant differences as determined by Student's t test (p≤0.05).
HssRS regulates hrtAB expression
Examination of the genomic context immediately adjacent to the hrtAB locus revealed the presence of two genes predicted to encode for a TCS (Figure 3A). This TCS designation is based on BLAST analyses which revealed that the closest annotated matches to these genes are the response regulator ompR (e-value 6 × 10−55) and histidine kinase baeS (e-value 8 × 10−32) of Escherichia coli (Nagasawa et al., 1993; Taylor et al., 1981). TCS sense environmental stimuli and regulate gene expression (Beier and Gross, 2006). On the basis of studies described below, we have named this newly identified TCS the Heme Sensor System Regulator and Sensor, HssRS (HssR-response regulator, HssS-histidine kinase). The predicted protein product of HssS contains two transmembrane regions flanking an extra-cytoplasmic ligand sensing domain. The cytoplasmic portion of HssS is predicted to be comprised of a HisKA dimerization/phosphoacceptor region linked to an ATPase domain (Figure S1A). BLAST analyses of full-length HssS as well as the predicted sensor domain demonstrate that the protein is conserved across many Gram positive bacteria; however this sensing domain is distinct from all previously described ligand binding domains of histidine kinases (Figure S1B). To evaluate the possibility that the heme-dependent expression of hrtAB is mediated by HssRS, we generated isogenic mutant strains in which either hssR or hssS were deleted (ΔhssR, ΔhssS). Growth curve analyses indicated that both HssR and HssS are required for S. aureus adaptation to heme toxicity (Figure 3B), a phenotype that was complemented by introducing a wildtype copy of hssR in trans into ΔhssR (Figure 3C). These data demonstrate that both HrtAB and HssRS are vital to staphylococcal heme adaptation.
Figure 3. HssRS is required for hrtA expression upon exposure to hemin.
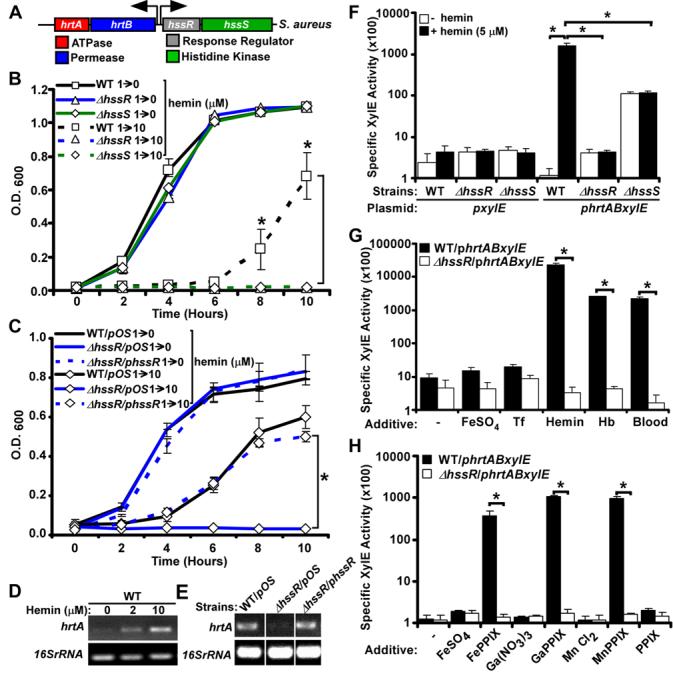
A) Schematic representation of the hrtAB and hssRS loci in S. aureus. B-C) Listed staphylococcal strains were grown O/N in TSB supplemented with 1 μM heme (1→) and then subcultured into TSB supplemented with 0 or 10 μM heme (→0 or →10). Growth rates were determined by measuring the absorbance (O.D.600) at the indicated time points (hrs). D-E) RT-PCR analyses. D) Total RNA was extracted from O/N cultures of S. aureus wildtype grown in TSB supplemented with 0, 2 or 10 μM hemin. cDNA was synthesized as described in Experimental Procedures and transcription of the hrtA gene and the 16sRNA (loading control) was assessed by PCR. E) Total RNA was extracted from O/N cultures of wildtype (WT/pOS), ΔhssR/pOS, and the complemented ΔhssR strain (ΔhssR/phssR) grown in TSB supplemented with 2 μM hemin. The cDNA was synthesized as described above and transcription of the hrtA gene and the 16sRNA (loading control) was determined as in panel D. Differences in the relative level of RTPCR product between panels D and E are likely a result of the required inclusion of chloramphenicol to the growth media in experiments shown in panel E. F-H) XylE fusion reporter assay. F) WT, ΔhssR, and ΔhssS transformed with the phrtABxylE or the pxylE plasmid were grown 2 hrs in TSB supplemented with 0 or 5 μM hemin and XlyE activity was determined as described in Experimental Procedures. G) Wildtype and ΔhssR harboring the phrtABxylE reporter plasmid were grown 2 hrs in TSB supplemented with FeSO4 (8 μM), transferrin (Tf; 8 μM), hemin (8 μM), hemoglobin (Hb; 2 μM), or mouse blood and XlyE activity was determined as in panel F. H) Wildtype and ΔhssR harboring the phrtABxylE reporter plasmid were grown 2 hrs in TSB supplemented with 1 μM of the indicated additives and the XlyE activity determined as in panel F. The results represent the mean ± S.D. from at least triplicate experiments. Asterisks denote statistically significant differences as determined by Student's t test (p<0.05).
In wildtype staphylococci, hrtA expression is induced by exogenous hemin (Figure 3D). This heme-dependent increase in expression is presumably responsible for the ability of wildtype staphylococci to grow in 10 μM hemin after a prolonged overnight incubation (data not shown). Considering the critical role HssRS plays in staphylococcal heme adaptation, we investigated whether HssRS is responsible for the heme-dependent up-regulation of hrtA transcript. We were unable to detect hrtA transcript in ΔhssR upon hemin exposure, a defect that was complemented by providing a wildtype copy of hssR in trans (Figure 3E). To confirm these data, we generated a reporter construct in which the predicted hrtAB promoter was fused to a xylE reporter gene (Chien et al., 1999). Wildtype and ΔhssR displayed background levels of reporter activity when the strains were grown in the absence of hemin (Figure 3F). In contrast, ΔhssS exhibited appreciable hrtAB expression in the absence of inducer. This result is consistent with the idea that, as with many histidine kinases, HssS is responsible for maintaining its cognate response regulator (HssR) in an unphosphorylated state in the absence of inducer (Mascher et al., 2006). When the strains harboring the phrtABxylE reporter were grown in media containing hemin, only the wildtype strain displayed a heme-dependent increase in reporter activity (Figure 3F). These data establish an absolute requirement for both HssR and HssS in the heme-dependent up-regulation of hrtAB. Notably, synthesis of endogenous heme by S. aureus is not sufficient for activation of hrtAB (Figure 3F), providing evidence that HssRS is triggered by exposure to exogenous heme.
