

Abstract
Hormonal signals activate trimeric G proteins by promoting exchange of GTP for GDP bound to the G protein’s α subunit (Gα). Here we describe a point mutation that impairs this activation mechanism in the α subunit of Gs, producing an inherited disorder of hormone responsiveness. Biochemical analysis reveals an activation defect that is paradoxically intensified by hormonal and other stimuli. By substituting histidine for a conserved arginine residue, the mutation removes an internal salt bridge (to a conserved glutamate) that normally acts as an intramolecular hasp to maintain tight binding of the γ-phosphate of GTP. In its basal, unperturbed state, the mutant αs binds guanosine 5′-[γ-thio]triphosphate (GTP[γS]), a GTP analog, slightly less tightly than does normal αs, but (in the GTP[γS]-bound form) can stimulate adenylyl cyclase. The activation defect becomes prominent only under conditions that destabilize binding of guanine nucleotide (receptor stimulation) or impair the ability of αs to bind the γ-phosphate of GTP (cholera toxin, AlF4− ion). Although GDP release is usually the rate-limiting step in nucleotide exchange, the biochemical phenotype of this mutant αs indicates that efficient G protein activation by receptors and other stimuli depends on the ability of Gα to clasp tightly the GTP molecule that enters the binding site.
Keywords: trimeric G proteins, Gs, pseudohypoparathyroidism, guanosine triphosphate, conformational change
The α subunits of heterotrimeric G proteins (Gα) are GTP-dependent molecular switches that relay signals from cell surface receptors to effector enzymes and ion channels (1, 2). Hormone-activated receptors turn αs on by catalyzing release of otherwise tightly bound GDP, which is rapidly replaced by GTP. The switch reverts to the “off” position when its intrinsic GTPase activity converts bound GTP to GDP. In this cycle the activity of Gα is directly controlled by the presence or absence of the γ-phosphate of bound guanine nucleotide. Although crystal structures of α-5′-[γ-thio]triphosphate (GTP[γS]) and α-GDP are similar (3–5), limited regions of Gα (switches 1, 2, and 3) undergo a GTP-dependent conformational change that is induced by interactions between Gα residues and the γ-phosphate of GTP.
This conformational change controls interactions of Gα with receptors, βγ, effectors, and regulator of G protein signaling proteins. The γ-phosphate also serves as a driving force to ensure unidirectionality of the Gα cycle. Gα binds GTP much more tightly than GDP because its interactions with the γ-phosphate provide additional binding energy. Consequently, GTP does not dissociate from Gα before it can be hydrolyzed to GDP.
In turning a G protein on, the hormone–receptor complex not only accelerates dissociation of GDP but also apparently accelerates GTP binding (2, 6), increasing the preference of Gα for binding GTP, relative to GDP. GDP competes against GTP more effectively in the G protein’s resting state than when GDP–GTP exchange is accelerated by receptor stimulation (7). Indeed, the concentration of GTP[γS] required for association with unstimulated pure Gα (8, 9) is much greater than that required for association with the receptor-activated G protein trimer (10).
We recently reported (11) that a new αs mutation, found in patients with pseudohypoparathyroidism, type I (PHP-I), impairs the ability of the mutant αs to mediate hormonal stimulation of cAMP accumulation in transiently transfected cells. Here we report a detailed biochemical analysis of the mutant αs phenotype, which shows that both GDP–GTP exchange and GTP-induced conformational change depend on the ability of Gα to clasp GTP tightly in the guanine nucleotide binding pocket. The mutation, which replaces Arg-231 with histidine, causes an activation defect that is paradoxically intensified by hormonal and other stimuli. Thus the mutant αs sabotages its own activation by making the activation process work against itself.
MATERIALS AND METHODS
Cell Culture and Transfection.
S49 cyc− cells, maintained in DMEM containing 10% horse serum, were transfected with the retroviral vector pMV7 containing DNA encoding hemagglutinin (HA)-tagged wild-type (wt) (12) or mutant αs (αs-R231H), and stable clones were selected as described (12, 13). The cyc− clones studied in these experiments expressed nearly identical amounts of normal or mutant αs. HEK293 cells were maintained in DMEM containing 10% fetal calf serum. Stable clones expressing the β2-adrenergic receptor (AR) (14) were cotransfected by calcium phosphate precipitation with pSVneo and the pcDNAI vector containing either HA-tagged wt or mutant αs, and stable clones were selected (15). COS-7 cells, maintained in DME-H21 medium containing 10% calf serum, were transiently transfected by the DEAE/adenovirus method (16) with pcDNAI vector containing either HA-tagged wt or mutant αs. Membranes of S49 cyc− cells or COS-7 cells were prepared after nitrogen cavitation as described (17).
Immunofluorescence.
Localization of HA-tagged αs and β2-AR was assessed in HEK293 cells as described (14, 15).
Purification of αs.
