

Abstract
Mutations in the yeast gene VPS41 give rise to poor growth on low iron medium, severe alterations in vacuolar morphology, and cause the missorting of membranous and soluble vacuolar proteins. Our studies predict that VPS41 encodes a hydrophilic protein of 992 amino acids that contains no obvious signal sequence or hydrophobic domains. The deduced Vps41p sequence contains a domain rich in glutamic and aspartic residues, as well as a domain with resemblance to a region of clathrin heavy chain. We have also identified and sequenced putative VPS41 homologues from Caenorhabditis elegans, plants, and humans. The VPS41 homologues (but not the yeast VPS41 itself) contain a conserved cysteine-rich RING-H2 zinc finger at their COOH termini. Biochemical experiments suggest that VPS41 functions in post-Golgi protein processing: the deletion mutant exhibits defective high affinity transport due to impaired Fet3p activity and also exhibits defects in the processing and sorting of multiple vacuolar hydrolases.
High-affinity iron uptake in Saccharomyces cerevisiae is mediated by the transmembrane transporter Ftr1p and the cell surface ferroxidase Fet3p (1). The multicopper oxidase Fet3p catalyzes the oxidation of ferrous iron to ferric iron, which is then transported across the membrane by Ftr1p. The catalytic activity of Fet3p requires the products of CTR1 (2), the cell surface copper transporter, and CCC2 (3), which transports copper into intracellular vesicles during the assembly of Fet3p. The CCC2 gene product is homologous to the mammalian Wilson disease and Menkes disease gene products (3). Mutations in either of the human genes result in defective copper transport and incomplete copper insertion into ceruloplasmin (4), a mammalian protein with homology to Fet3p (3).
To dissect the steps involved in the intracellular processing of Fet3p and other proteins involved in high-affinity iron transport, we have used a genetic screen that selects for mutants that are unable to grow on low-iron medium but can grow on high-iron medium (fet mutants, for ferrous transport). Growth on low-iron medium requires the proper assembly and localization of the Fet3p/Ftr1p transport system, but growth on iron-rich medium can occur by the low-affinity Fet4p iron transporter. From our fet screen, we isolated many mutants that had normal FET3 and FTR1 genes, but exhibited greatly reduced high-affinity iron transport. One such mutant was used to identify a gene required for the proper assembly and targeting of the high-affinity transport system. This gene, VPS41, which is essential for the functioning of Fet3p, is conserved across all species tested and is expressed in all human tissues. Yeast strains with a disrupted VPS41 show abnormalities in both vacuolar morphology and post-Golgi trafficking of vacuolar components. Our results suggest that the elements of the yeast high-affinity iron transport system require the post-Golgi-vacuolar pathway for proper functioning, and that the fet screen offers a facile way to identify new genes involved in the post-Golgi-vacuolar pathway.
MATERIALS AND METHODS
Strains and Media.
The S. cerevisiae strains used in this study were derived from SEY6210, DY150, and DY1457, as previously described (5, 6). Luria–Bertani medium was used to propagate Escherichia coli strains DH5α and HB101 and the strains were supplemented with antibiotics as required [Miller (7)]. Yeast extract-peptone-dextrose (YPD), or yeast nitrogen base was used for the standard growth of S. cerevisiae and supplemented as needed (8). Low-iron growth medium was made by the addition of bathophenanthroline sulfonate (BPS) to YPD (9). The fet2 mutant was isolated in a screen that selected for resistance to streptonigrin as described previously (5). To generate vps41Δ1, a NheI–PvuII fragment containing VPS41 in pBluescript KS(+) was digested with PstI and HindIII, and a HindIII/PstI fragment containing LEU2 was ligated into the gap. This construct was then digested with ScaI and BglII and transformed into SEY6210. To generate vps41Δ2, a NheI–NheI fragment containing VPS41 was digested with EcoNI and Sse8387I, filled in with Klenow, and a blunt-ended fragment containing URA3 was ligated into the gap and then transformed into DY150.
Reagents and Materials.
