

Abstract
Elevated extracellular glutamate, resulting from a loss of astrocytic glutamate transport capacity, may contribute to excitotoxic motor neuron (MN) damage in ALS. Accounting for their high excitotoxic vulnerability, MNs possess large numbers of unusual Ca2+ permeable AMPA channels (Ca-AMPA channels), the activation of which triggers mitochondrial Ca2+ overload and strong reactive oxygen species (ROS) generation. However, the causes of the astrocytic glutamate transport loss remain unexplained. To assess the role of Ca-AMPA channels on the evolution of pathology in vivo, we have examined effects of prolonged intrathecal infusion of the Ca-AMPA channel blocker, 1-naphthyl acetylspermine (NAS), in G93A transgenic rat models of ALS. In wild type animals, immunoreactivity for the astrocytic glutamate transporter, GLT-1, was particularly strong around ventral horn MNs. However, a marked loss of ventral horn GLT-1 was observed, along with substantial MN damage, prior to onset of symptoms (90-100 d) in the G93A rats. Conversely, labeling with the oxidative marker, nitrotyrosine, was increased in the neuropil surrounding MNs in the transgenic animals. Compared to sham treated G93A animals, 30 day NAS infusions (starting at 67±2 days of age) markedly diminished the loss of both MNs and of astrocytic GLT-1 labeling. These observations are compatible with the hypothesis that activation of Ca-AMPA channels on MNs contributes, likely in part through oxidative mechanisms, to loss of glutamate transporter in surrounding astrocytes.
Keywords: amyotrophic lateral sclerosis, G93A, excitotoxicity, EAAT2, motor neuron, 3-nitrotyrosine, 1-naphthyl acetylspermine
Amyotrophic lateral sclerosis (ALS) is an adult onset neurodegenerative disease characterized by the selective loss of upper and lower motor neurons (MNs). Although the cause of most cases is unknown, observations of deficiencies in glutamate uptake, resulting from a selective loss of the astrocytic glutamate transporter, GLT-1, suggested an excitotoxic contribution (Rothstein et al., 1992; Rothstein et al., 1995).
Much has been learned about factors that may make MNs particularly susceptible to excitotoxic injury. First, MNs are unusually sensitive to injury mediated through AMPA/kainate type glutamate receptors (Carriedo et al., 1995; Carriedo et al., 1996; Hugon et al., 1989; Rothstein and Kuncl, 1995), and blockers of these receptors protect MNs from injury caused by prolonged blockade of glutamate uptake (Carriedo et al., 1996; Rothstein et al., 1993).
Likely contributing to this vulnerability, MNs possess substantial numbers of unusual Ca2+ permeable AMPA type glutamate receptor channels (Ca-AMPA channels) (Carriedo et al., 1995; Carriedo et al., 1996; Van Den Bosch et al., 2000; Vandenberghe et al., 2000). Currently, the best animal models of ALS are provided by rodents harboring mutant forms of the enzyme Cu, Zn superoxide dismutase (SOD1), which are associated with familial ALS in humans. Solidifying the role of Ca-AMPA channels in in vivo models of ALS, recent studies indicate that the rate of progression of MN loss in SOD1 mutant mice varies bidirectionally with the level of expression of these channels (Kuner et al., 2005; Tateno et al., 2004; Van Damme et al., 2005). In addition, Ca-AMPA channels appear to contribute to MN loss in a distinct form of familial ALS, not linked to SOD1 (Lai et al., 2006) and a Ca-AMPA channel blocker was found to be protective in a model of virus induced MN degeneration (Darman et al., 2004).
Mechanisms through which Ca-AMPA channels mediate excitotoxic MN injury are also becoming elucidated. While these channels permit rapid Ca2+ entry, MNs buffer cytosolic Ca2+ loads poorly (Lips and Keller, 1998), with the consequence that much of the Ca2+ is readily taken up into mitochondria, resulting in strong ROS generation (Carriedo et al., 2000; Rao et al., 2003).
However, factors underlying the loss of astrocytic glutamate transport, which likely accounts for excitotoxic MN injury, have been unexplained. Providing a possible clue, recent in vitro studies indicated that the ROS produced in MNs in response to Ca-AMPA channel activation was capable of inducing oxidative disruption of glutamate transporters in surrounding astrocytes (Rao et al., 2003). If such a mechanism contributed to glutamate transport disruption in ALS, it could provide the basis for a feed forward cycle that could be integral to disease progression (Rao and Weiss, 2004).