In vertebrates, heme is predominantly bound to hemoglobin within erythrocytes. In this regard, hemoglobin exposure potently activates hrtAB expression (Figure 3G). Importantly, HssRS-dependent induction of hrtAB also occurs when staphylococci are exposed to blood (Figure 3G). Strains grown in medium supplemented with excess iron sulfate or the iron sequestering protein transferrin did not activate reporter expression, eliminating a role for iron in hrtAB activation (Figure 3G). Together, these results suggest that HssRS responds to heme as a component of vertebrate blood, resulting in the induction of hrtAB expression.
It is possible that HssRS senses heme through direct binding to HssS. Alternatively, HssRS may sense cellular stress mediated by excess heme exposure. Analyses of available genomic and proteomic screens of staphylococcal regulatory circuits that respond to environmental cues reveals that hrtAB is not induced upon iron starvation (Friedman et al., 2006), exposure to mild acid (Weinrick et al., 2004), or treatment with nitric oxide (Richardson et al., 2006). In addition, hrtA expression is not affected by cold shock, heat shock, the stringent response, or activation of the SOS response (Anderson et al., 2006). Moreover, hrtAB expression is not regulated by the global regulators of staphylococcal virulence MgrA (Luong et al., 2006), SigB, Agr, or SarA (Bischoff et al., 2004; Dunman et al., 2001). Proteins that directly complex heme typically recognize the encircled metal atom of the metalloporphyrin through coordination with an axial ligand. To test if S. aureus requires metal complexed porphyrins for HssRS-dependent hrtAB expression, we exposed staphylococci to a variety of protoporphyrin IX (PPIX) analogues. These emerging data revealed that HssRS up-regulates hrtAB expression upon exposure to Fe-PPIX (heme), Ga-PPIX, and Mn-PPIX. In contrast, exposure of S. aureus to metal-free PPIX does not result in activation of hrtAB. Furthermore, exposure of S. aureus to excess FeSO4, Ga(NO3)3, or MnCl2 does not result in hrtAB activation (Figure 3H). We conclude that metal-coordinated porphyrins are required for HssRS-dependent hrtAB activation and that HssRS does not indirectly recognize cellular stress associated with excess metal or porphyrin exposure. Moreover, although we cannot rule out the possibility that an as-yet-unidentified factor is transferring a signal from heme to HssRS, these data strongly support the model that HssRS senses heme to activate hrtA expression.
Inactivation of hssRS or hrtAB increases staphylococcal virulence
To test the role of HssRS heme sensing and concomitant HrtAB expression in S. aureus pathogenesis, mice were infected intravenously with S. aureus wildtype, ΔhrtA, or ΔhssR. Animals infected with wildtype S. aureus exhibited overt signs of disease characteristic of staphylococcal infection. Surprisingly, all animals infected with S. aureus ΔhrtA or ΔhssR appeared more moribund than those infected with wildtype as evidenced by a complete absence of mobility, a pronounced hunched posture, and extensive tremors. Autopsies conducted 96 hours postinfection revealed abscess formation in the kidneys of mice infected with any of the three staphylococcal strains (data not shown). In contrast, only mice infected with S. aureus ΔhrtA or ΔhssR developed abscesses in the liver (Figure 4). Enumeration of bacterial loads in the livers of infected animals revealed a 2-3 log increase in the number of mutant staphylococci as compared with wildtype (Figure 4B). This increase in virulence was liver-specific, since no difference was detected in the ability of the mutant strains to colonize the spleen or kidney compared to wildtype (Figure 4B). The increased liver-specific hypervirulence of S. aureus ΔhrtA and ΔhssR is not due to intrinsically faster growth rates, because mutant strains exhibit similar growth kinetics to wildtype in laboratory growth conditions (Figure 2 and 3). Histological examination of livers infected with the mutant staphylococci revealed that hepatic hypervirulence occurs despite the recruitment of polymorphonuclear (PMN) cells (Figure 4D). More specifically, the ΔhrtA or ΔhssR-induced abscesses were characterized by collections of purulent material containing PMNs, injured hepatocytes, and dense fibrous tissue (Figure 4D). Together, these findings demonstrate that S. aureus strains lacking HssRS or HrtAB exhibit increased hepatic virulence.
Figure 4. S. aureus ΔhssR and ΔhrtA exhibit liver-specific hypervirulence.
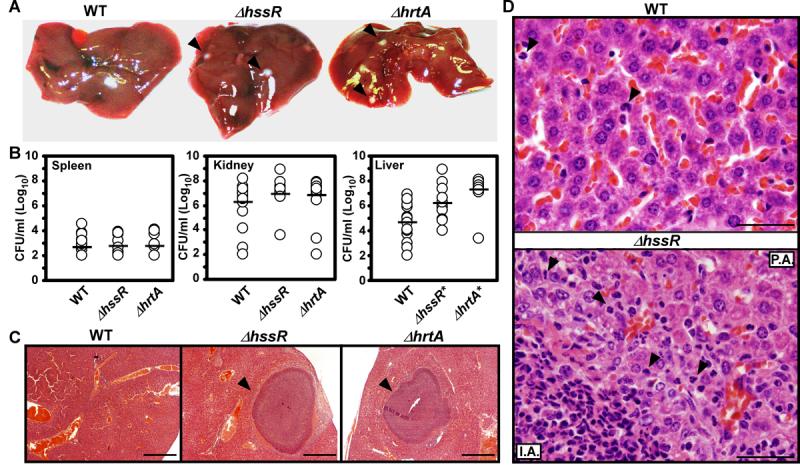
A) Photographs of livers dissected from BALB/c mice infected with wildtype and ΔhssR or ΔhrtA (1 × 106 CFUs for all strains) 96 hours post infection. Arrowheads mark ΔhssR and ΔhrtA-induced hepatic abscesses. Photographs are representative of all livers analyzed. Abscesses were visible in virtually all livers from ΔhssR and ΔhrtA infected mice, while none were found in wildtype infected mice. B) S. aureus multiplication in infected mouse organs as measured by tissue homogenization, dilution, and colony formation on agar media 96 hours post infection. Each symbol represents data from one infected animal. The limit of detection in these experiments is 100 CFUs. The horizontal line denotes the mean of the log and the asterisks denote statistically significant differences from wildtype as determined by Student's t test (p≤0.05). C) Representative Hematoxylin and Eosin (H&E) staining of liver sections infected with WT, ΔhrtA, or ΔhssR strains at 40X magnification. Arrowheads mark ΔhssR- and ΔhrtA-induced hepatic abscesses. D) Representative H&E staining of liver sections infected with WT or ΔhssR strains at 1,000X magnification. Arrowheads mark PMNs in the tissues. P.A; proximal to the abscess and I.A; inside the abscess.