DNA restriction and modifying enzymes were obtained from New England Biolabs, Boehringer Mannheim, or Promega. CDCFDA and FM4–64 were obtained from Molecular Probes. Trans 35S-label was obtained from ICN, and [α-32P]dCTP was from Amersham. The antiserum to carboxypeptidase Y has been described previously (10). All other reagents were from Sigma.
Cloning of VPS41 and Homologues.
DNA transformations of E. coli and S. cerevisiae were performed by standard procedures (11). The VPS41 gene was cloned by transformation of fet2 with a yeast genomic library and selection for complementation of the low-iron growth phenotype using techniques described previously (5). A single unique plasmid was isolated and then shown to be derived from chromosome 4 by sequencing the ends of the plasmid insert. Subcloning mapped a minimum fet2 complementing activity to a NheI–NheI fragment containing only the single, large ORF YDR080W. VPS41 was also cloned by complementation of the secretion of CPY-Inv from a suc2 version of one of the original vps41 mutant strains (vpl20–1) (12). A complementing fragment again was determined to be derived from chromosome 4, and a minimum complementing NheI–PvuII fragment again contained only YDR080W.
The human homologue of VPS41 (hVPS41) was identified by similarity of the predicted sequence of Vps41p with expressed sequence tag (EST) 127498 from the dbEST database, which was derived from normalized human fetal liver and spleen library (T. Newman, Washington University). This clone was obtained from the Lawrence Livermore National Lab and was found to contain a 1.6-kb insert. This cDNA was used to probe a λZAPII library that was oligo(dT)- and random-primed from human heart mRNA (Stratagene). Exhaustive probing of this library combined with a rapid amplification of cDNA ends (RACE) strategy was used to isolate the hVPS41 sequence.
Similarly, the tomato (Lycopersicon esculentum) homologue of VPS41 (tVPS41) was identified by similarity of the predicted sequence of Vps41p with EST 89D22T7 from the dbEST database, which was derived from an Arabidopsis thaliana library (T. Newman). The clone was obtained from the Arabidopsis Biological Resource Center at Ohio State and was found to contain a 1.2-kb insert, and was used to probe a λZAPII library that was oligo(dT)-primed from tomato mRNA. The library contained multiple, full-length copies of the tVPS41 sequence.
Iron Transport and Growth Assays.
Cultures were grown to midlog phase in YPD, either harvested or transferred to YPDBPS(0) for a 4-hr induction, and then harvested. Iron uptake was assessed essentially as previously described (5), in the presence of 1 μM ascorbate and in the absence of copper. For growth on synthetic LIM medium and LIM medium supplemented with FeCl3 and CuSO4, cells were washed with water and placed at a dilution of 1,000 cells per drop. The preparation of LIM is described in a previous report (9).
Cell Labeling and Immunoprecipitations.
Yeast cells were grown to an A600 of 0.6–0.7 in yeast nitrogen base containing the required amino acids. Cells were harvested, labeled with Trans 35S-label for 10 min at 30°C, chased for 0 min and 45 min, and converted to spheroplasts as described previously (13). Carboxypeptidase Y (CPY), proteinase A (PrA), and alkaline phosphatase (ALP) were immunoprecipitated by established methods (14, 15).
Vacuolar Staining with FM4–64 and Electron Microscopy.
Procedures for staining of vacuoles with FM4–64 have been described previously (16). For electron microscopy cells were grown at 26°C in log phase. Cells were harvested and prepared as described previously (13).
RESULTS
Characterization of VPS41.