αs was purified from cytosol of Sf9 cells infected with baculovirus encoding αs (18). Sf9 cells (1.5 × 106 cells per ml), maintained in Sf-900II medium at 28°C, were infected with baculoviruses encoding αs (three plaque-forming units per cell). After cell lysis using nitrogen cavitation, the supernatant fraction was sequentially chromatographed on columns of HiTrap Q (Pharmacia; 5 ml bed volume × 2) (13), Econo-Pac HTP (Bio-Rad; 5 ml bed volume) with a potassium phosphate gradient (19), and Mono Q (1 ml bed volume) (20) in the absence of detergents.
GTP[γS] Binding and GTPase Assays.
GTP[γS] binding and GTP hydrolysis were quantitated as described (13, 20). Apparent on rates (kapp) of GTP[γS] binding were determined as described (19).
cAMP Assay.
cAMP accumulation in intact S49 cyc− cells (21) and cAMP synthesis by recombinant αs in membranes of S49 cyc− cells (13) were assayed as described. Before the latter assay, the αs proteins were incubated with 100 μM GTP[γS] for 60 min.
Receptor Binding Assay.
Competition between isoproterenol and [125I]pindolol for binding to β-adrenergic receptors was determined in membranes of S49 cyc− cells expressing HA-tagged wt or mutant αs as described (22, 23).
Trypsin Protection Assay.
Hormonal activation of αs in COS-7 cell membranes was assessed as described for rhodopsin-dependent activation of αt (16), with modifications. Membranes (0.2 mg/ml) of COS-7 cells expressing HA-αs or HA-αs-R231H with the β2-AR and G protein β2 and γ2 subunits were incubated at 22°C for 3 min in buffer containing 20 mM Tris⋅HCl (pH 7.5), 100 mM NaCl, 2 mM MgCl2, 0.1 mM EDTA, and 1 mM dithiothreitol, in the presence or in the absence of 10 μM isoproterenol before the reaction was initiated by adding 10 μM GTP[γS]. At the times indicated, the samples were mixed with 0.6 mg/ml trypsin, 1% Lubrol-PX (Sigma), and 0.5 mM GDP and further incubated on ice for 60 min. Trypsin-resistant fragments of αs were visualized and quantitated by SDS/PAGE and Western blot analysis using 12CA5 antibody (12).
To assess protection of recombinant protein from trypsin, 0.7 μM αs or αs-R231H were incubated at 22°C for 60 min in buffer containing 20 mM Na-Hepes, 2 mM MgSO4, 0.1 mM EDTA, 3 mM 2-mercaptoethanol, 0.025% Lubrol, and the indicated guanine nucleotides. Samples were then incubated on ice for 60 min with 0.1 mg/ml of trypsin and trypsin-resistant fragments of αs were visualized by SDS/PAGE followed by Coomassie blue staining.
RESULTS
R231H Mutation Impairs Activation.
Activation of αs-R231H is impaired in intact cells, as assessed after stable expression in S49 cyc− cells, which genetically lack endogenous αs. Stimulation of cAMP accumulation by isoproterenol and cholera toxin was reduced by 80% relative to cAMP accumulation in cells expressing similar amounts of αs-wt (Fig. 1A). The mutant protein’s abundance (not shown) and plasma membrane localization (see below) were similar to αs-wt, however.
Figure 1.
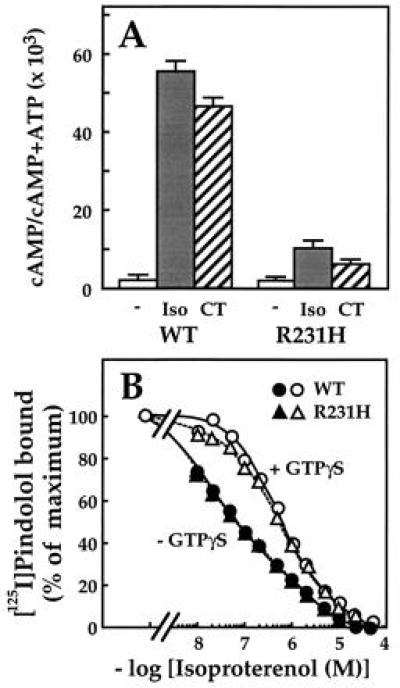
cAMP accumulation and β-adrenergic receptor binding in S49 cyc− cells transfected with wild type or mutant αs. (A) cAMP accumulation in intact cells. S49 cyc− cells stably transfected with HA-αs or HA-αs-R231H were incubated at 37°C for 30 min with 100 nM 3-isobutyl-1-methylxanthine and 10 μM isoproterenol (▪) or no drug (□ and ▨) and cAMP accumulation was measured (21). Cholera toxin (1 μg/ml; ▨) was added to the culture medium 3 h before adding IBMX. Values represent mean ± SD of triplicate determinations. (B) Competition between isoproterenol and [125I]pindolol for binding the β-adrenergic receptor. Membranes (0.15 mg/ml) of S49 cyc− cells stably transfected with HA-αs or HA-αs-R231H were incubated with [125I]pindolol (72 pM) and the indicated concentrations of isoproterenol in the presence (open symbols) or absence (filled symbols) of 30 μM GTP[γS]; incubations and binding assays were performed as described (22).
