

Precis
Cardiac development and postnatal growth depend upon activation of AKT, but initial strategies to improve myocardial repair utilizing AKT were stymied by undesirable corollary alterations in myocardial structure and function. These unfortunate precedents were based upon high level expression of constitutively activated AKT, predominantly in the cytoplasm of the cell. Based upon subsequent studies establishing that activated AKT accumulates in the nucleus we reasoned that the location of AKT, not simply the activity level, would be a critical determinant of the phenotypic outcome resulting from AKT activation. Using myocardial–specific expression of nuclear-targeted AKT (AKT/nuc), the proliferation of myocardial stem and progenitor cell populations is enhanced, casting new light upon the implementation of AKT activity as a molecular interventional approach for treatment of cardiomyopathic damage resulting from acute injury, chronic stress, or the debilitating changes of aging.
AKT properties: a multifunctional player in cardiomyocytes and the myocardium.
Ask a dozen cardiovascular researchers what they find most intriguing about AKT signaling and you're likely to get a dozen different responses. Whether studying cell survival, growth, or metabolism, the involvement of AKT is inescapable. As a critical nodal point in signal transduction, AKT continues to be a popular target for researchers looking to manipulate cellular responses involving growth factors, apoptotic stimuli, mechanical stress, hormones, energy utilization, protein synthesis, proliferation, differentiation, motility and gene transcription (to name a select few). Thus far this dizzying array of consequences for altering AKT activity has been somewhat easier to follow in the context of mature cardiomyocytes that behave as post-mitotic, highly organized, non-motile, and non-polarized cells. Recognition of cell death as a contributing factor in heart failure prompted studies to promote survival signaling in cardiomyocytes. Indeed, AKT is responsible for salutary effects of insulin-like growth factor-1 (IGF-1) or estrogen that have both been touted as cardioprotective agents to blunt apoptotic cell death, reduce maladaptive remodeling, and enhance hemodynamic performance in response to cardiomyopathic challenge. The spectrum of phenotypic effects resulting from AKT activity can vary widely depending upon duration, intensity, and localization as demonstrated in several elegant studies that document profound hypertrophic remodeling (Matsui et al. 2002, Shiojima et al. 2005, Taniyama et al. 2005), increased contractility (Condorelli et al. 2002), altered glucose metabolism (Matsui et al. 2006), and cardiomyopathic challenge resulting in suppressed (Fujio et al. 2000) or enhanced (Nagoshi et al. 2005) susceptibility to apoptotic cell death. In addition to these diverse effects of AKT activation, the recent advent of regenerative medicine in myocardial biology has uncovered yet another facet where AKT-mediated effects could potentially benefit the myocardium by promoting survival, proliferation, and trafficking of stem cells and cardiac progenitor cell populations. And, although the subject of AKT's influence upon cardiac stem cell populations is a relatively nascent topic, signposts are springing up in cardiovascular literature and elsewhere to support a role for AKT in regulation of cardiomyocyte cycling and cardiac progenitor cell proliferation which is the primary focus of this brief review.
AKT in stem cells: helping the garden grow
A decade ago connections were established between stimulation of hematopoetic stem cells and AKT activation (Testa and Bellacosa 1997, Zhang and Vik 1997), with many additional regulators of stem cell renewal identified in subsequent years(Akala and Clarke 2006). AKT shares responsibility for enhancing stem cell growth and survival with another serine / threonine kinase called Pim that possesses similar target substrate specificity (Bachmann and Moroy 2005, Hammerman et al. 2005, Wang et al. 2001). AKT activity maintains stem cells by promoting both viability and proliferation (Hammerman et al. 2005, Kim et al. 2005), which is counterbalanced by equally important inhibitory molecules such as PTEN or PHLPP to prevent oncogenesis (Brognard et al. 2007, Gao et al. 2005, Groszer et al. 2001, Yilmaz et al. 2006, Zhang et al. 2006) and promote differentiation (Otaegi et al. 2006). Concurrent findings appear in the cardiovascular literature where AKT has been linked to proliferation of human cardiac progenitor cells (Tateishi et al. 2007) as well as cardiomyocytes derived from embryonic stem cells (McDevitt et al. 2005, Roggia et al. 2007). In turn, activation of AKT in stem cells is associated with c-met (the receptor for hepatocyte growth factor) (Forte et al. 2006, Okano et al. 2003, Roggia et al. 2007), IGF-1 (Ye and D'Ercole 2006), estrogen (Imanishi et al. 2005), and Notch receptor (Androutsellis-Theotokis et al. 2006). Perhaps not coincidentally, these factors and their corresponding signaling cascades also share the characteristic of conferring protection in the face of cardiomyopathic insults.