The liver-specific immune response is inhibited against staphylococci inactivated for hssRS or hrtAB
The hepatic hypervirulence exhibited by S. aureus ΔhrtA or ΔhssR could be the consequence of (i) a defective immune response to mutant staphylococci, (ii) an increase in the expression of staphylococcal virulence factors, (iii) enhanced bacterial resistance to immune clearance, or (iv) increased tissue tropism of S. aureus ΔhssR and ΔhrtA. To begin distinguishing between these possibilities, we characterized the immune cell profiles in organs from mice infected with S. aureus wildtype, ΔhrtA, or ΔhssR via multiparametric flow cytometric analyses. We did not detect changes in the adaptive immune response to these strains, as measured by equivalent numbers of resident or infiltrating B cells, CD4+ T cells, or CD8+ T cells between tissues from infected or uninfected mice (data not shown).
The organ-specific innate immune response to staphylococcal infection is not well defined. Therefore, we initially analyzed the innate immune cell populations in infected versus uninfected tissues. We found increased numbers of phagocytes (CD11b+/CD11c−) in the spleens of infected animals relative to uninfected controls (Figure 5A). We did not detect significant infiltration of dendritic cells, natural killer (NK) cells, or invariant-natural killer T (iNKT) cells into this organ (Figure 5 B-D). In contrast, we were able to detect infiltration of NK cells, iNKT cells, phagocytes and granulocytes into the kidneys and livers of infected animals (Figure 5 A-D), organs containing a higher bacterial load than the spleen (Figure 4B). Importantly, the NK and iNKT cells detected in the livers and kidneys expressed activation markers (i.e., CD69), suggesting that these lymphocytes were activated and proliferated in response to staphylococcal infection (data not shown). Together, these results imply that differences in bacterial loads across organs influence the degree to which innate immune cells are recruited and activated in staphylococcal infected tissue.
Figure 5. Infection with S. aureus ΔhssR or ΔhrtA inhibits innate immune responses.
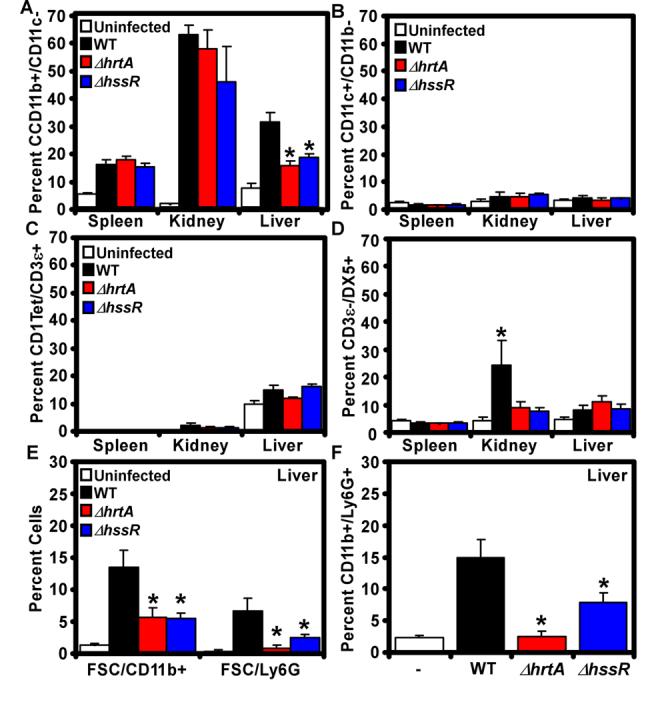
BALB/c animals were left uninfected or infected with wildtype, ΔhssR or ΔhrtA. Four days postinfection (A-D) or two days post infection (E-F) organs were dissected, homogenized, and the infiltration of the indicated immune cells was determined by multiparametric FACS analysis as described in Experimental Procedures. Isolated cells were stained for the detection of: A) phagocytes (B220−/CD11b+/CD11c−), B) dendritic cells (D11c+/CD11b−), C) invariant natural killer T cells (iNKT: CD1 Tetramer (tet)+/B220−/CD3ε+), D) natural killer cells (DX5+/B220−/CD3ε−), E) large CD11b+ and Ly6G+ cells (FSC/CD11b+ and FSC/Ly6G+), and F) granulocytes (B220−/CD11b+/Ly6G+). Results represent the mean ± S.E. from at least three independent animals. Asterisks denote a statistically significant reduction in the detected cells compared to animals infected with the wildtype strain as determined by Student's t test (p<0.05).