Immunofluorescence showed that the R231H mutation blocks a second effect of αs activation, agonist-induced translocation from plasma membrane to cytosol (Fig. 2). Stimulation of the β2-AR in HEK293 cells stably expressing the receptor and HA-tagged αs caused αs-wt (15), but not the mutant protein, to appear in cytosol; the agonist caused the β2AR to translocate normally (14, 15) in cells expressed either wt or mutant αs. Cholera toxin also failed to cause the cytosolic translocation of αs-R231H that was observed with αs-wt (15) (not shown).
Figure 2.
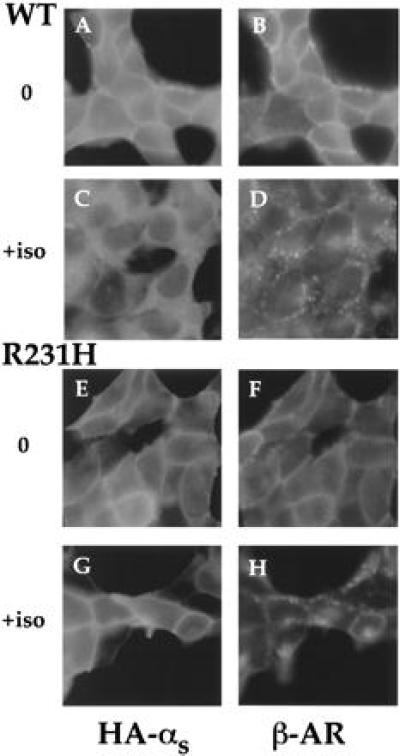
Immunofluorescence of wt or mutant αs. HEK293 cells stably expressing the β2-AR and HA-αs or HA-αs-R231H were incubated with (C, D, G, and H) or without (A, B, E, and F) 10 μM isoproterenol for 20 min and then fixed with 4% formaldehyde. αs was detected by incubation with mAb 12CA5, followed by fluorescein isothiocyanate-conjugated anti-mouse antibody (15). The β2-AR was detected by incubation with a polyclonal rabbit antiserum against its C terminus (14), followed by Texas red-conjugated anti-rabbit antibody (15). Images were visualized as described (14, 15).
Despite its loss of function, αs-R231H interacts normally with receptors, as assessed by its ability to enhance the affinity of the β2-AR for binding the β-adrenergic agonist, isoproterenol. In membranes of cells expressing αs-wt, the receptor shows high affinity for binding the agonist, and GTP[γS], a hydrolysis-resistant analog of GTP, abolishes high affinity binding (Fig. 1B), as described (22, 23). The high affinity binding of agonist is thought to reflect interaction of the receptor with Gs in the empty state, and is prevented by either binding of guanine nucleotide or absence of αs (e.g., in cyc− membranes). The ability of αs-R231H to increase the affinity of agonist binding was identical to that of αs-wt (Fig. 1B), indicating that the receptor-Gs interaction is largely intact. Because receptor binding is measured at equilibrium, however, this result does not necessarily reflect possible changes in agonist-induced rates of GDP dissociation or GTP[γS] binding. In addition, this assay is not a valid measure of conformational change in Gα, because both GDP and GTP[γS] abolish high affinity binding (24).
Behavior of Pure αs-R231H Under Basal Conditions.
Despite its functional defect in cells, pure recombinant αs-R231H can bind GTP and undergo GTP-induced conformational change. GTP[γS] associated with αs-R231H almost as rapidly as with αs-wt (kapp = 0.13 min−1 vs. 0.28 min−1; Fig. 3A). Because the rate of association of GTP[γS] is limited by dissociation of GDP, this result suggests that the R231H mutation does not accelerate dissociation of GDP from αs.
Figure 3.