The aim of the present study was to gain insights into ways in which Ca-AMPA channels may contribute to the pathological processes leading to MN degeneration in an in vivo animal model of ALS. Specifically, in light of culture studies suggesting that ROS produced in MNs in response to excitotoxic activation might contribute to astrocytic dysfunction, our aim was to examine effects of prolonged Ca-AMPA channel blockade not only on MN loss, but on oxidative changes and transporter levels in surrounding astrocytes as well. As there are presently no CaAMPA channel blockers that can be administered systemically, we have carried out prolonged (1 month) intrathecal infusions of the Ca-AMPA channel blocker, 1-naphthyl acetylspermine (NAS) in G93A transgenic SOD1 rats. We find that this treatment slows not only MN loss in these animals, but also slows the loss of GLT-1 glutamate transporter in ventral horn regions near MNs, consistent with the idea that Ca-AMPA channel activation on MNs contributes to the loss of astrocytic glutamate transport.
Materials and Methods
Animals
Male hemizygous SOD1 G93A transgenic rats [Tac:N:(SD)-TgN(SOD1G93A)L26H, Emerging Models Program sponsored by Amyotrophic Lateral Sclerosis Association, Taconic labs, Germantown, NY] were bred with wild type females, and offspring genotyped by PCR analysis (Howland et al., 2002); wild type siblings serve as controls for mutant animals. Animals are killed when they can no longer right themselves within 10 seconds of being pushed on their side. All animal procedures were approved by the Institutional Animal Care and Use Committee.
Surgical procedure
Intrathecal infusion studies used rats at 65-70 days of age (body weight ∼200-300 grams). Anesthesia was induced using 5% isoflurane, and maintained at 2.5% at a flow rate of 1 L/min. An incision was made through the skin of the dorsal head and the atlanto-occipital membrane, through which a PE5 catheter was inserted into the subarachnoid space and advanced 6.5-8.5 cm to the lumbar enlargement, as previously described (Hayes et al., 2003). The catheter was connected to an Alzet mini-osmotic pump (model 2004; 200 μl volume, 0.25 μl/hr x 30 d) which was pre-filled with NAS (21mM), or saline, as described (Darman et al., 2004). After surgery, animals were housed individually and body weight recorded daily. In addition, evidence of pain or infection and motor dysfunction were closely monitored. Animals were sacrificed if they appeared distressed or if after 5 days, motor function remained impaired or body weight had not recovered to pre surgery levels.
Tissue Preparation and staining
30 days after the start of intrathecal infusions, the animals were anesthetized with ketamine, and perfused transcardially with PBS, followed by 4% paraformaldehyde (PFA) for 10 minutes. The lumbar enlargements of spinal cords were dissected and post-fixed in 4% PFA for 24 hrs and removed to 30% sucrose/PBS for another 2 days. Serial 25 μm frozen sections were cut from the middle of the lumbar enlargement for ∼ 5 mm in the caudal direction.
After every 10 sections, 3-4 serial sections were set aside for staining. Immunohistochemical stains were carried out on floating sections, blocked (10% FBS, 1h), and exposed to primary antibody in 10% FBS, 0.3% Triton-X 100 (SMI-32, 1:8000 ip, 1:2000 if, Sternberger Monoclonals, Berkeley, CA; GLT-1, 1: 1000, Chemicon, Temecula, CA; 3-Nitrotyrosine, 10 μg/ml, Upstate Biotechnology, Waltham, MA). Labeling was visualized either by routine ABC immunoperoxidase techniques or under fluorescence using secondary antibodies linked to fluorophores (Alexafluor 488, Molecular Probes, Eugene, OR; or Cy3, Jackson immunoResearch, West Grove, PA).