Comparison of the immune cell profiles in the spleens and kidneys of animals infected with wildtype, ΔhrtA, or ΔhssR staphylococci revealed minimal differences, with the exception of NK cells (Figure 5A-D). Indeed, there was a significant increase in the number of NK cells detected in the kidneys of wildtype infected animals compared to those infected with ΔhrtA or ΔhssR (Figure 5D). Although interesting, we did not investigate this finding further as the effect on NK cells was specific to the kidney and therefore not likely to play a role in the observed liver hypervirulence. Importantly, livers from ΔhrtA or ΔhssR infected mice contained approximately half the number of CD11b+/CD11c− phagocytes found in livers from mice infected with wildtype S. aureus (Figure 5A). More rigorous analysis at 48 hours post-infection revealed that this decreased population is comprised primarily of CD11b+/Ly6G+ granulocytes (Figure 5EF). Based on our visualization of PMNs in histological preparations of abscessed livers from mice infected with ΔhssR, it is possible that this quantitative decrease is due to increased granulocyte death in these organs. Alternatively, the non-quantitative nature of our histological analyses (Figure 4D) does not rule out the possibility that there is a decreased recruitment of granulocytes to the livers of ΔhssR/ΔhrtA infected mice. Thus, we conclude that infection with ΔhssR or ΔhrtA inhibits the innate immune response to S. aureus infection.
Inactivation of HrtAB alters expression and secretion of S. aureus virulence factors
The hepatic hypervirulence of S. aureus ΔhrtA and ΔhssR suggests a tip in the balance of the bacteria-phagocyte interaction to favor S. aureus. S. aureus pathogenesis is characterized by the secretion of numerous virulence factors that defend against immune cell killing (Foster, 2005). Thus, we investigated whether ΔhrtA differs from wildtype in its secreted protein profile following growth in medium with or without hemin. As shown in Figure 6, exposure to hemin induced changes in the abundance of multiple secreted proteins in ΔhrtA compared to wildtype. In contrast, the complemented ΔhrtA mutant strain (ΔhrtA/phrtA) displayed a secreted protein profile similar to the wildtype strain (Figure 6). Mass spectrometry-based identification of the proteins overrepresented in the supernatants of ΔhrtA grown in hemin revealed the increased expression and/or secretion of at least 8 staphylococcal proteins. All proteins identified in this analysis are secreted or contain putative N-terminal secretion signals, eliminating the possibility that the integrity of the bacterial membrane is compromised upon inactivation of hrtA. The seven proteins found more abundantly in the supernatant of hemin-exposed S. aureus ΔhrtA were proteins with known roles in immunomodulation (exotoxin, exotoxin-3, -5 and -8) (Williams et al., 2000), inhibition of phagocyte recruitment (Map-w) (Chavakis et al., 2002), inhibition of opsonophagocytosis (fibrinogen binding protein) (Foster, 2005; Lee et al., 2004), and inhibition of neutrophil activation and chemotaxis (FLIPr) (Prat et al., 2006). Transcriptional analyses demonstrated that the increased presence of the majority of these virulence factors in the supernatants of heme-exposed ΔhrtA is due to increased transcription of the corresponding genes (Figure 6C). Together these data provide a potential mechanistic explanation for the decrease in phagocytes at hepatic sites of infection with ΔhrtA staphylococci. It is compelling to speculate that an increased expression of genes encoding for secreted proteins with known immunomodulatory functions is responsible for the hypervirulence of staphylococcal strains unable to sense and excrete surplus internalized heme.
Figure 6. Inactivation of the HrtAB system results in increased expression of secreted virulence factors.
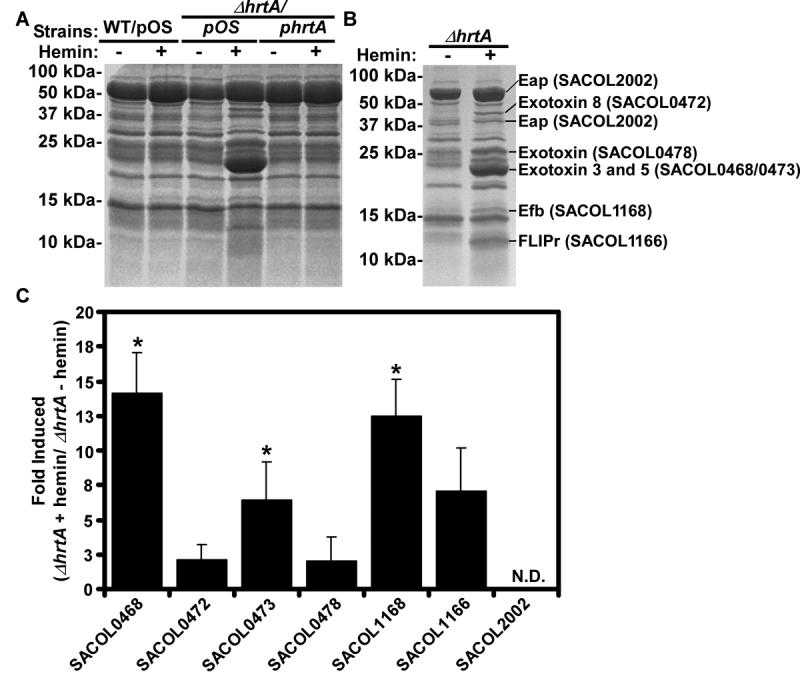
A) S. aureus wildtype (WT/pOS1), ΔhrtA mutant (ΔhrtA/pOS1), or ΔhrtA complemented strain (ΔhrtA/phrtA) were grown O/N at 37 °C with aeration for 15 hrs in RPMI supplemented with 0 or 1 μM hemin. Culture supernatants were collected, filtered, precipitated, and separated on 15% SDS-PAGE gels. Proteins were stained with colloidal blue. B) Indicated proteins were excised from the gel and subjected to mass spectrometry-based identification. The identities of the proteins are indicated with corresponding gene numbers from S. aureus strain COL shown in parentheses. C) Fold induction of the indicated genes as determined by transcriptional analyses comparing changes between ΔhrtA grown in the presence or absence of 1 μM hemin. Transcript levels not determined due to saturating levels of expression marked with N.D.. Experiments were performed in triplicate and asterisks denote statistical significance as determined by Student's t test (p<0.05).
HrtAB and HssRS are conserved across Gram positive pathogens
Genomic analyses of the hrt and hss loci indicate that these systems are highly conserved across Gram positive bacteria, including the important human pathogens Staphylococcus epidermidis, Bacillus anthracis, Listeria monocytogenes, and Enterococcus faecalis (Figure 7A). As an indirect measure of the functional conservation of these systems, we investigated whether S. epidermidis and B. anthracis adapt to hemin toxicity. Similar to S. aureus, S. epidermidis and B. anthracis adapt and avoid hemin toxicity when pre-exposed to sub-inhibitory concentrations of hemin (Figure S2). These functional data suggest that orthologous Hss and Hrt systems act across genera to coordinate a response to excess hemin exposure.