AKT to the rescue: myocardial repair and regeneration
Cell death was recognized as a contributing factor in the pathogenesis of cardiomyopathic injury and heart failure over a decade ago (Cheng et al. 1995, Gottlieb et al. 1994). Ensuing research identified and characterized numerous molecular pathways that control apoptotic cell death. Despite frustrating problems with use of AKT for treatment of heart failure, beneficial effects of IGF-1 were recognized both in vitro (Fujio et al. 2000, Wang et al. 1998) and in vivo (Chao et al. 2003, Li et al. 1997). Paradoxically, IGF-1 exerted anti-apoptotic activity via AKT activation but did not provoke the hypertrophic and cardiomyopathic effects that had plagued efforts to use AKT (Li et al. 1997, Reiss et al. 1996). IGF-1 even showed promise as a treatment regimen for patients in heart failure (Donath et al. 1998, Donath and Zapf 1999, Fazio et al. 1996). Unfortunately, IGF-1 exerts multiple effects in addition to inhibiting cell death and as a paracrine factor can alter cell metabolism and function throughout the body. Ideally, an optimal approach to therapeutic intervention would combine the protective effect of AKT kinase with the activation properties of IGF within the local environment of the myocardium. To reconcile the discrepancy between the IGF-1 action and AKT activation, the cellular action of AKT constructs would need to be modified.
AKT activation by cardioprotective agents such as IGF-1 or estrogen shows one important distinction from the mutant-activated AKT constructs: nuclear accumulation. Physiologic AKT activation provokes transient membrane association, phosphorylation by PI3-K, and subsequent accumulation of active AKT within the nucleus of the cell. Artificially created activated constructs (such as myristolated or phosphomimetic mutants) do not undergo this temporal redistribution and instead accumulate predominantly throughout the cytoplasm, in proximity to the membrane and, to a lesser extent, within the nucleus. Transgenic mouse models employing these constitutively active AKT constructs reveal important effects upon hypertrophic cardiac remodeling, angiogenesis, and enhanced contractility. While both valuable and informative from an experimental standpoint, these observations made with constitutively active AKT mutants contrasts sharply with the behavior of AKT in response to the paracrine stimuli that normally regulate activation, such as IGF-1. Cardioprotective effects of IGF-1 in studies of infarction and ischemia-reperfusion damage (Fujio et al. 2000, Li et al. 1997, Su et al. 2003) set the stage for demonstrating the ability of IGF-1 to inhibit development of heart failure (Welch et al. 2002). Collective evidence was pointing to a beneficial effect of AKT activity under appropriate stimulation. Thus, my laboratory pursued the postulate that nuclear targeting of wild-type AKT kinase activated by endogenous cellular phosphorylation mechanisms would provide beneficial cardioprotective effects without promoting maladaptive remodeling typical of mutant AKT constructs.
Nuclear AKT: the good without the bad or the ugly
If our postulate was correct, then nuclear targeting of AKT would confer beneficial phenotypic characteristics of IGF-1 treatment without cardiomyopathic side effects observed with previously created mutant AKT constructs. In the first of a series of nuclear-targeted AKT-related publications, a wild-type AKT was used to maintain near-physiologic levels of kinase activity with targeting mediated by a concatameric nuclear localization sequence. Nuclear accumulation of AKT produced profound anti-apoptotic activity without evidence of hypertrophic growth in either cultured cardiomyocytes or genetically engineered mice that specifically expressed nuclear targeted AKT (Shiraishi et al. 2004). Inhibition of apoptosis met or exceeded that of myristolated AKT, and prevention of ischemia/reperfusion damage in vivo was comparable to the potent effect of preconditioning. Striking similarities between cardiac-specific expression of nuclear-targeted AKT or IGF indicated the identification of a pivotal requirement for AKT activation, allowing for beneficial characteristics of IGF-mediated protection without maladaptive hypertrophy or undesirable paracrine-signaling side effects. Indeed, subsequent publications have demonstrated that nuclear accumulation of AKT is actually anti-hypertrophic (Tsujita et al. 2006), in agreement with findings obtained with AKT knockout mice (DeBosch et al. 2006).