We selected the mutant fet2, which showed a growth defect on low-iron medium, and identified VPS41 by complementation of the low-iron growth phenotype. The deletion strain vps41Δ2 showed a more severe growth defect than fet2, with deficient growth on both high- and low-iron medium. That VPS41 was the normal allele of fet2 was confirmed by demonstrating that vps41Δ2 cells complimented with the rescued fet2 allele showed normal growth on iron-replete medium, but defective growth on low-iron medium. The different phenotypes observed in the fet2 and vps41Δ2 alleles suggests that the original fet2 mutant may retain some function, although the mutant allele has not been genetically characterized. The phenotype of poor growth on low iron can be caused either by a deficiency in iron transport activity or by a metabolic defect that confers sensitivity to low-iron conditions. A role in high-affinity iron transport for VPS41 was demonstrated by comparison of iron uptake between wild-type and vps41Δ2 deletion strains (Fig. 1). In high-iron conditions (YPD), iron transport is mediated primarily by Fet4p and is unaffected by the deletion of VPS41. However, incubation in iron-deficient media (BPS) produced a marked induction of transporter activity in wild-type cells, whereas the deletion strain exhibited a defect in iron transport.
Figure 1.
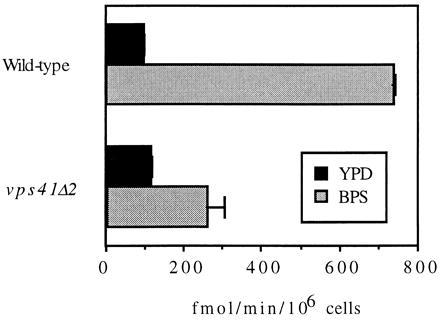
Iron transport in wild-type yeast and the vps41Δ2 strain. Cells were grown to midlog phase in iron replete (YPD) media, and then assayed immediately or incubated for 4 hr in iron-limited (BPS) media. The cells were harvested and iron transport was assayed using 0.5 μM iron in the absence of ascorbate and copper.
Identification of VPS41 Homologues.
A tblastn computer search of the swissprot database revealed several high-probability matches to the predicted amino acid sequence of VPS41. One ORF, designated as F32A6.3, published as part of the C. elegans sequencing project (17), codes for a protein of unknown function. This sequence is homologous throughout its length to VPS41; we will subsequently refer to this gene as cVPS41.
A search of dbEST also identified partial human EST clones, including S53, a cDNA in the region of the early-onset Alzheimer disease gene (18). As the database sequence of hVPS41 was incomplete, multiple rounds of cloning from a human heart λZAPII library and RACE were necessary to obtain a full-length 3.5-kb sequence coding for hVPS41. This insert was used to probe a multiple-tissue Northern blot (CLONTECH) and detected 3.5-kb transcripts equally expressed in all tissues tested. Additionally, since the EST S53 has been mapped, we know that hVPS41 is on human chromosome 14q23 (18).
The National Center for Biotechnology Information search also identified an A. thaliana EST (19). Only a few hundred nucleotides were available from the database for this sequence, so we obtained the clone, sequenced it, and used the insert to probe a tomato λZAPII library. From this library, we obtained a clone with a 3.3-kb insert containing tVPS41.
Comparison of VPS41Homologues.
Sequence alignment of the predicted sequences of Vps41p, cVps41p, tVps41p, and hVps41p revealed significant structural similarity across the length of the protein (Fig. 2), although hVPS41 did not complement the phenotype of vps41Δ2. Most interesting is the region between amino acids 806 and 882 of the Vps41p predicted sequence: across all four species, the sequence similarity exceeds 70%, and identity exceeds 30%. This highly conserved sequence in Vps41p and homologues is also similar to residues 1036–1118 in human clathrin heavy chain (50% similarity between Vps41p homologues and clathrin). A sequence motif found in the Vps41p homologues but not in Vps41p itself is the RING-H2 motif, a zinc finger-like motif previously identified in VPS8, VPS18, and VPS11 (20). Other proteins containing this motif have been found to associate with endosomes, the vacuole, or other vesicular structures (21).
Figure 2.

Alignment of Vps41p, cVps41p, tVps41p, and hVps41p.
Another striking region was a 19-residue sequence containing only glutamic and aspartic acids (positions 78–97 in Vps41p). More than 600 known protein sequences contain such glu, asp-rich domains, and although these domains have no known function, many proteins containing such acidic regions bind calcium or other metals. This motif is found in Vps41p, tVps41p, and hVps41p, but not cVps41p. Interestingly, a second glu, asp-rich sequence is also found between the second and third metal binding motifs in the RING-H2 finger of tVps41p; a similar motif exists in the homologous region of our partial A. thaliana Vps41p sequence (data not shown).