Figure 7. Model for the role of HrtAB and HssRS in S. aureus pathogenesis.
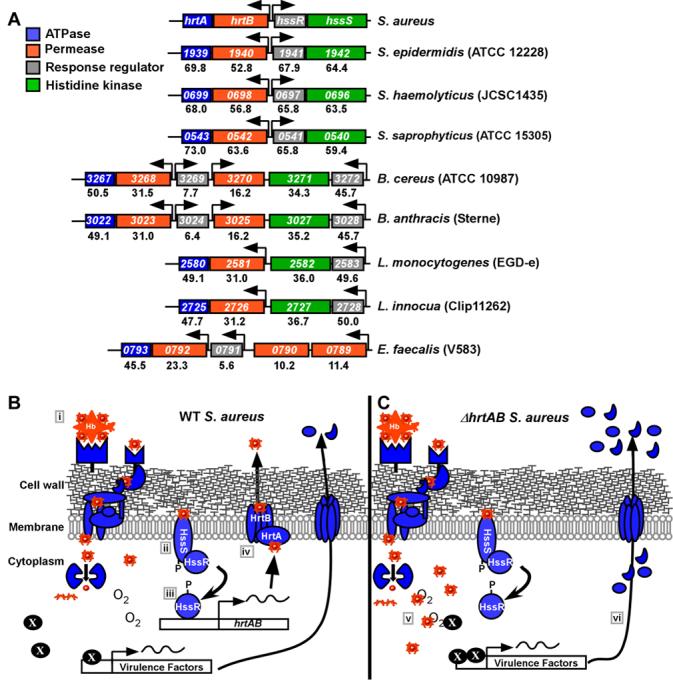
A) The Hrt and Hss systems are conserved across several Gram positive bacteria. Alignment of genomic sequences among Gram positive bacteria that contain orthologous hrtAB and hssRS systems. The numbers within each box represent corresponding gene numbers in the listed annotation. The numbers underneath each gene correspond to the percent amino acid identity to the representative S. aureus genes. Arrows denote the predicted direction of transcription. B) In S. aureus, heme internalized through cell wall anchored proteins (i), is sensed by HssS which subsequently activates HssR (ii). HssR then binds the promoter region upstream of hrtAB (iii), leading to increased expression and elaboration of the HrtAB efflux pump (iv). HrtAB then pumps surplus cytoplasmic heme out of the bacterium. C) Inactivation of hrtAB leads to the cytoplasmic accumulation of heme which increases cellular stress (v). Staphylococcal stress sensing systems are activated leading to an increase in the expression and/or secretion of virulence factors including exotoxins 3, 5 and 8, Map-w, fibronectin binding protein and FLIPr (vi), which increase liver-specific hypervirulence through inhibiting immune cell recruitment.
DISCUSSION
During commensal colonization of the skin, S. aureus is exposed to host tissues that are low in heme and heme binding proteins. However, once the initial colonization sites are breached, S. aureus encounters elevated levels of hemoglobin-containing erythrocytes and myoglobin-containing myocytes in the vasculature and musculature, respectively. Herein we describe the identification of HssRS, a staphylococcal two-component system responsible for sensing heme as a component of these abundant host proteins and modulating virulence. One model to explain the data described in this manuscript is presented in Figure 7. Upon erythrocyte and myocyte lysis, S. aureus encounters high concentrations of hemoglobin and myoglobin at the infection site. Due to the efficiency of the staphylococcal heme uptake systems (Mazmanian et al., 2003; Skaar et al., 2004), heme is removed from these proteins and rapidly transported into the bacterial cytoplasm. Upon transport into the bacterium, heme is recognized by the HssS histidine kinase resulting in the activation of HssR. Alternatively, HssS may indirectly sense heme through an as-yet-unidentified intermediary and subsequently activate HssR. Activated HssR then binds to the hrtAB promoter inducing the expression of HrtAB. Heme that is not mobilized for cellular iron or porphyrin needs is excreted via HrtAB resulting in the avoidance of heme toxicity. Our model envisions that S. aureus strains unable to elaborate HrtAB accumulate intracellular heme due to a block in heme excretion. In turn, the accumulation of intracellular heme activates staphylococcal stress sensing systems responsible for increasing the transcription of genes encoding for virulence factors with potent immunomodulatory functions. In all, the coordinated activity of Hss and Hrt allow S. aureus to sense internal host tissues, resulting in the tempering of virulence to avoid excessive host tissue damage.
The enhanced virulence exhibited by S. aureus ΔhrtA or ΔhssR could be due to a combination of factors. However, the observed hypervirulence correlates with a profound decrease in viable granulocytes in the infected liver. This decrease is potentially caused by the increased expression and secretion of Map-w, fibrinogen binding protein, FLIPr, and exotoxins 3, 5 and 8. Map-w is a staphylococcal factor that inhibits the interaction of ICAM-1 with integrins required for functional leukocyte adhesion systems, resulting in reduced phagocyte recruitment to the site of infection (Chavakis et al., 2005). Fibrinogen binding protein binds complement factor C3 and blocks its deposition on the bacterial cell surface, thereby inhibiting opsonization (Lee et al., 2004). FLIPr inhibits the neutrophil response to formyl peptide receptor inhibiting neutrophil activation and chemotaxis (Prat et al., 2006). Moreover, the staphylococcal exotoxins identified here belong to a family of cytotoxins with potent cytokine modulating characteristics (Williams et al., 2000). It is possible that the combined effect of the increased secretion of these virulence factors inhibits phagocyte migration to infected livers. The liver-specific nature of the increased virulence is potentially due to the abundance of heme in this organ.
The contribution of phagocytes to the avoidance of staphylococcal infections is exemplified in humans with chronic granulomatous disease (CGD), a rare inherited immunodeficiency that results in a defective nicotinamide dinucleotide phosphate (NADPH) oxidase complex (Segal et al., 2000). Phagocytes from CGD patients are defective in the generation of an effective oxidative burst (Segal et al., 2000). Hence, patients with CGD suffer from recurrent, life threatening infections by catalase positive microorganisms (Segal et al., 2000). A common infectious complication experienced by CGD patients is the development of hepatic-abscesses, which are most frequently caused by S. aureus (Lublin et al., 2002). These clinical data support our model that an impaired phagocytic response to the livers of animals infected with S. aureus ΔhrtA or ΔhssR is responsible for the observed hypervirulence and hepatic abscess formation reported here.