Pursuant to these studies, it seemed plausible that there was more to nuclear AKT accumulation than just inhibition of cell death. Morphometric analyses of hearts from transgenic mice created by α-myosin heavy chain-driven expression of nuclear-targeted AKT showed an increase in the number of cardiomyocytes that were smaller in volume, likely owing in part to the aforementioned anti-hypertrophic effects of nuclear AKT accumulation. The combination of more but smaller cardiomyocytes preserved overall cardiac mass and geometry in nuclear-AKT transgenic mice, but what was the mechanistic basis for the hypercellular phenotype resulting from nuclear AKT expression? In comparison, cardiac-specific expression of IGF-1B also led to hypercelluarity but no reduction in cell volume, resulting in increased cardiac mass. The commonality of increased cardiomyocyte numbers in both nuclear-targeted AKT and IGF-1B transgenic mice pointed toward examination of cellular proliferation. Since mature cardiomyocytes are notoriously reluctant to undergo mitotic replication, efforts were directed toward characterizing the effect of nuclear AKT upon not just cardiomyocytes but also the cardiac progenitor cell population. There was reason to believe that nuclear AKT was fundamental to the effects mediated by IGF-1B, since cause and effect relationships linking enhanced telomerase activity to nuclear accumulation of AKT induced by IGF-1B had been established, with telomerase activity increased following expression of nuclear-targeted AKT construct in cardiomyocytes (Torella et al. 2004). Since telomerase maintains cell replication, antagonizes cell death, and is expressed predominantly in germ cells and progenitor / stem cells (Flores et al. 2006), the underlying basis for myocardial hypercellularity could be linked to potentiation of progenitor cell proliferation in our nuclear-targeted AKT transgenic mice. The timing was fortunate for this hypothesis, as the concept of myocardial stem cells and resident progenitor cell populations responsible for generating new myocytes was commanding increasing attention and acceptance by the cardiovascular community.
The heart of nuclear-targeted AKT: cardiac progenitor cells
Presumably the nuclear-targeted AKT construct could promote hypercellularity by a combination of increased cell survival as well as enhanced cell cycling. Previous studies had already established the anti-apoptotic action of nuclear-targeted AKT for cardiomyocytes, but the impact of the nuclear-targeted AKT construct upon stem / progenitor cell proliferation in the myocardium had not been examined. Therefore, a study was fashioned to assess myocardial cell proliferation in nuclear-targeted AKT transgenic mouse hearts relative to nontransgenic controls (Gude et al. 2006). The presence of c-kit antigen identified stem cells; cycling cells were assessed by immunolabeling for Ki67 (a marker of cells typically undergoing mitotic replication), and cardiac lineage commitment was confirmed by coincident expression of the transcription factor GATA4. The postnatal period is a time of rapid cardiac growth and cellular proliferation, wherefore samples from young postnatal myocardium were examined to validate the experimental design and provide further insight into this critical period of myocardial growth. Indeed, neonatal two day old myocardium is enriched for c-kit cells as well as cycling cardiac progenitor cells, as revealed by coincident labeling for both Ki67 and GATA4 that declines rapidly by 2-3 weeks of age. As this age-associated reduction occurs within the first few weeks after birth, the number of cycling cardiac progenitor cells was doubled in our nuclear-targeted AKT transgenic heart samples relative to nontransgenic controls from 2 weeks on to at least 6 weeks (the latest time point assessed in our study). Furthermore, the number of cardiomyocyte progenitor cells in young adult mice co-expressing c-kit as well as cardiomyocyte-specific transcription factors Nkx 2.5 or MEF2C were increased approximately three fold. Collectively, these findings indicate that nuclear-targeted AKT expressed in the heart expands the population of cycling cardiac progenitor cells. Upon examination of cytokine and growth factor expression induced by nuclear-targeted AKT, a panoply of mRNA inductions were observed in transgenic mice, with an overall expression profile reminiscent of the neonatal myocardium suggesting that nuclear-targeted AKT evokes a phenotypic effect similar to that of a young growing postnatal heart (Gude et al. 2006).
Mechanistically speaking, there are a few ways to account for expansion of the cardiac progenitor and cardiomyocyte population by nuclear-targeted AKT: 1) influencing cell cycle regulatory proteins to favor myocyte proliferation, 2) inhibition of myocyte cell death or enhancement of cell survival, and 3) increasing proliferation of the early committed cardiomyocyte progenitor cell pool. Expansion of the cardiac progenitor population could be due to induction of transgene expression from a very early stage of cardiomyocyte lineage commitment since transgenic mice expressing cardiac-specific nuclear-targeted AKT were created with the α-myosin heavy chain promoter. The coincidence of nuclear-targeted AKT transgene in cells expressing c-kit antigen is indicative of progenitor cardiomyocyte status, and extensive analyses revealed a small percentage of such cells representing only a few percent of the total c-kit+ population examined in the myocardium. Nuclear-targeted AKT expression in these myocyte progenitor cells presumably serves to increase intracellular proliferative and survival signaling, thereby increasing their number as well as that of their progeny that will ultimately lose c-kit expression upon continued progression to cardiomyocyte commitment. These early myocyte precursors may undergo additional rounds of replication, further expanding the number of cells entering the myocyte pool. Once entrenched in the cardiomyocyte lineage, the continued expression of nuclear-targeted AKT promotes survival and inhibits hypertrophy. The combination of increasing proliferation of the precursor pool and blunting hypertrophic growth together account for the ultimate phenotype of the nuclear-targeted AKT transgenic heart: smaller and more numerous myocytes with preservation of cardiac size and mass that is capable of enhanced resistance to cardiomyopathic injury and augmented hemodynamic function.