Quantification of histopathological changes
Surviving MNs were counted in ventral horn of lumbar spinal slices from each condition (15-20 slices per animal). MNs with pyknotic nuclei, or with a markedly atrophic, irregular or fragmented soma were not counted as alive. For examination of NT and GLT-1 labeling staining in the neuropil surrounding ventral horn MNs, care was taken to ensure that all slices from each experiment were labeled using identical primary and secondary antibody exposures, and fluorescence photographs taken with identical camera settings so that labeling could be compared. For quantification, photographs were imported into an image analysis package (Metamorph software, Molecular Devices Corp., Downington, PA) as 8 bit gray scale images, and regions of neuropil marked surrounding all readily identifiable surviving ventral horn MNs, extending outward from the border of the soma but masking out evident neurons or neuronal processes. For NT, fluorescence intensity was measured in 25 μm zones surrounding identified MNs, and the raw mean intensity value surrounding each MN normalized to that surrounding MNs in the WT condition of the same experiment. For GLT-1, fluorescence was measured in 5 μm zones surrounding the MNs. For this measure, non-specific background fluorescence (from the center of a neuron, a region lacking specific GLT-1 labeling) was subtracted prior to normalization of values to WT as above.
Chemicals and Reagents
NAS was kindly provided by Daicel Chemical Corporation (Tokyo, Japan). Antibodies were from the following sources: SMI-32, Sternberger Monoclonals, Berkeley, CA; GLT-1, Chemicon, Temecula, CA; 3-Nitrotyrosine, Upstate Biotechnology, Waltham, MA. For fluorescence labeling, we used secondary antibodies linked to the Alexafluor 488(Molecular Probes, Eugene, OR) or Cy3 (Jackson ImmunoResearch, West grove, PA). All other chemicals and reagents were obtained from common commercial sources.
Results
NAS slows MN loss in G93A SOD1 transgenic rats
As previously reported, hemizygous G93A SOD1 transgenic rats generally develop symptoms between ∼115 and 130 day of age and progress rapidly to substantial paralysis (generally within 7-10 days) (Howland et al., 2002). In initial studies we examined pathological indices in lumbar spinal cord at late presymptomatic ages (95-100 days), in comparison to both age matched controls (littermates lacking the transgene) and “end stage” transgenic animals (defined as onset of symptomatic gait impediment). MNs were labeled with Nissl stain, and immunocytochemically using an antibody to non-phosphorylated nerurofilament epitopes, SMI-32, which strongly labels MNs as well as their neuritic processes (Carriedo et al., 1996). In the WTs, virtually all MNs appeared healthy. However, in the transgenics, there were many MNs in differing stages of degeneration, as indicated by cellular and nuclear constriction, eccentric nuclei, vacuolar changes, and fragmentation of dendrites. In some cases, degenerated MNs were replaced by glial nodules.
We next set out to examine the effect of prolonged Ca-AMPA channel inhibition on this MN damage. The Ca-AMPA channel blocker, NAS, was delivered intrathecally over 30 days, via an Alzet minipump. Each experiment comprised 3 groups: wild type animals fitted with a minipump and infused with saline (WT), transgenic animals infused with saline (Tg-S), and sibling transgenic animals infused with NAS (Tg-NAS). Infusions were started at 67±2 days of age. At the end of the infusion, animals were perfused and spinal cords removed for pathological examination as described. In order to compare tissue from these experimental treatment animals with end stage (symptomatic) animals, spinal cords from some end stage animals (Tg-ES) were fixed and kept frozen for sectioning, staining and photographing in parallel with experimental treatment animals.
Surviving MNs were counted in slices from each condition. In WTs, there were an average of ∼ 50 MNs / slice (comprising 2 ventral horns), with ∼ 40% loss in the Tg-S condition, and substantially greater MN loss in the end stage (Tg-ES) animals. However, in transgenics infused with NAS (Tg-NAS), MN loss was significantly decreased (Fig. 1).