Numerous molecules that have been described as TCS activators can be found in vertebrates, however very few of these molecules are associated specifically with internal tissues of the host. Some examples of host specific molecules that activate TCS include antimicrobial peptides which activate PhoPQ of Salmonella typhimurium (Bader et al., 2005), and epinephrine/norepinephrine, which activate QseCB of E. coli O157:H7 (Clarke et al., 2006). Staphylococcal regulatory systems that alter virulence gene expression may be the most well studied area of staphylococcal pathogenesis; however host molecules that activate these systems are not well defined. The identification and functional characterization of HssRS as a heme sensing system fills a gap in our knowledge of the host molecule sensing systems of S. aureus. Although S. aureus is capable of endogenous heme production, HssRS is not activated in the absence of extracellular heme, suggesting that this system specifically recognizes host-derived heme. The identification of systems that contribute to S. aureus virulence factor production and immune cell modulation in the host could lead to the development of novel therapeutics targeting staphylococcal gene regulation. In light of the increased prevalence of S. aureus strains resistant to virtually all relevant antimicrobials, the design of novel therapeutics is paramount toward combating the inevitable increase in severe staphylococcal infections.
EXPERIMENTAL PROCEDURES
Bacterial Strains
S. aureus Newman, a human clinical isolate, was used in this study (Duthie and Lorenz, 1952). The ΔsrtA mutant strain has been previously described (Mazmanian et al., 2000; Mazmanian et al., 1999). Erythromycin cassette insertion mutants of the hrtA, hrtB, and hssS genes were obtained from the Phoenix (N) library, clones PhiNE 03177 (SAV2359), PhiNE 01762 (SAV2360), PhiNE 01562 (SAV2362), and PhiNE 07744 (SAV2362) (Bae et al., 2004). All the Phoenix (N) library mutants were transduced into Newman. The ΔhrtA and ΔhssR isogenic mutant strains were generated by deletion of the genes following a protocol described by Bae and Schneewind (Bae and Schneewind, 2005). To create a complementation vector coding for wildtype hrtA, the hrtAB intergenic region containing the predicted promoter sequence for hrtAB was fused to the hrtA coding sequence by polymerase chain reaction sequence overlap extension (PCR-SOE) (Horton et al., 1990). Details for the creation of these strains are available in supplemental information.
Analyses of Secreted Proteins
S. aureus cultures were grown O/N at 37 °C with shaking in 5 mls of RPMI containing 1 % casamino acids with or without 1 μM hemin. Cultures were then sedimented and the supernatant collected. 1.2 ml of each supernatant was then precipitated by adding 10% TCA (v/v) and incubating the samples for ∼ 15 hrs at 4 °C. The precipitated proteins were then sedimented and washed twice with 100 % ethanol. Proteins were dried and then resuspended with 30 μl of a SDS-loading buffer and boiled at 95 °C for 10 minutes. Protein samples were separated on 15 % SDS-PAGE gels and stained with colloidal blue (Invitrogen). Proteins of interest were excised from the gel, in-gel digested into peptides and the peptides analyzed by matrix assisted laser desorption/ionization, time-of-flight mass spectrometry (MALDI-TOF MS) and data-dependant TOF/TOF tandem MS/MS as described previously (Friedman et al., 2006). The resulting peptide mass maps and the associated fragmentation spectra were collectively used to interrogate S. aureus Mu50 sequences as described previously (Friedman et al., 2006).
Hemin Cytotoxicity Assays
S. aureus cultures were grown O/N at 37 °C with shaking in TSB with or without hemin. Cultures were then diluted 1:75 and inoculated into round-bottom 96-well plates in a final volume of 150 μl of medium supplemented with different concentrations of hemin. Cultures were grown at 37 °C with aeration for 2-3 hrs. Bacterial viability was determined by serial dilution and plating on solid agar.
XylE Reporter Assay
Details on the construction of the reporter vectors are available in supplemental methods. S. aureus strains harboring the appropriate reporter construct were inoculated into 500 μl of TSB containing 10 μg/ml chloramphenicol in 1.5 ml tubes. Cultures were grown O/N at 37°C with shaking. Bacteria were pelleted by centrifugation and spent medium was aspirated. The pellet was washed once with 500 μl of 20 mM potassium phosphate, pH 7.6, and resuspended in 150 μl of 100 mM potassium phosphate buffer, pH 8.0, 10% (v/v) acetone, 25 μg/ml lysostaphin. After 20 minute incubation at 37°C and 5 minute incubation on ice, samples were centrifuged at 20,000 g for 30 minutes at 4°C. 1-10 μl of supernatant was added to a 96-well plate and 200 μl of 100 mM potassium phosphate, pH 8.0, 0.2 mM pyrocatechol was added to each well. Formation of 2-hydroxymuconic semialdehyde was tracked by measuring the absorbance at 375 nm every minute for 30 minutes on a Varian MP 50 microplate reader. Protein concentration in samples was determined by BCA (Pierce). One unit of specific activity of XylE in a sample is defined as the formation of 1 nmol of 2-hydroxymuconic semialdehyde per minute per milligram of cellular protein at 30°C (Chien et al., 1999). Metalloporphyrins were purchased from Frontier Biosciences.
Histological Tissue Analysis
Parafin-embeded mouse tissues were stained with H&E. Sections were evaluated by a single pathologist (J. Iturregui, MD).
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Skaar lab and Drs. David G. Russell, Timothy L. Cover, Hank S. Seifert, Dean Ballard, and Clarence B. Creech for critical reading of this manuscript. We would also like to thank the Vanderbilt pathology core and Dr. David Friedman for technical assistance, Dr. Ambrose Cheung for the gift of pALC1639, Dr. Olaf Schneewind for the gift of the ΔsrtA strain and the pOS-1 plasmid, and Dr. Dominique Missiakas for Phoenix Library Derivatives. This research was supported by Vanderbilt University Medical Center Development funds, the Searle Scholars Program, and United States Public Health Service Grant AI69233 (EPS) and AI042284 (SJ) from the National Institute of Allergy and Infectious Diseases. EPS holds an Investigator in Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. VJT was supported by Ruth L. Kirschstein NRSA AI071487 postdoctoral fellowship, and DLS was supported by T32 HL069765, from the National Institute of Allergy and Infectious Diseases. P.M.D. and K.L.A. were supported by American Heart Association grant # 0535037N.