Healing a broken heart: prospects for enhancing stem cell therapies with AKT
Revolutionary ideas of stem cell biology are challenging perceptions of the cardiovascular community toward myocardial remodeling, repair, and aging (reviewed in (Anversa et al. 2005, Bruneau and Black 2007, Cho et al. 2006, Christoforou and Gearhart 2007, Evans et al. 2007, Germani et al. 2007, Gupta et al. 2007, Liao et al. 2007, Lyon and Harding 2007, Mummery 2007, Pallante and Edelberg 2006, Schuleri et al. 2007, Sohn et al. 2007). This recent departure from established precepts of cardiac biology is being driven by the discovery of stem cells with the capacity to differentiate and integrate into the functioning myocardium. With the demonstration that such cells exist, longstanding beliefs in cardiac biology become open to reinterpretation, and novel directions for therapeutic intervention in heart failure have been proposed. Of course, controversies related to the efficiency of engraftment, transdifferentiation, interactions with endogenous cell populations, and contributions of cell fusion continue to be played out in the literature (reviewed in Ang et al. 2006, Balsam and Robbins 2005, Kocher et al. 2007, Kucia et al. 2004, Mathur and Martin 2004, Taylor 2004), but these issues are more relevant to mechanism and efficacy and not to the existence of progenitor cells with regenerative potential. Thus, if the heart is an organ capable of self-renewal, then the possibility exists to harness this potential for improving myocardial structure and function. To achieve this goal, the biology of myocardial stem cells needs to be understood and treatment approaches optimized by judicious manipulation of the stem cell population and their environment. Predictably, the initial flurry of experiments to restore cardiac function by adoptive transfer of stem cells yielded mixed results, owing in part to our relatively meager understanding of the mechanistic basis for myocardial regeneration. Chasing the ultimate goal of stem cell-based repair will require augmentation of normal cellular function to achieve optimal results, with manipulation of signal transduction as a promising strategy for improving efficiency of myocardial regeneration and repair. Cardioprotection is enhanced by induction of AKT in adoptive transfer of either bone marrow stem cells exposed to hypoxia (Uemura et al. 2006) or mesenchymal stem cells genetically modified to constitutively express activated AKT (Gnecchi et al. 2005) in the infarcted heart. A follow-up to the latter study invoked a requirement for secreted frizzled-related protein-2 (Mirotsou et al. 2007) and in both cases the donated cell population is presumed to generate paracrine factor(s) that inhibit cell death and maladaptive remodeling. While AKT activation may prolong donated cell survival or perhaps increase proliferation for a few days, the lack of persistence or conversion to a cardiogenic phenotype for transferred cells indicates that AKT is not promoting retention of donated cells in these studies. However, it is important to bear in mind that these were relatively short-term experimental designs, the potential contribution of endogenous stem / progenitor populations recruited by the donated cell population was not examined, and the AKT was a constitutively activated type rather than nuclear targeted. Moreover, the type of AKT activation employed was deliberately transient rather than engineered for endurance.
Multiple studies have implicated AKT as a critical mediator of cardiac regeneration in the damaged heart (Elmadbouh et al. 2007, Gude et al. 2006, Hur et al. 2007, McDevitt et al. 2005, Tateishi et al. 2007). However, a measured approach to cell-based regenerative treatment involving AKT needs to consider and incorporate multiple criteria such as: 1) AKT activation (type, duration, intensity), 2) characteristics of the cells being used for therapeutic intervention, 3) type and extent of cardiomyopathic injury, 4) timing and dosage of treatment, and 5) direct (e.g. engraftment) versus indirect (e.g. paracrine) mechanisms. Of course, any evaluation of AKT as a therapy (either as a target of gene therapy or an element of cell-based therapy) needs to be tested in real disease models beyond transgenic and small animal experimental systems. Furthermore, ongoing investigations of AKT-mediated effects will undoubtedly uncover additional downstream mediators of protective and proliferative signaling with a narrower scope than the diverse cellular functions of AKT. The foreseeable future will require progress toward AKT activity that is both regulated and focused, involving carefully selected cells with permanent genomic incorporation of optimally designed AKT purposefully created to facilitate cardiac repair. Toward that end, the engineering of cardiac stem cells modified to incorporate nuclear-targeted AKT into the genome results in markedly increased proliferation in vitro as well as enhanced regenerative activity upon reintroduction to the infarcted myocardium (Figure 1). Ex vivo manipulation of stem cells will enable the use of optimized populations of cells to rebuild and repair the damaged myocardium. All this is not as far-fetched as it may sound, as selective salutary effects of nuclear-targeted AKT upon survival and proliferation of cardiomyocytes / cardiac progenitor cells raise optimism that we are closer than ever to the goal of regenerating the heart.