1. Progressive motor neuron degeneration in G93A rats: attenuation by Ca-AMPA channel blockade.
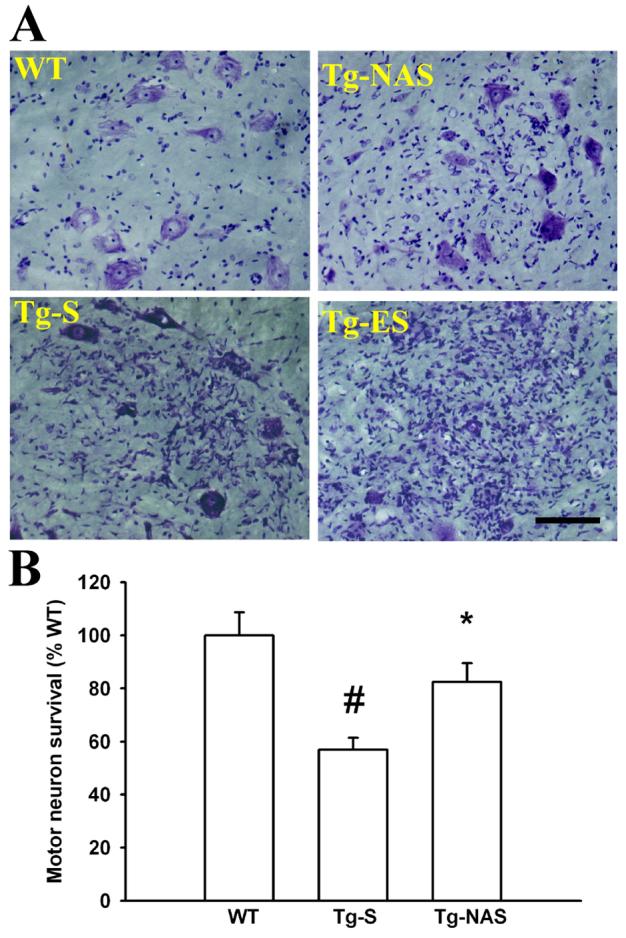
A. Motor neuron morphology: Photomicrographs (200x) show ventral horn regions of lumbar spinal cord sections derived from 97±2 day old rats (upon termination of the 30 day intrathecal infusions), stained for Nissl substance. In contrast to the healthy appearing MNs in the control (WT) condition, in transgenic animals treated with saline infusion (Tg-S), there is moderate MN injury characterized variably by cell shrinkage and fragmentation, eccentric nuclei, and microglial infiltration. The infusion of NAS into the transgenic animals (Tg-NAS) decreased MN injury. In symptomatic transgenic animals (Tg-ES), MN injury was far greater than in the presymptomatic animals, with >70% MN loss or severe injury. Bar = 100 μm.
B. Quantification of injury: Graph shown quantification of MN injury, based upon cell counts from 7 independent experiments (12-13 animals, > 10 slices counted / animal each condition). # indicates difference from WT, * indicates difference from Tg-S by 2-tailed t test (p<0.01).
Effects of NAS on glial pathology in G93A SOD1 transgenic rats
Further pathological examination was focused on characterizing associated changes in nonneuronal cells, most prominently astrocytes. Previous reports have indicated progressive reactive astrogliosis, nitrotyrosine labeling and loss of astrocytic glutamate transport in SOD1 mutant rodent models of ALS (Alexander et al., 2000; Ferrante et al., 1997; Tu et al., 1996), and a specific decrease of the GLT-1 glutamate transporter has been reported in ventral horn of the G93A rats that are the subject of this study (Howland et al., 2002). Consistent with these prior studies we see a dramatic increase in astrogliosis, as indicated by GFAP labeling, most prominent initially in ventral horn, and extending throughout the gray matter of the spinal cord by the onset of symptoms (Fig 2A, 3). Counts in ventral horn revealed an increase in both numbers and intensity of labeling of large GFAP-positive astrocytes in Tg-S, mild attenuation of this astrocytosis in the Tg-NAS condition, and markedly increased astrocytosis in Tg-ES (Fig. 3B).