Characterization of the vpsPhenotype.
Originally, vps41 alleles were obtained from screens of mutants that secrete resident vacuolar proteins. We examined the sorting of three vacuolar hydrolases, ALP, CPY, and PrA in yeast strains that were wild-type, vps41Δ1, and vps41Δ1 complemented with centromeric VPS41 (Fig. 3). Wild-type cells and vps41Δ1 complemented by VPS41 showed nearly identical sorting and processing of all hydrolases examined. In contrast, vps41Δ1 strains showed no maturation of CPY or ALP even after a 40-min chase. In addition, approximately 60% of the newly synthesized CPY was secreted as the Golgi modified p2 form, indicating a partial defect for vacuolar protein sorting in the Golgi. ALP is a type II integral membrane protein and is not released from whole cells; its presence in the intracellular fractions is therefore expected even when vacuolar localization is not achieved. PrA, the other hydrolase examined, shows significant intracellular accumulation of precursor PrA and some mature protein. A significant portion of PrA is released to the extracellular media following the 40-min chase indicating a defect in vps41Δ1 strains for the sorting of PrA from the Golgi to the vacuole.
Figure 3.
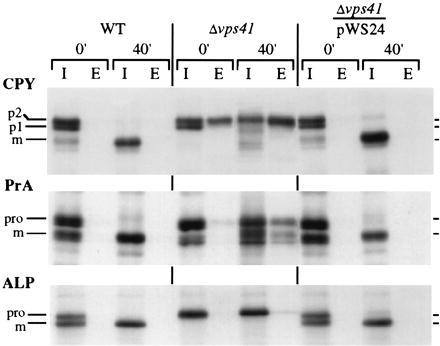
Pulse–chase analysis of vacuolar hydrolases. Wild-type cells, vps41Δ1 deletion mutants, and vps41Δ1 cells complemented with VPS41 (pWS24) were grown to midlog phase at 30°C, pulse-labeled with Trans 35S-label for 10 min, and chased with excess unlabeled cysteine and methionine, and cells were harvested 0 min and 40 min after the addition of the chase medium. The samples were then separated into intracellular (I) and extracellular (E) fractions, and specific proteins were immunoprecipitated and applied to SDS/PAGE and autoradiography.
A common consequence of certain mutations that impair trafficking of vacuolar proteins is abnormal vacuolar morphology (12). Examination of vps41Δ1 mutants stained with the fluorescent dye FM4–64, a useful membrane stain for the vacuole (16), revealed extensive fragmentation of the vacuole (Fig. 4 A and B). The observation of 20–50 small, brightly staining compartments in vps41Δ1 mutants is consistent with its previous classification as a class B vps mutant (12). Vacuolar fragmentation is not seen in vps41Δ1 mutants complemented with a centromeric plasmid containing VPS41 (Fig. 4 C and D). Examination of vps41Δ1 mutants by electron microscopy revealed the presence of many small membrane-bound compartments (Fig. 4E). Wild-type or complemented deletion mutants have 1–3 electron dense vacuolar compartments (Fig. 4F; for wild types, see refs. 13 and 22–24). The membrane-bound compartments in vps41Δ1 cells likely correspond to the brightly staining compartments observed in Fig. 4A, the fragmented vacuoles, and related structures that accumulate in the absence of Vps41p.
Figure 4.
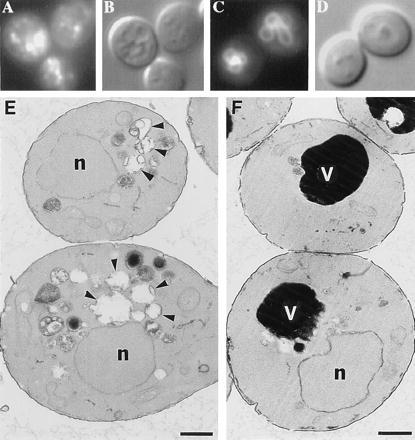
Vacuolar morphology examined by staining with the vacuole-specific dye FM4–64 (A and C), by Nomarski imaging (B and D), or by electron microscopy (E and F) of vps41Δ1 deletion mutants (A, B, and E) or vps41Δ1 complemented with VPS41 (C, D, and F).