Figure 1.
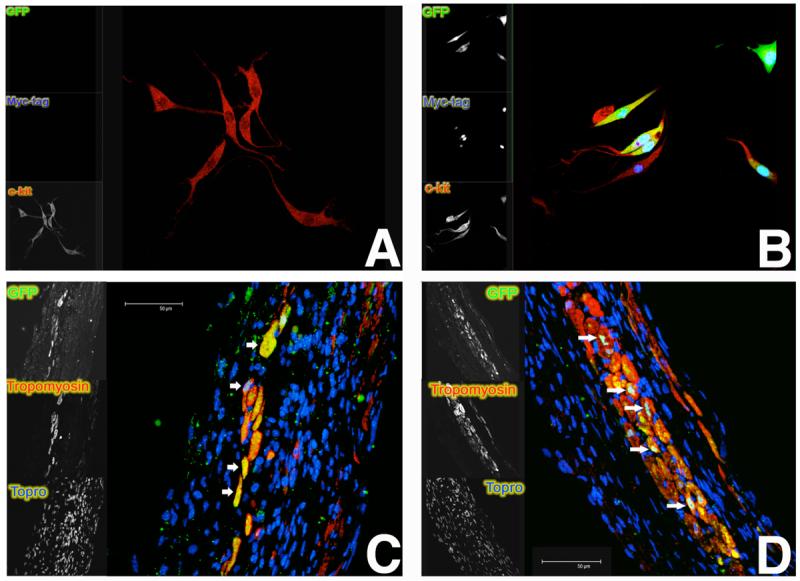
Cardiac stem cells genetically modified to express nuclear-targeted AKT at one month post-infarction. Such genetic modifications by AKT may pave the road to enhancing cardiac progenitor cell proliferation and survival. Unmodified c-kit+ cardiac stem cells (red in overlay) under normal conditions (A) or genetically engineered to express nuclear-targeted AKT (myc-tagged; blue in overlay) together with bicistronic green fluorescent protein (GFP) in culture (B). Genetically modified cells as shown in (B) adoptively transferred to infarcted recipient mice persist and commit to the myocyte lineage after adoptive transfer to syngeneic mice (C and D) as identified by presence of GFP (green in overlay). Cardiac commitment is indicated by expression of tropomyosin (red in overlay) together with GFP (arrows). Blue in tissue sections represents nuclei.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akala OO, Clarke MF. Hematopoietic stem cell self-renewal. Curr Opin Genet Dev. 2006;16:496–501. doi: 10.1016/j.gde.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442:823–826. doi: 10.1038/nature04940. [DOI] [PubMed] [Google Scholar]
- Ang KL, Shenje LT, Srinivasan L, Galinanes M. Repair of the damaged heart by bone marrow cells: from experimental evidence to clinical hope. Ann Thorac Surg. 2006;82:1549–1558. doi: 10.1016/j.athoracsur.2006.05.047. [DOI] [PubMed] [Google Scholar]
- Anversa P, Rota M, Urbanek K, Hosoda T, Sonnenblick EH, Leri A, et al. Myocardial aging--a stem cell problem. Basic Res Cardiol. 2005;100:482–493. doi: 10.1007/s00395-005-0554-3. [DOI] [PubMed] [Google Scholar]
- Bachmann M, Moroy T. The serine/threonine kinase Pim-1. Int J Biochem Cell Biol. 2005;37:726–730. doi: 10.1016/j.biocel.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Balsam LB, Robbins RC. Haematopoietic stem cells and repair of the ischaemic heart. Clin Sci (Lond) 2005;109:483–492. doi: 10.1042/CS20050087. [DOI] [PubMed] [Google Scholar]
- Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;25:917–931. doi: 10.1016/j.molcel.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Bruneau BG, Black BL. The heart's Da Vinci code: a Renaissance at Keystone. Development. 2007;134:1631–1633. doi: 10.1242/dev.002014. [DOI] [PubMed] [Google Scholar]
- Chao W, Matsui T, Novikov MS, Tao J, Li L, Liu H, et al. Strategic advantages of insulin-like growth factor-I expression for cardioprotection. J Gene Med. 2003;5:277–286. doi: 10.1002/jgm.347. [DOI] [PubMed] [Google Scholar]
- Cheng W, Li B, Kajstura J, Li P, Wolin MS, Sonnenblick EH, et al. Stretch-induced programmed myocyte cell death. J Clin Invest. 1995;96:2247–2259. doi: 10.1172/JCI118280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HJ, Lee J, Wecker A, Yoon YS. Bone marrow-derived stem cell therapy in ischemic heart disease. Regen Med. 2006;1:337–345. doi: 10.2217/17460751.1.3.337. [DOI] [PubMed] [Google Scholar]
- Christoforou N, Gearhart JD. Stem cells and their potential in cell-based cardiac therapies. Prog Cardiovasc Dis. 2007;49:396–413. doi: 10.1016/j.pcad.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Condorelli G, Drusco A, Stassi G, Bellacosa A, Roncarati R, Iaccarino G, et al. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:12333–12338. doi: 10.1073/pnas.172376399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, et al. Akt1 is required for physiological cardiac growth. Circulation. 2006;113:2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- Donath MY, Sutsch G, Yan XW, Piva B, Brunner HP, Glatz Y, et al. Acute cardiovascular effects of insulin-like growth factor I in patients with chronic heart failure. J Clin Endocrinol Metab. 1998;83:3177–3183. doi: 10.1210/jcem.83.9.5122. [DOI] [PubMed] [Google Scholar]