2. Marked changes in astrocytes surrounding ventral horn MNs in presymptomatic G93A rats.
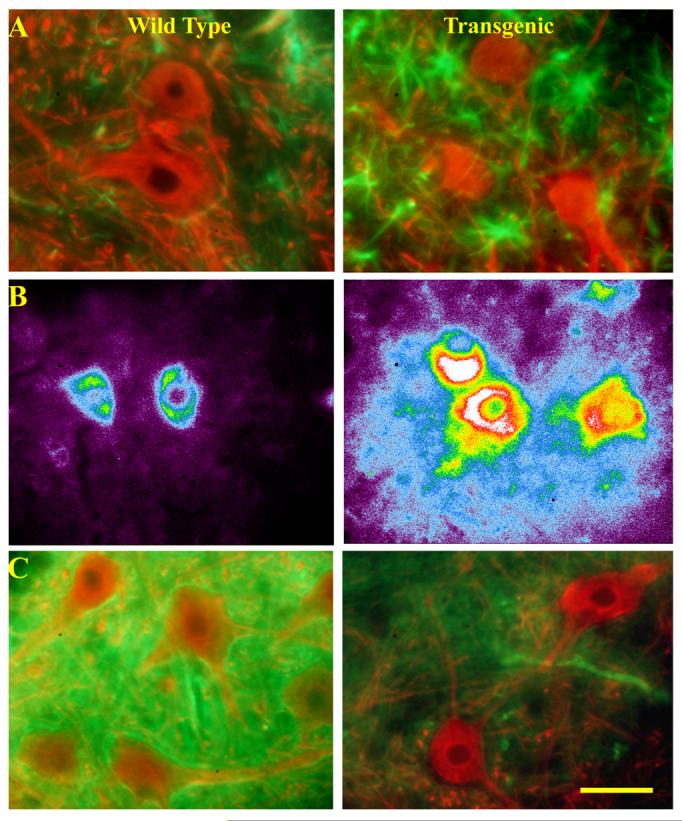
Lumbar spinal cord sections were obtained from 97±2 day old wild type and G93A transgenic rats and stained for glial fibrillary acidic protein (GFAP, green) and SMI-32 (red, A), 3-nitrotyrosine (B), or the astrocytic glutamate transporter, GLT-1 (green) along with SMI-32 (red, C). Note the increased number of reactive astrocytes near transgenic MNs indicated by strong GFAP labeling. Further note the nitrotyrosine labeling immediately surrounding transgenic MNs, consistent with the possibility that the labeling is the result of ROS produced within MNs (the image is shown in pseudocolor, with white and red highest and purple lowest in order to highlight the gradient in labeling intensity). Finally note the rim of strong GLT-1 labeling surrounding MNs in wild type animals and the distinct loss of labeling surrounding MNs in the transgenics.
3. Progressive astrogliosis and nitrotyrosine labeling of G93A spinal cord.
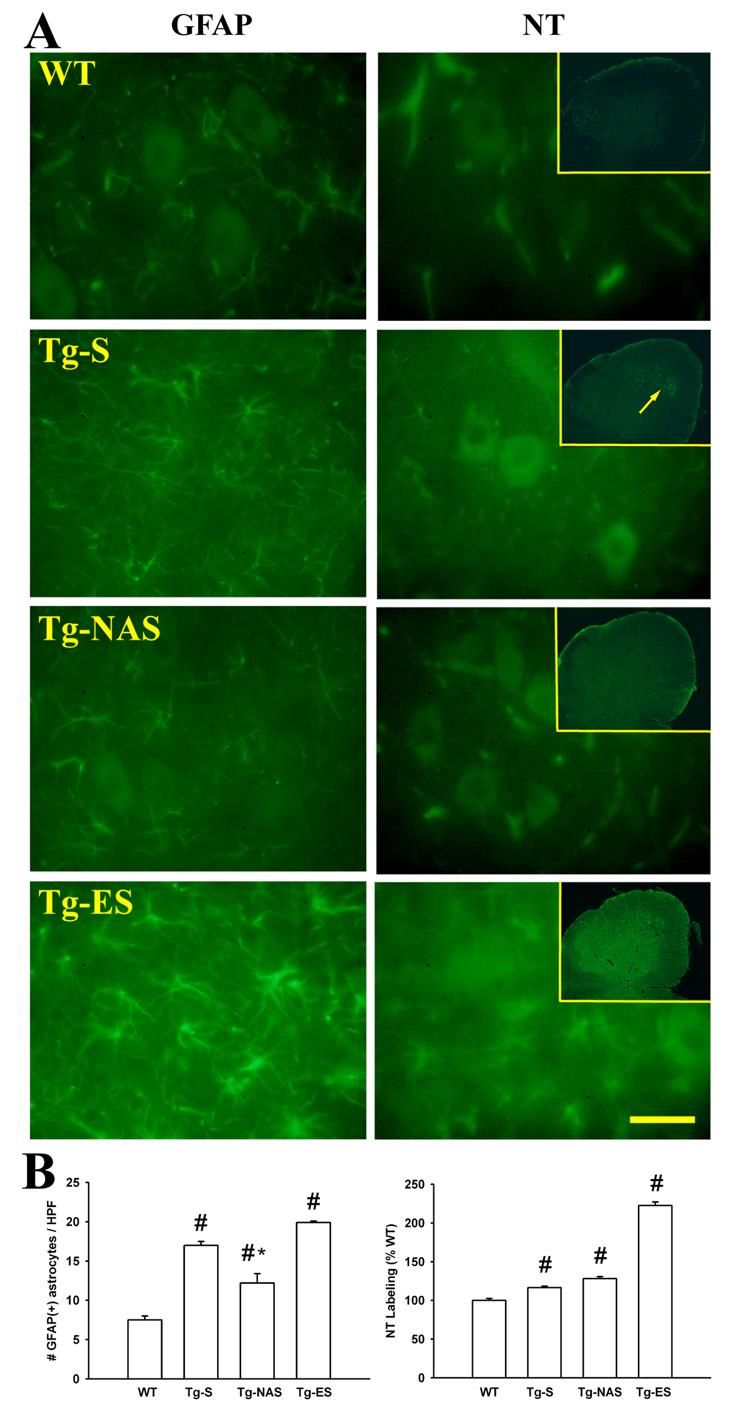
A. Photomicrographs show the ventral horn region of lumbar spinal cord sections derived from 97±2 day old rats (upon termination of the 30 day intrathecal infusions), stained for glial fibrillary acidic protein (GFAP; 400x) or for 3-nitrotyrosine (NT; 400x, inserts 40x). Note the marked increase in numbers of GFAP-positive reactive astrocytes in transgenic animals (Tg-S), the modest attenuation of this astrogliosis in the presence of NAS (Tg-NAS), and the sharp further increase in astrogliosis in symptomatic (Tg-ES) animals.
The irregular areas of NT staining in wild type animals (WT) largely represent capillary endothelium. Note the marked increase in NT labeling in the transgenic animals (Tg-S), initially most prominent within and surrounding ventral horn MNs, with a further increase in staining of neuropil and of reactive astrocytes throughout the gray matter in symptomatic (Tg-ES) animals. Arrow shows early staining in ventral horn of transgenic animals. Bar = 50 μm.
B. Quantification of labeling: Graphs show quantification of GFAP and NT labeling. GFAP labeling was quantified as the number of distinct GFAP-positive astrocytes per high power (400x) field, whereas NT staining was quantified as the mean labeling intensity in 25 μm zones surrounding each MN (GFAP counts based upon 6 independent experiments, > 20 fields for WT, Tg-S and Tg-NAS conditions; 3 experiments, 12 fields for Tg-ES. NT values based upon cell counts from 3 independent experiments, >70 surround regions for WT and Tg-S conditions; 3 animals, >70 regions for Tg-ES condition), # indicates difference from WT, * indicates difference from Tg-S by 2-tailed t test (p<0.01).