Cause of Low-Iron Growth Defect.
Because vps41Δ1 interferes with the processing of vacuolar hydrolases, and because Fet3p requires several distinct posttranslational processing events, including glycosylation and copper loading, it seemed possible that the defect in high-affinity iron transport in vps41 deletion cells was due in part to improperly processed, and thus inactive, Fet3p. Northern blot analysis showed that the concentration of VPS41 mRNA was independent of iron content in the media, and that wild-type, fet2, and vps41Δ2 cells had normal low-iron induction of FET3 mRNA (data not shown). Western blot analysis of both wild-type and vps41Δ2 cells induced for expression of the high-affinity transport system revealed a strongly staining glycosylated Fet3p band as well as a weakly staining unglycosylated Fet3p band (data not shown; ref. 2). Oxidase activity of Fet3p was tested by assaying the oxidation of paraphenylene diamine (PPD), an organic substrate used as an oxidase indicator (3). Oxidation of PPD by membranes isolated from vps41Δ2 cells was found to be reduced to the level of fet3Δ cells (Fig. 5a). That Fet3p from vps41Δ2 cells was normally glycosylated but lacked PPD activity suggested that the deletion mutant might be blocked in the processing step in which copper is incorporated into apoFet3p. Consistent with this hypothesis, we found that the low-iron growth defect of vps41Δ2 could be overcome by growth in the presence of high copper (Fig. 5b). High concentrations of copper also rescued the low-iron growth defect of ccc2Δ (3), which is known to be defective in intracellular Fet3p copper incorporation, but did not rescue fet3Δ.
Figure 5.
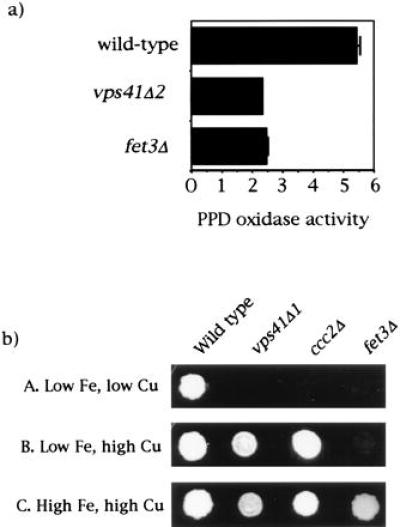
Effect of VPS41 deletion on Fet3p activity. (a) PPD oxidase activity of wild-type, vps41Δ2, and fet3Δ cells. Five milligrams of a crude membrane fraction obtained from cells grown in low-iron medium was incubated with 0.05% PPD in 100 mM NaOAc (pH 5.7) for 12 hr and quantified by A570. (b) Metal-dependent growth phenotypes of vps41Δ1. The effect of added iron or copper on growth on LIM medium was determined for the indicated yeast strains. A, 5 μM Fe, 0.125 μM Cu; B, 5 μM Fe, 500 μM Cu; C, 50 μM Fe, 500 μM Cu.
DISCUSSION
We have identified VPS41, which is required for growth on low-iron medium, correct vacuolar protein sorting, and normal vacuolar morphology. Cells containing deletions of VPS41 show normal induction of FET3 mRNA and protein but defective Fet3p function, both in oxidase activity and in iron transport. The reduced PPD oxidase activity in the vps41Δ2 deletion suggests that its low-iron growth phenotype results from defective functioning of Fet3p. The observation that high copper can rescue the mutant indicates that the defect is due to a lack of copper incorporation into Fet3p.