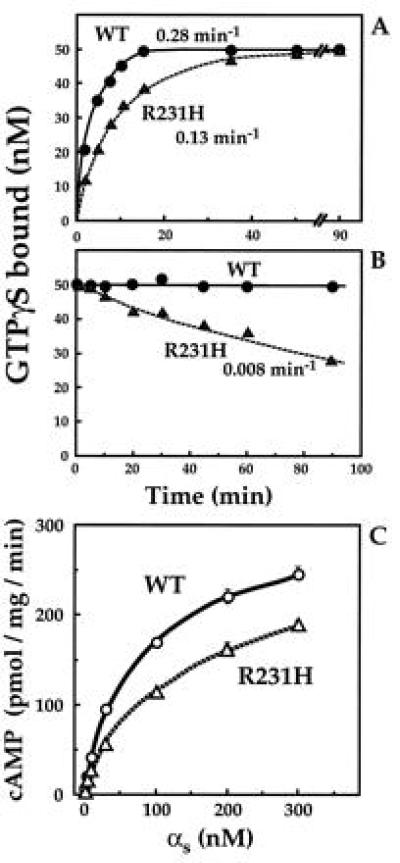
Biochemical properties of recombinant wt and mutant αs. (A) Rates of GTP[γS] binding. αs or αs-R231H (• and ▴, respectively; 50 nM each) were incubated at 22°C with 1 μM [35S]GTP[γS] (3 × 105 cpm/pmol). At the times indicated, the reaction was terminated and GTP[γS] binding was quantitated by filtration on nitrocellulose filters (13, 20) and apparent on-rates of GTP[γS] binding (kapp) were calculated (19). (B) Rates of dissociation of GTP[γS]. αs or αs-R231H (• and ▴, respectively; 50 nM each) were incubated at 22°C with 1 μM [35S]GTP[γS] as described in A for 45 min. Dissociation of bound [35S]GTP[γS] was assessed by adding 200 μM unlabeled GTP[γS] (at time zero in B). At the times indicated, the reaction was terminated and GTP[γS] binding was quantitated as described in A. (C) cAMP synthesis stimulated by different concentrations of αs or αs-R231H in the presence of GTP[γS]. Reactions were conducted at 22°C for 15 min in 50 μl volumes containing 15 μg cyc− membranes, as described (13). Before the assay, the αs proteins were incubated with 100 μM GTP[γS] for 60 min.
Although the mutation apparently does not destabilize binding of GDP, it does impair the stability of GTP[γS] binding. Although GTP[γS] did not measurably dissociate from αs-wt (19), GTP[γS] dissociated from αs-R231H at a low but easily measurable rate (0.008 min−1; Fig. 3B), as assessed by measuring the rate at which nonradioactive GTP[γS] (200 μM) replaced [35S]GTP[γS] bound to recombinant protein.
αs-R231H can nonetheless assume an active conformation, as indicated by resistance to proteolysis and ability to activate effector. When activated by GTP[γS], Gα proteins are cleaved by trypsin near their N termini but the proteolytic products are resistant to further proteolysis. GTP[γS] protected αs-R231H and αs-wt from trypsin, while GDP did not (Fig. 4C). We also tested activation of adenylyl cyclase by adding αs to cyc− membranes. In the presence of GTP[γS], αs-R231H activated adenylyl cyclase almost as effectively as αs-wt, over αs concentrations from 0–300 nM (Fig. 3C).
Figure 4.
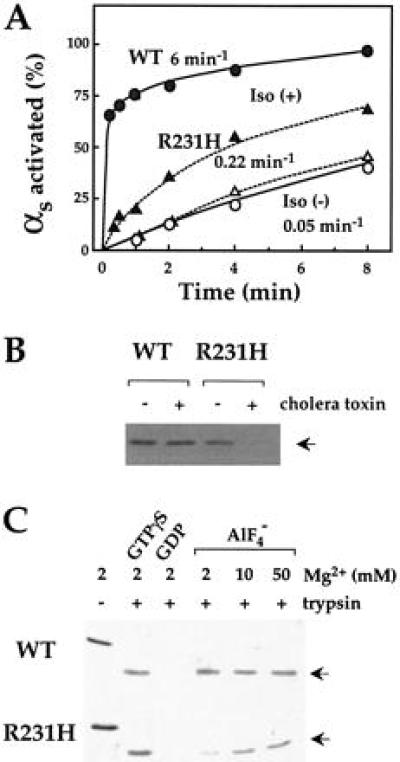
Effect of activation on tryptic cleavage of wt and mutant αs. (A) Receptor dependent activation of wt and mutant αs. Membranes (0.2 mg/ml) of COS-7 cells expressing recombinant HA-αs (•) or HA-αs-R231H (▴) plus the β2-AR and G protein β2 and γ2 subunits were incubated at 22°C with (filled symbols) or without (open symbols) 10 μM isoproterenol plus 10 μM GTP[γS]. At the times indicated, the reaction was terminated and samples were treated with trypsin (0.6 mg/ml) as described in Materials and Methods. Trypsin-resistant fragments of αs were visualized and quantitated by Western blot analysis using 12CA5 antibody (12). (B) Effect of modification by cholera toxin on protection by GTP[γS] against cleavage by trypsin. HEK293 cells stably transfected with HA-αs or HA-αs-R231H were cultured in the absence or in the presence of 1 μg/ml of cholera toxin for 3 h. Membranes were incubated with 10 μM GTP[γS] at 22°C for 10 min. Samples were incubated with trypsin (10 μg/ml) and trypsin-resistant fragments of αs (indicated by arrow) were visualized by Western blot analysis as described in A. (C) Effect of GTP[γS] and GDP/AlF4− on tryptic cleavage. αs or αs-R231H (0.7 μM each) were incubated with 10 μM GTP[γS], 10 μM GDP, or 10 μM GDP plus 20 μM AlCl3 and 10 mM NaF at 22°C for 60 min. Samples were further incubated in the absence or presence of trypsin (0.1 mg/ml) on ice for 60 min and trypsin-resistant fragments of αs (arrows) were visualized by SDS/PAGE followed by Coomassie blue staining.