- Donath MY, Zapf J. Insulin-like growth factor I: an attractive option for chronic heart failure? Drugs Aging. 1999;15:251–254. doi: 10.2165/00002512-199915040-00001. [DOI] [PubMed] [Google Scholar]
- Elmadbouh I, Haider H, Jiang S, Idris NM, Lu G, Ashraf M. Ex vivo delivered stromal cell-derived factor-1alpha promotes stem cell homing and induces angiomyogenesis in the infarcted myocardium. J Mol Cell Cardiol. 2007;42:792–803. doi: 10.1016/j.yjmcc.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans SM, Mummery C, Doevendans PA. Progenitor cells for cardiac repair. Semin Cell Dev Biol. 2007;18:153–160. doi: 10.1016/j.semcdb.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Fazio S, Sabatini D, Capaldo B, Vigorito C, Giordano A, Guida R, et al. A preliminary study of growth hormone in the treatment of dilated cardiomyopathy. N Engl J Med. 1996;334:809–814. doi: 10.1056/NEJM199603283341301. [DOI] [PubMed] [Google Scholar]
- Flores I, Benetti R, Blasco MA. Telomerase regulation and stem cell behaviour. Curr Opin Cell Biol. 2006;18:254–260. doi: 10.1016/j.ceb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Forte G, Minieri M, Cossa P, Antenucci D, Sala M, Gnocchi V, et al. Hepatocyte growth factor effects on mesenchymal stem cells: proliferation, migration, and differentiation. Stem Cells. 2006;24:23–33. doi: 10.1634/stemcells.2004-0176. [DOI] [PubMed] [Google Scholar]
- Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101:660–667. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Germani A, Di Rocco G, Limana F, Martelli F, Capogrossi MC. Molecular mechanisms of cardiomyocyte regeneration and therapeutic outlook. Trends Mol Med. 2007;13:125–133. doi: 10.1016/j.molmed.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Gnecchi M, He H, Liang OD, Melo LG, Morello F, Mu H, et al. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat Med. 2005;11:367–368. doi: 10.1038/nm0405-367. [DOI] [PubMed] [Google Scholar]
- Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94:1621–1628. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- Gude N, Muraski J, Rubio M, Kajstura J, Schaefer E, Anversa P, et al. Akt promotes increased cardiomyocyte cycling and expansion of the cardiac progenitor cell population. Circ Res. 2006;99:381–388. doi: 10.1161/01.RES.0000236754.21499.1c. [DOI] [PubMed] [Google Scholar]
- Gupta S, Das B, Sen S. Cardiac hypertrophy: mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2007;9:623–652. doi: 10.1089/ars.2007.1474. [DOI] [PubMed] [Google Scholar]
- Hammerman PS, Fox CJ, Birnbaum MJ, Thompson CB. Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Blood. 2005;105:4477–4483. doi: 10.1182/blood-2004-09-3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur J, Yoon CH, Lee CS, Kim TY, Oh IY, Park KW, et al. Akt is a Key Modulator of Endothelial Progenitor Cell Trafficking in Ischemic Muscle. Stem Cells. 2007 doi: 10.1634/stemcells.2006-0385. [DOI] [PubMed] [Google Scholar]
- Imanishi T, Hano T, Nishio I. Estrogen reduces endothelial progenitor cell senescence through augmentation of telomerase activity. J Hypertens. 2005;23:1699–1706. doi: 10.1097/01.hjh.0000176788.12376.20. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Cheon SH, Yoo SJ, Kwon J, Park JH, Kim CG, et al. Contribution of the PI3K/Akt/PKB signal pathway to maintenance of self-renewal in human embryonic stem cells. FEBS Lett. 2005;579:534–540. doi: 10.1016/j.febslet.2004.12.024. [DOI] [PubMed] [Google Scholar]
- Kocher AA, Schlechta B, Gasparovicova A, Wolner E, Bonaros N, Laufer G. Stem cells and cardiac regeneration. Transpl Int. 2007;20:731–746. doi: 10.1111/j.1432-2277.2007.00493.x. [DOI] [PubMed] [Google Scholar]