The activity of Fet3p requires intracellular copper incorporation, which is dependent on the copper transporter Ccc2p. Analogously, copper incorporation into ceruloplasmin, the mammalian Fet3p homologue, occurs in an intracellular compartment and requires the products of the Menkes and Wilson disease genes (4), which are copper transporters homologous to Ccc2p. The observation that high-copper medium can rescue the low-iron growth phenotype of vps41 deletion mutants suggests that the compromised high-affinity iron uptake in these cells results from a lack of copper incorporation into Fet3p, possibly because VPS41 is required for proper sorting of Fet3p or Ccc2p. These proteins may require the environment of the prevacuolar endosome for their function. Alteration of delivery to, or function of, this compartment in vps41 mutants would result in the observed defects in Fet3p function.
In the absence of VPS41, Fet3p and CPY are glycosylated, indicating that the processing defect occurs after the trans-Golgi network. Class E vps mutants, which show defects in vesicular traffic from the prevacuolar endosome to the vacuole (12), show no deficiency of growth on low-iron medium, and therefore, these mutants must process Fet3p normally. The prevacuolar endosome is an organelle analogous to the mammalian late endosome, which may be one of the post-Golgi vesicles identified to contain the Menkes gene product (25). On the basis of these observations, we surmise that the Fet3p defect in VPS41 deletion cells is due to a defect in the function of the prevacuolar endosome. This hypothesis is consistent with the observed defects in vacuolar protein sorting. It is possible that the primary defect in VPS41 deletion cells is assembly of the iron uptake system and that high-affinity iron uptake (or at least a functional Fet3p) is necessary for vacuolar protein sorting, although this hypothesis is unlikely, as fet3 mutants do not display vps phenotypes.
Homologues of VPS41 have been found in all species examined, and the mammalian hVPS41 mRNA was found in all tissues examined. Vps41p and its homologues have similarity with other proteins involved in vesicular traffic. The most conserved region between Vps41p and its homologues is also similar to a region of clathrin heavy chain, which is involved in protein targeting from the trans-Golgi network to the vacuole (26). On the basis of this homology and the biochemical defects in vps41 mutants, we hypothesize that Vps41p may function in the packaging of some vacuolar proteins into transport intermediates such as vesicles.
It is striking that while all of the homologues contain a RING-H2 finger, this motif is lacking in yeast Vps41p. Since it seems very unlikely that a motif conserved across species as diverse as C. elegans and humans should be unnecessary for the function of Vps41p, it seems probable that the RING-H2 motif in yeast is provided by an unidentified protein complexed to Vps41p. Further support for the hypothesis that functional regions may be conserved within a Vps41p–protein complex can be inferred from the lack of a glu, asp-rich domain in cVps41p; we predict that the C. elegans equivalent of the Vps41p-associated protein will be found to contain an acidic region. Our fet screen may identify the yeast Vps41p-associated protein, since a mutation in such a gene might produce the same phenotype as fet2. Examination of fet mutants stained with FM4–64 for fet2-like vacuolar appearance may identify mutations in the Vps41p-associated protein, and complementation of the low-iron phenotype of fet mutants will identify the defective gene. Indeed, other fet mutants in our collection have been found to exhibit the vps phenotype, showing defects in both protein sorting and vacuolar morphology (data not shown).
Acknowledgments
We thank J. Michael McCaffery for electron microscopy analysis, Tama Fox for assistance with cloning tVPS41, and members of the J.K. and S.D.E. lab for helpful discussion. W.B.S. is supported as a postdoctoral fellow of the American Cancer Society. This work was supported by grants from the National Institutes of Health (HL26922 and DK30534 to J.K., GM32703 and CA58689 to S.D.E.). S.D.E. is supported as an investigator of the Howard Hughes Medical Institute.