The R231H mutation does not impair the interaction of αs with βγ, as assessed by measuring the ability of βγ to reduce the rate of association of GTP[γS] with purified αs. βγ inhibited the apparent rates of association of GTP[γS] with αs-wt and αs-R231H at similar concentrations and to a similar extent (data not shown).
Activation Defect of αs-R231H.
Despite normal activation by GTP[γS] under basal conditions in vitro, αs-R231H showed defective activation by receptors, cholera toxin, and AlF4−.
Because isoproterenol failed to stimulate cAMP accumulation or translocation to the cytosol in intact cells expressing αs-R231H, we asked more directly whether the mutation impairs receptor-mediated activation of αs. As assessed by a trypsin protection assay (16) in membranes from COS-7 cells cotransfected with the β2-AR and Gs subunits, isoproterenol promoted binding of GTP[γS] to αs-R231H, but at a rate 30-fold less than αs-wt (Fig. 4A). In contrast, GTP[γS] protected mutant and wt αs at similar (slower) rates in the basal state, in the absence of isoproterenol.
Because cholera toxin failed to stimulate cAMP accumulation in intact cells expressing αs-R231H, we used the trypsin protection assay to test whether R231H mutation impairs activation by cholera toxin. HEK293 cells stably expressing αs-wt or αs-R231H were cultured in the presence or absence of cholera toxin. Treatment with cholera toxin prevented GTP[γS] from protecting αs-R231H but not αs-wt from trypsin (Fig. 4B). The toxin catalyzed ADP ribosylation of αs-R231H to the same extent as αs-wt, however, in membranes exposed to activated toxin and radioactive NAD+ (result not shown).
AlF4−, which activates Gα by mimicking the γ-phosphate of GTP, protected pure recombinant αs-R231H only weakly from tryptic proteolysis (Fig. 4C), in contrast to αs-wt. Because Gα crystal structures show that Mg2+ links AlF4− to Gα residues in the nucleotide binding pocket (25, 26), we tested the effect of increasing the Mg2+ concentration. High concentrations of Mg2+ restored protection (Fig. 4C). This result was in accord with measurements of adenylyl cyclase activity stimulated by αs-R231H in cyc− membranes; although the mutant protein stimulated cAMP synthesis weakly in the presence of AlF4−, increasing the Mg2+ concentration restored activation to a level comparable to that seen with AlF4−-stimulated αs-wt (not shown).
DISCUSSION
In this report we show that Gα activation requires a tight grasp of the protein on bound GTP. The R231H mutation causes a conditional defect that is intensified by activating stimuli. In its basal state, activation of αs-R231H is almost normal, because the mutant protein can assume an active conformation, even though it binds GTP[γS] less rapidly and less tightly than does αs-wt. Upon stimulation by receptor, cholera toxin, or AlF4−, however, the activation defect becomes much more prominent. Here we discuss how the R231H mutation impairs tight binding of GTP, how this causes a conditional activation defect, and implications of these findings for PHP-I.
An Intramolecular Hasp.
R231 in αs is cognate to R204 of αt, which is located in the switch 2 region (the α2 helix) of Gα. Upon binding of GTP, switch 2 undergoes a conformational change, forming a coordinated complex with residues in the α3 helix and the switch 3 region, as previously noted (3, 4). R204 in αt, and by inference R231 in αs, is one of the residues that interacts with α3, forming a salt bridge with a conserved glutamate (E241 in αt, E268 in αs) in the α3 helix. The biochemical phenotype of αs-R231H suggests that this salt bridge serves as an intramolecular hasp to fasten switch 2 to the α3 helix; this hasp helps Gα to hold GTP more tightly and to maintain an active conformation (Fig. 5).
Figure 5.
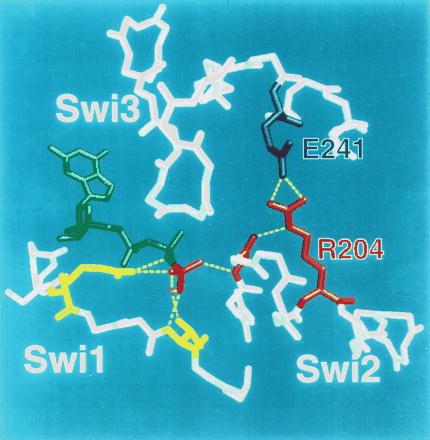
The arginine-glutamate hasp of Gα. Switch (Swi) regions 1–3 in the crystal structure of αt⋅GTP[γS] (3) are shown in relation to bound GTP (green; γ-phosphate colored red) and Mg2+ ion (magenta). Except for side chains of key amino acids, only main chains are depicted. The γ-phosphate of GTP (red) is linked to two amino acids (yellow, not labeled) in switch 1; these include R174 (on the left side of the picture) and T177. R174 corresponds to R201 of αs, the target of ADP ribosylation by cholera toxin. T177 (T 204 in αs) is linked to the γ-phosphate via bound Mg2+ (magenta). The γ-phosphate is linked to switch 2 by the main chain amide of G199 in αt (red, not labeled; corresponding to G226 in αs). The intramolecular hasp is the salt bridge between R204 (red; cognate to R231 in αs) in switch 2 (the α2 helix) and E241 (blue; E268 of αs) in α3. The hasp helps to maintain both the tight binding of GTP and the GTP-induced active conformation by fastening switch 2 and α3, thus reinforcing linkage of the glycine residue in switch 2 to the γ-phosphate of GTP.