Abbreviations
- TCS
two-component system
- Hrt
heme regulated transporter
- Hss
heme sensor system
- WT
wildtype
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Anderson KL, Roberts C, Disz T, Vonstein V, Hwang K, Overbeek R, Olson PD, Projan SJ, Dunman PM. Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J. Bacteriol. 2006;188:6739–6756. doi: 10.1128/JB.00609-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader MW, Sanowar S, Daley ME, Schneider AR, Cho U, Xu W, Klevit RE, Le Moual H, Miller SI. Recognition of antimicrobial peptides by a bacterial sensor kinase. Cell. 2005;122:461–472. doi: 10.1016/j.cell.2005.05.030. [DOI] [PubMed] [Google Scholar]
- Bae T, Banger AK, Wallace A, Glass EM, Aslund F, Schneewind O, Missiakas DM. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc. Natl. Acad. Sci. U S A. 2004;101:12312–12317. doi: 10.1073/pnas.0404728101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae T, Schneewind O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid. 2005 doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Beier D, Gross R. Regulation of bacterial virulence by two-component systems. Curr. Opin. Microbiol. 2006;9:143–152. doi: 10.1016/j.mib.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Bernheimer AW, Avigad LS, Grushoff P. Lytic effects of staphylococcal alpha-toxin and delta-hemolysin. J. Bacteriol. 1968;96:487–491. doi: 10.1128/jb.96.2.487-491.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff M, Dunman P, Kormanec J, Macapagal D, Murphy E, Mounts W, Berger-Bachi B, Projan S. Microarray-based analysis of the Staphylococcus aureus sigmaB regulon. J. Bacteriol. 2004;186:4085–4099. doi: 10.1128/JB.186.13.4085-4099.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner S, Monteil H, Prevost G. Regulation of virulence determinants in Staphylococcus aureus: complexity and applications. FEMS Microbiol. Rev. 2004;28:183–200. doi: 10.1016/j.femsre.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Brook I. Secondary bacterial infections complicating skin lesions. J. Med. Microbiol. 2002;51:808–812. doi: 10.1099/0022-1317-51-10-808. [DOI] [PubMed] [Google Scholar]
- Bullen JJ, Griffiths E. Iron and Infection: Molecular, Physiological and Clinical Aspects. John Wiley and Sons; New York: 1999. [Google Scholar]
- Chavakis T, Hussain M, Kanse SM, Peters G, Bretzel RG, Flock JI, Herrmann M, Preissner KT. Staphylococcus aureus extracellular adherence protein serves as anti-inflammatory factor by inhibiting the recruitment of host leukocytes. Nat. Med. 2002;8:687–693. doi: 10.1038/nm728. [DOI] [PubMed] [Google Scholar]
- Chavakis T, Wiechmann K, Preissner KT, Herrmann M. Staphylococcus aureus interactions with the endothelium: the role of bacterial “secretable expanded repertoire adhesive molecules” (SERAM) in disturbing host defense systems. Thromb. Haemost. 2005;94:278–285. doi: 10.1160/TH05-05-0306. [DOI] [PubMed] [Google Scholar]
- Cheung AL, Bayer AS, Zhang G, Gresham H, Xiong YQ. Regulation of virulence determinants in vitro and in vivo in Staphylococcus aureus. FEMS Immunol. Med. Microbiol. 2004;40:1–9. doi: 10.1016/S0928-8244(03)00309-2. [DOI] [PubMed] [Google Scholar]
- Chien Y, Manna AC, Projan SJ, Cheung AL. SarA, a global regulator of virulence determinants in Staphylococcus aureus, binds to a conserved motif essential for sar-dependent gene regulation. J. Biol. Chem. 1999;274:37169–37176. doi: 10.1074/jbc.274.52.37169. [DOI] [PubMed] [Google Scholar]
- Clarke MB, Hughes DT, Zhu C, Boedeker EC, Sperandio V. The QseC sensor kinase: a bacterial adrenergic receptor. Proc. Natl. Acad. Sci. U S A. 2006;103:10420–10425. doi: 10.1073/pnas.0604343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiss A. Iron metabolism in reticuloendothelial cells. Semin. Hematol. 1983;20:81–90. [PubMed] [Google Scholar]
- Dunman PM, Murphy E, Haney S, Palacios D, Tucker-Kellogg G, Wu S, Brown EL, Zagursky RJ, Shlaes D, Projan SJ. Transcription profiling-based identification of Staphylococcus aureus genes regulated by the agr and/or sarA loci. J. Bacteriol. 2001;183:7341–7353. doi: 10.1128/JB.183.24.7341-7353.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duthie ES, Lorenz LL. Staphylococcal coagulase; mode of action and antigenicity. J. Gen. Microbiol. 1952;6:95–107. doi: 10.1099/00221287-6-1-2-95. [DOI] [PubMed] [Google Scholar]
- Everse J, Hsia N. The toxicities of native and modified hemoglobins. Free Radic. Biol. Med. 1997;22:1075–1099. doi: 10.1016/s0891-5849(96)00499-6. [DOI] [PubMed] [Google Scholar]
- Foster TJ. Immune evasion by staphylococci. Nat. Rev. Microbiol. 2005;3:948–958. doi: 10.1038/nrmicro1289. [DOI] [PubMed] [Google Scholar]
- Fowler VG, Jr., Miro JM, Hoen B, Cabell CH, Abrutyn E, Rubinstein E, Corey GR, Spelman D, Bradley SF, Barsic B, et al. Staphylococcus aureus endocarditis: a consequence of medical progress. JAMA. 2005;293:3012–3021. doi: 10.1001/jama.293.24.3012. [DOI] [PubMed] [Google Scholar]
- Fridkin SK, Hageman JC, Morrison M, Sanza LT, Como-Sabetti K, Jernigan JA, Harriman K, Harrison LH, Lynfield R, Farley MM. Methicillin-resistant Staphylococcus aureus disease in three communities. N. Engl. J. Med. 2005;352:1436–1444. doi: 10.1056/NEJMoa043252. [DOI] [PubMed] [Google Scholar]