ABBREVIATIONS
- CPY
carboxypeptidase Y
- PrA
proteinase A
- ALP
alkaline phosphatase
- YPD
yeast extract-peptone-dextrose
- PPD
paraphenylene diamine
Footnotes
References
- 1.Stearman R, Yuan D S, Yamaguchi-Iwai Y, Klausner R D, Dancis A. Science. 1996;271:1552–1557. doi: 10.1126/science.271.5255.1552. [DOI] [PubMed] [Google Scholar]
- 2.Dancis A, Yuan D S, Haile D, Askwith C, Eide D, Moehle C, Kaplan J, Klausner R D. Cell. 1994;76:393–402. doi: 10.1016/0092-8674(94)90345-x. [DOI] [PubMed] [Google Scholar]
- 3.Yuan D S, Stearman R, Dancis A, Dunn T, Beeler T, Klausner R D. Proc Natl Acad Sci USA. 1995;92:2632–2636. doi: 10.1073/pnas.92.7.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris Z L, Gitlin J D. Am J Clin Nutr. 1996;63:836S–841S. doi: 10.1093/ajcn/63.5.836. [DOI] [PubMed] [Google Scholar]
- 5.Askwith C, Eide D, Ho A V, Bernard P S, Li L, Davis-Kaplan S, Sipe D M, Kaplan J. Cell. 1994;76:3018–3023. doi: 10.1016/0092-8674(94)90346-8. [DOI] [PubMed] [Google Scholar]
- 6.Robinson J S, Klionsky D J, Banta L M, Emr S D. Mol Cell Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller J. Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press; 1972. [Google Scholar]
- 8.Sherman F, Fink G R, Lawrence L W. Laboratory Course Manual for Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press; 1986. [Google Scholar]
- 9.Eide D, Guarante L. J Gen Microbiol. 1992;138:34–54. doi: 10.1099/00221287-138-2-347. [DOI] [PubMed] [Google Scholar]
- 10.Klionsky D J, Emr S D. EMBO J. 1989;8:2241–2250. doi: 10.1002/j.1460-2075.1989.tb08348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 12.Raymond C K, Howald-Stevenson I, Vater C A, Stevens T H. Mol Biol Cell. 1992;3:1389–1402. doi: 10.1091/mbc.3.12.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cowles C R, Emr S D, Horazdovsky B F. J Cell Sci. 1994;107:3449–3459. doi: 10.1242/jcs.107.12.3449. [DOI] [PubMed] [Google Scholar]
- 14.Klionsky D J, Banta L M, Emr S D. Mol Cell Biol. 1988;8:2105–2116. doi: 10.1128/mcb.8.5.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klionsky D J, Emr S D. EMBO J. 1989;8:2241–2250. doi: 10.1002/j.1460-2075.1989.tb08348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vita T A, Emr S D. J Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson R, Ainscough R, Anderson K, Baynes C, Berks M, et al. Nature (London) 1994;368:32–38. doi: 10.1038/368032a0. [DOI] [PubMed] [Google Scholar]
- 18.Sherington R, Rogaev E I, Liang Y, Rogaeva E A, Levesque G, et al. Nature (London) 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 19.Newman T, de Bruijn F J, Green P, Keegstra K, Kende H, McIntosh L, Ohlrogge J, Raikhel N, Somerville S, Tomashow M, Retzel E, Somerville C. Plant Physiol. 1994;106:1241–1255. doi: 10.1104/pp.106.4.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horazdovsky B F, Cowles C R, Mustol P, Holmes M, Emr S D. J Biol Chem. 1996;271:33607–33615. doi: 10.1074/jbc.271.52.33607. [DOI] [PubMed] [Google Scholar]
- 21.Saurin A G, Borden K L B, Boddy M D, Freemont P S. Trends Biochem Sci. 1996;21:208–214. [PubMed] [Google Scholar]
- 22.Burd C R, Mustol P A, Schu P V, Emr S D. Mol Cell Biol. 1996;16:2369–2377. doi: 10.1128/mcb.16.5.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horazdovsky B R, Busch G R, Emr S D. EMBO J. 1994;13:1297–1309. doi: 10.1002/j.1460-2075.1994.tb06382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rieder S E, Banta L M, Köhrer K, McCaffery J M, Emr S D. Mol Biol Cell. 1996;7:985–999. doi: 10.1091/mbc.7.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamaguchi Y, Heiny M E, Suzuki M, Gitlin J D. Proc Natl Acad Sci USA. 1996;93:14030–14055. doi: 10.1073/pnas.93.24.14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scheckman R, Orci L. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]