In keeping with the postulated essential function of the intramolecular hasp in G protein signaling, this arginine-glutamate pair is completely conserved among Gα proteins. In addition, mutational replacement of the glutamate member of this pair impairs G protein signaling in Saccharomyces cerevisiae. The E364K mutation in the yeast Gα subunit (E364 is cognate to E241 in αt) makes yeast cells resistant to otherwise lethal doses of pheromones that act on G protein-coupled receptors (27). Biochemical analysis (David Stone, personal communication) suggests that the putative hasp in the yeast Gα stabilizes binding of GTP and the GTP-induced active conformation: The yeast mutant binds GTP[γS] less tightly at low concentrations of Mg2+ than does the wt protein.
The position of R204 in the crystal structure of αt also explains why mutation of the cognate arginine in αs does not prevent the mutant protein from interacting normally with βγ, the β2-AR, or the effector. The side chain of this arginine does not interact with βγ in crystals of αtβγ-GDP, despite its location in the region that provides most of the Gα surface for interacting with βγ (28). In the same crystals (28), the location of this arginine relative to the presumed plane of the plasma membrane (28–30) makes an interaction with the receptor very unlikely. Finally, participation of the arginine in a salt bridge with its paired glutamate (3, 4) should prevent it from forming part of an effector-interacting surface.
The Activation Defect.
Absence of the intramolecular hasp loosens the grip of αs-R231H on GTP, slowing the rate at which the mutant protein binds and is activated by GTP[γS] (Fig. 3A). In response to stimulation by cholera toxin, AlF4−, or hormone receptor, the activation defect becomes much more severe (Figs. 1A, 2, and 4). This is because each of these stimuli activates αs by a mechanism (different for each stimulus; see below) that also destabilizes the binding of guanine nucleotide by normal or mutant αs. Synergy between this stimulus-induced instability and the intrinsic instability of GTP binding in αs-R231H causes conditional exacerbation of the activation defect because the intramolecular hasp is not available to reinforce binding of the γ-phosphate.
From crystal structures we infer that both cholera toxin and AlF4− would destabilize binding of GTP, by interfering with the ability of Gα to bind the γ-phosphate of GTP (Fig. 5). In the crystal structure of αt (3), the arginine that is modified by cholera toxin directly interacts with the γ-phosphate of GTP. Although AlF4− mimics the γ-phosphate of GTP, the GDP⋅AlF4−-bound form of Gα is inevitably less stable than the GTP[γS]-bound form, simply because AlF4− is not covalently attached to GDP. Thus the combination of the R231H mutation with either of these stimuli would severely destabilize the interaction of αs with GTP, even though neither the mutation nor the stimuli would do so by themselves.
Conversely, a higher concentration of Mg2+ (another link between the γ-phosphate and Gα; Fig. 5) is expected to counteract the destabilizing effect of the R231H mutation on activation by AlF4−. This proved to be the case (Fig. 4C). The idea that two destabilizing influences may potentiate one another explains the earlier observation (31) that AlF4− activates adenylyl cyclase weakly when αs has been modified by cholera toxin. In this case toxin-induced modification of a residue that interacts directly with the γ-phosphate presumably reduces stability of the αs⋅GDP⋅AlF4− complex, thereby inhibiting its ability to activate the effector.
To promote release of bound GDP, receptors must destabilize the guanine nucleotide binding site also. We have postulated (13, 30) that receptors do so by altering interactions between Gα and the guanine ring of GDP. The receptor interaction should similarly destabilize binding of GTP; indeed, receptors can increase rates of dissociation of GTP analogs from G proteins (10, 32). GTP can replace GDP in the binding site, however, because GTP rescues normal Gα from receptor-induced instability, in two ways: First, the instability is offset by the additional binding energy furnished by the γ-phosphate, allowing Gα to bind GTP in preference to GDP. Second, GTP (but not GDP) induces a conformational change in Gα that causes it to dissociate from βγ and from the receptor; separation from the receptor definitively removes its destabilizing effect. Efficiency of both mechanisms in αs-R231H will be reduced, because the disrupted arginine-glutamate hasp weakens the protein’s ability to clasp GTP and to stabilize the conformation that disengages it from βγ and receptor.
An alternative possibility is that the R231H mutation inhibits receptor-induced release of GDP rather than binding of GTP. Because it fails to explain the defective responses to cholera toxin and AlF4−, we infer that this explanation is not correct.
Implications for PHP-I.
Finally, the biochemical phenotype of the αs-R231H mutant suggests a possible modification in the criteria we and others have used as a basis for inferring the locus of genetic mutations in families of PHP-I patients.