- Friedman DB, Stauff DL, Pishchany G, Whitwell CW, Torres VJ, Skaar EP. Staphylococcus aureus Redirects Central Metabolism to Increase Iron Availability. PLoS Pathog. 2006;2 doi: 10.1371/journal.ppat.0020087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM, Cai ZL, Ho SN, Pease LR. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques. 1990;8:528–535. [PubMed] [Google Scholar]
- Kobayashi N, Nishino K, Yamaguchi A. Novel macrolide-specific ABC-type efflux transporter in Escherichia coli. J. Bacteriol. 2001;183:5639–5644. doi: 10.1128/JB.183.19.5639-5644.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LY, Hook M, Haviland D, Wetsel RA, Yonter EO, Syribeys P, Vernachio J, Brown EL. Inhibition of complement activation by a secreted Staphylococcus aureus protein. J Infect Dis. 2004;190:571–579. doi: 10.1086/422259. [DOI] [PubMed] [Google Scholar]
- Lublin M, Bartlett DL, Danforth DN, Kauffman H, Gallin JI, Malech HL, Shawker T, Choyke P, Kleiner DE, Schwartzentruber DJ, et al. Hepatic abscess in patients with chronic granulomatous disease. Ann. Surg. 2002;235:383–391. doi: 10.1097/00000658-200203000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong TT, Dunman PM, Murphy E, Projan SJ, Lee CY. Transcription Profiling of the mgrA Regulon in Staphylococcus aureus. J. Bacteriol. 2006;188:1899–1910. doi: 10.1128/JB.188.5.1899-1910.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher T, Helmann JD, Unden G. Stimulus perception in bacterial signal-transducing histidine kinases. Microbiol. Mol. Biol. Rev. 2006;70:910–938. doi: 10.1128/MMBR.00020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Liu G, Jensen ER, Lenoy E, Schneewind O. Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. U S A. 2000;97:5510–5515. doi: 10.1073/pnas.080520697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Liu G, Ton-That H, Schneewind O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science. 1999;285:760–763. doi: 10.1126/science.285.5428.760. [DOI] [PubMed] [Google Scholar]
- Mazmanian SK, Skaar EP, Gaspar AH, Humayun M, Gornicki P, Jelenska J, Joachmiak A, Missiakas DM, Schneewind O. Passage of heme-iron across the envelope of Staphylococcus aureus. Science. 2003;299:906–909. doi: 10.1126/science.1081147. [DOI] [PubMed] [Google Scholar]
- Nagasawa S, Ishige K, Mizuno T. Novel members of the two-component signal transduction genes in Escherichia coli. J. Biochem. (Tokyo) 1993;114:350–357. doi: 10.1093/oxfordjournals.jbchem.a124180. [DOI] [PubMed] [Google Scholar]
- Novick RP. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 2003;48:1429–1449. doi: 10.1046/j.1365-2958.2003.03526.x. [DOI] [PubMed] [Google Scholar]
- Prat C, Bestebroer J, de Haas CJ, van Strijp JA, van Kessel KP. A new staphylococcal anti-inflammatory protein that antagonizes the formyl peptide receptor-like 1. J. Immunol. 2006;177:8017–8026. doi: 10.4049/jimmunol.177.11.8017. [DOI] [PubMed] [Google Scholar]
- Richardson AR, Dunman PM, Fang FC. The nitrosative stress response of Staphylococcus aureus is required for resistance to innate immunity. Mol. Microbiol. 2006;61:927–939. doi: 10.1111/j.1365-2958.2006.05290.x. [DOI] [PubMed] [Google Scholar]
- Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 2000;79:170–200. doi: 10.1097/00005792-200005000-00004. [DOI] [PubMed] [Google Scholar]
- Skaar EP, Humayun M, Bae T, DeBord KL, Schneewind O. Iron-source preference of Staphylococcus aureus infections. Science. 2004;305:1626–1628. doi: 10.1126/science.1099930. [DOI] [PubMed] [Google Scholar]
- Skaar EP, Schneewind O. Iron-regulated surface determinants (Isd) of Staphylococcus aureus: stealing iron from heme. Microbes Infect. 2004;6:390–397. doi: 10.1016/j.micinf.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Taylor RK, Hall MN, Enquist L, Silhavy TJ. Identification of OmpR: a positive regulatory protein controlling expression of the major outer membrane matrix porin proteins of Escherichia coli K-12. J. Bacteriol. 1981;147:255–258. doi: 10.1128/jb.147.1.255-258.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres VJ, Pishchany G, Humayun M, Schneewind O, Skaar EP. IsdB is a staphylococcal hemoglobin receptor required for heme-iron utilization. J. Bacteriol. 2006;188:8421–9. doi: 10.1128/JB.01335-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeiren CL, Pluym M, Mack J, Heinrichs DE, Stillman MJ. Characterization of the heme binding properties of Staphylococcus aureus IsdA. Biochemistry. 2006;45:12867–12875. doi: 10.1021/bi0607711. [DOI] [PubMed] [Google Scholar]
- Weinrick B, Dunman PM, McAleese F, Murphy E, Projan SJ, Fang Y, Novick RP. Effect of mild acid on gene expression in Staphylococcus aureus. J Bacteriol. 2004;186:8407–8423. doi: 10.1128/JB.186.24.8407-8423.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim HF, Vos MC, Ott A, van Belkum A, Voss A, Kluytmans JA, van Keulen PH, Vandenbroucke-Grauls CM, Meester MH, Verbrugh HA. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet. 2004;364:703–705. doi: 10.1016/S0140-6736(04)16897-9. [DOI] [PubMed] [Google Scholar]
- Williams RJ, Ward JM, Henderson B, Poole S, O'Hara BP, Wilson M, Nair SP. Identification of a novel gene cluster encoding staphylococcal exotoxin-like proteins: characterization of the prototypic gene and its protein product, SET1. Infect. Immun. 2000;68:4407–4415. doi: 10.1128/iai.68.8.4407-4415.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.