- Kucia M, Dawn B, Hunt G, Guo Y, Wysoczynski M, Majka M, et al. Cells expressing early cardiac markers reside in the bone marrow and are mobilized into the peripheral blood after myocardial infarction. Circ Res. 2004;95:1191–1199. doi: 10.1161/01.RES.0000150856.47324.5b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Li B, Wang X, Leri A, Jana KP, Liu Y, et al. Overexpression of insulin-like growth factor-1 in mice protects from myocyte death after infarction, attenuating ventricular dilation, wall stress, and cardiac hypertrophy. J Clin Invest. 1997;100:1991–1999. doi: 10.1172/JCI119730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao R, Pfister O, Jain M, Mouquet F. The bone marrow--cardiac axis of myocardial regeneration. Prog Cardiovasc Dis. 2007;50:18–30. doi: 10.1016/j.pcad.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon A, Harding S. The potential of cardiac stem cell therapy for heart failure. Curr Opin Pharmacol. 2007;7:164–170. doi: 10.1016/j.coph.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Mathur A, Martin JF. Stem cells and repair of the heart. Lancet. 2004;364:183–192. doi: 10.1016/S0140-6736(04)16632-4. [DOI] [PubMed] [Google Scholar]
- Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH, et al. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J Biol Chem. 2002;277:22896–22901. doi: 10.1074/jbc.M200347200. [DOI] [PubMed] [Google Scholar]
- Matsui T, Nagoshi T, Hong EG, Luptak I, Hartil K, Li L, et al. Effects of chronic Akt activation on glucose uptake in the heart. Am J Physiol Endocrinol Metab. 2006;290:E789–797. doi: 10.1152/ajpendo.00564.2004. [DOI] [PubMed] [Google Scholar]
- McDevitt TC, Laflamme MA, Murry CE. Proliferation of cardiomyocytes derived from human embryonic stem cells is mediated via the IGF/PI 3-kinase/Akt signaling pathway. J Mol Cell Cardiol. 2005;39:865–873. doi: 10.1016/j.yjmcc.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirotsou M, Zhang Z, Deb A, Zhang L, Gnecchi M, Noiseux N, et al. Secreted frizzled related protein 2 (Sfrp2) is the key Akt-mesenchymal stem cell-released paracrine factor mediating myocardial survival and repair. Proc Natl Acad Sci U S A. 2007;104:1643–1648. doi: 10.1073/pnas.0610024104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummery C. Cardiomyocytes from human embryonic stem cells: more than heart repair alone. Bioessays. 2007;29:572–579. doi: 10.1002/bies.20583. [DOI] [PubMed] [Google Scholar]
- Nagoshi T, Matsui T, Aoyama T, Leri A, Anversa P, Li L, et al. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J Clin Invest. 2005;115:2128–2138. doi: 10.1172/JCI23073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano J, Shiota G, Matsumoto K, Yasui S, Kurimasa A, Hisatome I, et al. Hepatocyte growth factor exerts a proliferative effect on oval cells through the PI3K/AKT signaling pathway. Biochem Biophys Res Commun. 2003;309:298–304. doi: 10.1016/j.bbrc.2003.04.002. [DOI] [PubMed] [Google Scholar]
- Otaegi G, Yusta-Boyo MJ, Vergano-Vera E, Mendez-Gomez HR, Carrera AC, Abad JL, et al. Modulation of the PI 3-kinase-Akt signalling pathway by IGF-I and PTEN regulates the differentiation of neural stem/precursor cells. J Cell Sci. 2006;119:2739–2748. doi: 10.1242/jcs.03012. [DOI] [PubMed] [Google Scholar]
- Pallante BA, Edelberg JM. Realizing the cardiac stem cell promise: a case for trophism. Regen Med. 2006;1:217–221. doi: 10.2217/17460751.1.2.217. [DOI] [PubMed] [Google Scholar]
- Reiss K, Cheng W, Ferber A, Kajstura J, Li P, Li B, et al. Overexpression of insulin-like growth factor-1 in the heart is coupled with myocyte proliferation in transgenic mice. Proc Natl Acad Sci U S A. 1996;93:8630–8635. doi: 10.1073/pnas.93.16.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggia C, Ukena C, Bohm M, Kilter H. Hepatocyte growth factor (HGF) enhances cardiac commitment of differentiating embryonic stem cells by activating PI3 kinase. Exp Cell Res. 2007;313:921–930. doi: 10.1016/j.yexcr.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Schuleri KH, Boyle AJ, Hare JM. Mesenchymal stem cells for cardiac regenerative therapy. Handb Exp Pharmacol. 2007:195–218. doi: 10.1007/978-3-540-68976-8_9. [DOI] [PubMed] [Google Scholar]
- Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi I, Melendez J, Ahn Y, Skavdahl M, Murphy E, Welch S, et al. Nuclear targeting of Akt enhances kinase activity and survival of cardiomyocytes. Circ Res. 2004;94:884–891. doi: 10.1161/01.RES.0000124394.01180.BE. [DOI] [PubMed] [Google Scholar]
- Sohn RL, Jain M, Liao R. Adult stem cells and heart regeneration. Expert Rev Cardiovasc Ther. 2007;5:507–517. doi: 10.1586/14779072.5.3.507. [DOI] [PubMed] [Google Scholar]
- Su EJ, Cioffi CL, Stefansson S, Mittereder N, Garay M, Hreniuk D, et al. Gene therapy vector-mediated expression of insulin-like growth factors protects cardiomyocytes from apoptosis and enhances neovascularization. Am J Physiol Heart Circ Physiol. 2003;284:H1429–1440. doi: 10.1152/ajpheart.00885.2002. [DOI] [PubMed] [Google Scholar]
- Taniyama Y, Ito M, Sato K, Kuester C, Veit K, Tremp G, et al. Akt3 overexpression in the heart results in progression from adaptive to maladaptive hypertrophy. J Mol Cell Cardiol. 2005;38:375–385. doi: 10.1016/j.yjmcc.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Tateishi K, Ashihara E, Honsho S, Takehara N, Nomura T, Takahashi T, et al. Human cardiac stem cells exhibit mesenchymal features and are maintained through Akt/GSK-3beta signaling. Biochem Biophys Res Commun. 2007;352:635–641. doi: 10.1016/j.bbrc.2006.11.096. [DOI] [PubMed] [Google Scholar]
- Taylor DA. Cell-based myocardial repair: how should we proceed? Int J Cardiol. 2004;95(Suppl 1):S8–12. doi: 10.1016/s0167-5273(04)90003-4. [DOI] [PubMed] [Google Scholar]
- Testa JR, Bellacosa A. Membrane translocation and activation of the Akt kinase in growth factor-stimulated hematopoietic cells. Leuk Res. 1997;21:1027–1031. doi: 10.1016/s0145-2126(97)00093-3. [DOI] [PubMed] [Google Scholar]
- Torella D, Rota M, Nurzynska D, Musso E, Monsen A, Shiraishi I, et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res. 2004;94:514–524. doi: 10.1161/01.RES.0000117306.10142.50. [DOI] [PubMed] [Google Scholar]
- Tsujita Y, Muraski J, Shiraishi I, Kato T, Kajstura J, Anversa P, et al. Nuclear targeting of Akt antagonizes aspects of cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2006;103:11946–11951. doi: 10.1073/pnas.0510138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura R, Xu M, Ahmad N, Ashraf M. Bone marrow stem cells prevent left ventricular remodeling of ischemic heart through paracrine signaling. Circ Res. 2006;98:1414–1421. doi: 10.1161/01.RES.0000225952.61196.39. [DOI] [PubMed] [Google Scholar]
- Wang L, Ma W, Markovich R, Chen JW, Wang PH. Regulation of cardiomyocyte apoptotic signaling by insulin-like growth factor I. Circ Res. 1998;83:516–522. doi: 10.1161/01.res.83.5.516. [DOI] [PubMed] [Google Scholar]
- Wang Z, Bhattacharya N, Weaver M, Petersen K, Meyer M, Gapter L, et al. Pim-1: a serine/threonine kinase with a role in cell survival, proliferation, differentiation and tumorigenesis. J Vet Sci. 2001;2:167–179. [PubMed] [Google Scholar]
- Welch S, Plank D, Witt S, Glascock B, Schaefer E, Chimenti S, et al. Cardiac-specific IGF-1 expression attenuates dilated cardiomyopathy in tropomodulin-overexpressing transgenic mice. Circ Res. 2002;90:641–648. doi: 10.1161/01.res.0000013780.77774.75. [DOI] [PubMed] [Google Scholar]
- Ye P, D'Ercole AJ. Insulin-like growth factor actions during development of neural stem cells and progenitors in the central nervous system. J Neurosci Res. 2006;83:1–6. doi: 10.1002/jnr.20688. [DOI] [PubMed] [Google Scholar]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- Zhang X, Vik TA. Growth factor stimulation of hematopoietic cells leads to membrane translocation of AKT1 protein kinase. Leuk Res. 1997;21:849–856. doi: 10.1016/s0145-2126(97)00055-6. [DOI] [PubMed] [Google Scholar]