Classical PHP-I is a syndrome characterized by multiple abnormalities, including Albright hereditary osteodystrophy and resistance to several hormones that act by stimulating Gs (33–35). Erythrocyte membranes of most PHP-I patients contain only 50% of the normal complement of Gs activity (36, 37). The Gs-deficient phenotype, classified as PHP-Ia (36), indicates that the affected patients carry inactivating mutations in an αs gene (the residual activity represents the intact αs allele); DNA sequencing has revealed αs mutations in many PHP-Ia patients (11, 35). In contrast, the PHP-Ib phenotype is assigned to a smaller number of PHP-I patients whose erythrocytes contain normal (or nearly normal) Gs activity (36). Most PHP-Ib patients, but not all (33), lack several of the clinical features of classical PHP-I, including Albright hereditary osteodystrophy; PHP-Ib patients have been thought to carry mutations in genes other than αs.
The three affected patients in the family carrying the αs-R231H mutation showed classical clinical features of the disease, including Albright hereditary osteodystrophy, but Gs activities in their erythrocytes were nearly normal (ranging between 60–90% of normal; results not shown). The Gs assay (36, 38) measures the ability of erythrocyte extracts to stimulate the adenylyl cyclase of cyc− membranes in the presence of isoproterenol and GTP[γS]. Because Gs activity in this crude assay reflects stimulation by GTP[γS] as much as that induced by the hormonal agonist (38), αs-R231H activity was scored as nearly normal, despite the dramatic hormone-response defect produced by the mutant protein in cultured cells and in affected patients.
Thus the αs-R231H patients show that results of the erythrocyte Gs assay can lead to an incorrect inference with respect to the genetic basis of the disease. Instead, PHP-I patients with apparently normal or nearly normal erythrocyte Gs activities merit careful investigation, especially when they display the classical clinical phenotype, including Albright hereditary osteodystrophy. Although such patients may inherit mutations in genes distinct from αs, their αs genes may encode mutant proteins with instructive qualitative defects, including impairment of conformational change, subcellular localization, or interaction with other proteins, including receptors, βγ, effectors, and regulator of G protein signaling proteins.
Acknowledgments
We thank Mark von Zastrow for anti β-AR antibody, Philip B. Wedegaertner and members of the Bourne laboratory for useful advice, and Helen Czerwonka for secretarial support. This work was supported by an National Institutes of Health (National Institute of General Medical Sciences) Grant GM27800 (to H.R.B.). T.I. was supported by a Julius H. Comroe Jr. Award by the Cardiovascular Research Institute. Z.F. was supported by a grant from the Israeli Ministry of Health.
ABBREVIATIONS
- Gα
α subunits of heterotrimeric G proteins
- PHP-1
pseudohypoparathyroidism, type 1
- AR
adrenergic receptor
- wt
wild type
- HA
hemagglutinin
- GTP[γS]
guanosine 5′-[γ-thio]triphosphate
References
- 1.Conklin B R, Bourne H R. Cell. 1993;73:631–641. doi: 10.1016/0092-8674(93)90245-l. [DOI] [PubMed] [Google Scholar]
- 2.Gilman A G. Annu Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 3.Noel J P, Hamm H E, Sigler P B. Nature (London) 1993;366:654–663. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- 4.Lambright D G, Noel J P, Hamm H E, Sigler P B. Nature (London) 1994;369:621–628. doi: 10.1038/369621a0. [DOI] [PubMed] [Google Scholar]
- 5.Mixon M B, Lee E, Coleman D E, Berghuis A M, Gilman A G, Sprang S R. Science. 1995;270:954–960. doi: 10.1126/science.270.5238.954. [DOI] [PubMed] [Google Scholar]
- 6.Brandt D R, Ross E M. J Biol Chem. 1986;261:1656–1664. [PubMed] [Google Scholar]
- 7.Florio V A, Sternweis P C. J Biol Chem. 1989;264:3909–3915. [PubMed] [Google Scholar]
- 8.Hepler J R, Kozasa T, Smrcka A V, Simon M I, Rhee S G, Sternweis P C, Gilman A G. J Biol Chem. 1993;268:14367–14375. [PubMed] [Google Scholar]
- 9.Singer W D, Miller R T, Sternweis P C. J Biol Chem. 1994;269:19796–19802. [PubMed] [Google Scholar]
- 10.Berstein G, Blank J L, Smrcka A V, Higashijima T, Sternweis P C, Exton J H, Ross E M. J Biol Chem. 1992;267:8081–8088. [PubMed] [Google Scholar]
- 11.Farfel Z, Iiri T, Shapira H, Roitman Z, Mouallem M, Bourne H R. J Biol Chem. 1996;271:19653–19655. doi: 10.1074/jbc.271.33.19653. [DOI] [PubMed] [Google Scholar]
- 12.Levis M J, Bourne H R. J Cell Biol. 1992;119:1297–1307. doi: 10.1083/jcb.119.5.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iiri T, Herzmark P, Nakamoto J M, Van Dop C, Bourne H R. Nature (London) 1994;371:164–168. doi: 10.1038/371164a0. [DOI] [PubMed] [Google Scholar]
- 14.von Zastrow M, Kobilka B K. J Biol Chem. 1992;267:3530–3538. [PubMed] [Google Scholar]
- 15.Wedegaertner P B, Bourne H R, von Zastrow M. Mol Biol Cell. 1996;7:1225–1233. doi: 10.1091/mbc.7.8.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia P D, Onrust R, Bell S M, Sakmar T P, Bourne H R. EMBO J. 1995;14:4460–4469. doi: 10.1002/j.1460-2075.1995.tb00125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iiri T, Homma Y, Ohoka Y, Robishaw J D, Katada T, Bourne H R. J Biol Chem. 1995;270:5901–5908. doi: 10.1074/jbc.270.11.5901. [DOI] [PubMed] [Google Scholar]
- 18.Iiri T, Backlund P B, Jones T L Z, Wedegaertner P B, Bourne H R. Proc Natl Acad Sci USA. 1996;93:14592–14597. doi: 10.1073/pnas.93.25.14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graziano M P, Freissmuth M, Gilman A G. J Biol Chem. 1989;264:409–418. [PubMed] [Google Scholar]
- 20.Iiri T, Ohoka Y, Ui M, Katada T. J Biol Chem. 1992;267:1020–1026. [PubMed] [Google Scholar]
- 21.Wong Y H, Federman A, Pace A M, Zachary I, Evans T, Pouysségur J, Bourne H R. Nature (London) 1991;351:63–65. doi: 10.1038/351063a0. [DOI] [PubMed] [Google Scholar]
- 22.Bourne H R, Kaslow D, Kaslow H R, Salomon M R, Licko V. Mol Pharmacol. 1981;20:435–441. [PubMed] [Google Scholar]
- 23.Miller R T, Masters S B, Sullivan K A, Beiderman B, Bourne H R. Nature (London) 1988;334:712–715. doi: 10.1038/334712a0. [DOI] [PubMed] [Google Scholar]
- 24.Haga K, Haga T, Ichiyama A. J Biol Chem. 1986;261:10133–10140. [PubMed] [Google Scholar]
- 25.Coleman D E, Berghuis A M, Lee E, Linder M E, Gilman A G, Sprang S R. Science. 1994;265:1405–1412. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- 26.Sondek J, Lambright D G, Noel J P, Hamm H E, Sigler P B. Nature (London) 1994;372:276–279. doi: 10.1038/372276a0. [DOI] [PubMed] [Google Scholar]
- 27.Stratton H F, Zhou J, Reed S I, Stone D E. Mol Cell Biol. 1996;16:6325–6337. doi: 10.1128/mcb.16.11.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lambright D G, Sondek J, Bohm A, Skiba N P, Hamm H E, Sigler P B. Nature (London) 1996;379:311–9. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- 29.Lichtarge O, Bourne H R, Cohen F E. Proc Natl Acad Sci USA. 1996;93:7507–7511. doi: 10.1073/pnas.93.15.7507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Onrust R, Herzmark P, Chi P, Garcia P D, Lichtarge O, Kingsley C, Bourne H R. Science. 1997;275:381–384. doi: 10.1126/science.275.5298.381. [DOI] [PubMed] [Google Scholar]
- 31.Cassel D, Selinger Z. Proc Natl Acad Sci USA. 1977;74:3307–3311. doi: 10.1073/pnas.74.8.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cassel D, Selinger Z. J Cyclic Nucleotide Res. 1977;3:11–22. [PubMed] [Google Scholar]
- 33.Farfel Z, Bourne H R. Miner Electrolyte Metab. 1982;8:227–236. [PubMed] [Google Scholar]
- 34.Spiegel A M, Shenker A, Weinstein L S. Endocr Rev. 1992;13:536–565. doi: 10.1210/edrv-13-3-536. [DOI] [PubMed] [Google Scholar]
- 35.Ringel M D, Schwindinger W F, Levine M A. Medicine (Baltimore) 1996;75:171–184. doi: 10.1097/00005792-199607000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Farfel Z, Brickman A S, Kaslow H R, Brothers V M, Bourne H R. N Engl J Med. 1980;303:237–242. doi: 10.1056/NEJM198007313030501. [DOI] [PubMed] [Google Scholar]
- 37.Levine M A, Downs R W, Jr, Singer M, Marx S J, Aurbach G D, Spiegel A M. Biochem Biophys Res Commun. 1980;94:1319–1324. doi: 10.1016/0006-291x(80)90563-x. [DOI] [PubMed] [Google Scholar]
- 38.Farfel Z, Brothers V M, Brickman A S, Conte F, Neer R, Bourne H R. Proc Natl Acad Sci USA. 1981;78:3098–3102. doi: 10.1073/pnas.78.5.3098. [DOI] [PMC free article] [PubMed] [Google Scholar]