Paralleling this increased astrogliosis, we also note marked increases in 3-nitrotyrosine (NT) labeling. This was initially particularly prominent in ventral horn with increased labeling of MNs as well as increased diffuse staining in the neuropil surrounding and between many MNs. Of note, this NT labeling in the presymptomatic animals was often particularly strong in the close proximity to MN somata, falling off sharply with distance, possibly consistent with a role of MN ROS in the astrocyte damage (Fig. 2B).
We next attempted to obtain quantitative measures of the effects of NAS treatment on the intensity of NT staining in the neuropil surrounding ventral horn MNs (see Materials and Methods). There was a modest increase in NT labeling in the Tg-S conditions, which, however, was not decreased by NAS. The reason for the lack of effect of NAS on NT labeling are uncertain, but might reflect a narrow temporal window of strong peri-motor neuronal NT labeling, due to both the non-uniform nature of the early disease, and possibly partial resolution of the staining when MNs become severely dysfunctional or die. However, NT labeling was markedly stronger in the Tg-ES condition, with a widespread increase in labeling of astrocyte somata throughout the gray matter of the spinal cord (Fig. 3).
Finally, there was a marked and consistent decrease in GLT-1 staining, most prominent in ventral horn. Notably, in wild type animals, GLT-1 labeling often showed a rim of particularly strong labeling immediately surrounding MN somata, whereas even in the presymptomatic animals this rim was often conspicuously absent (Fig. 2C). Conversely, strong staining in dorsal horn persisted even in symptomatic animals (Fig. 4). Quantification of GLT-1 labeling in the neuropil surrounding MNs revealed a sharp decrease in GLT-1 labeling surrounding MNs in sham treated transgenic animals (Tg-S). However, in contrast to the paucity of effect of NAS on NT labeling, NAS completely prevented the loss of GLT-1 labeling (Fig. 4).
4. Loss of GLT-1 in ventral horn astrocytes of G93A rats and preservation by NAS.
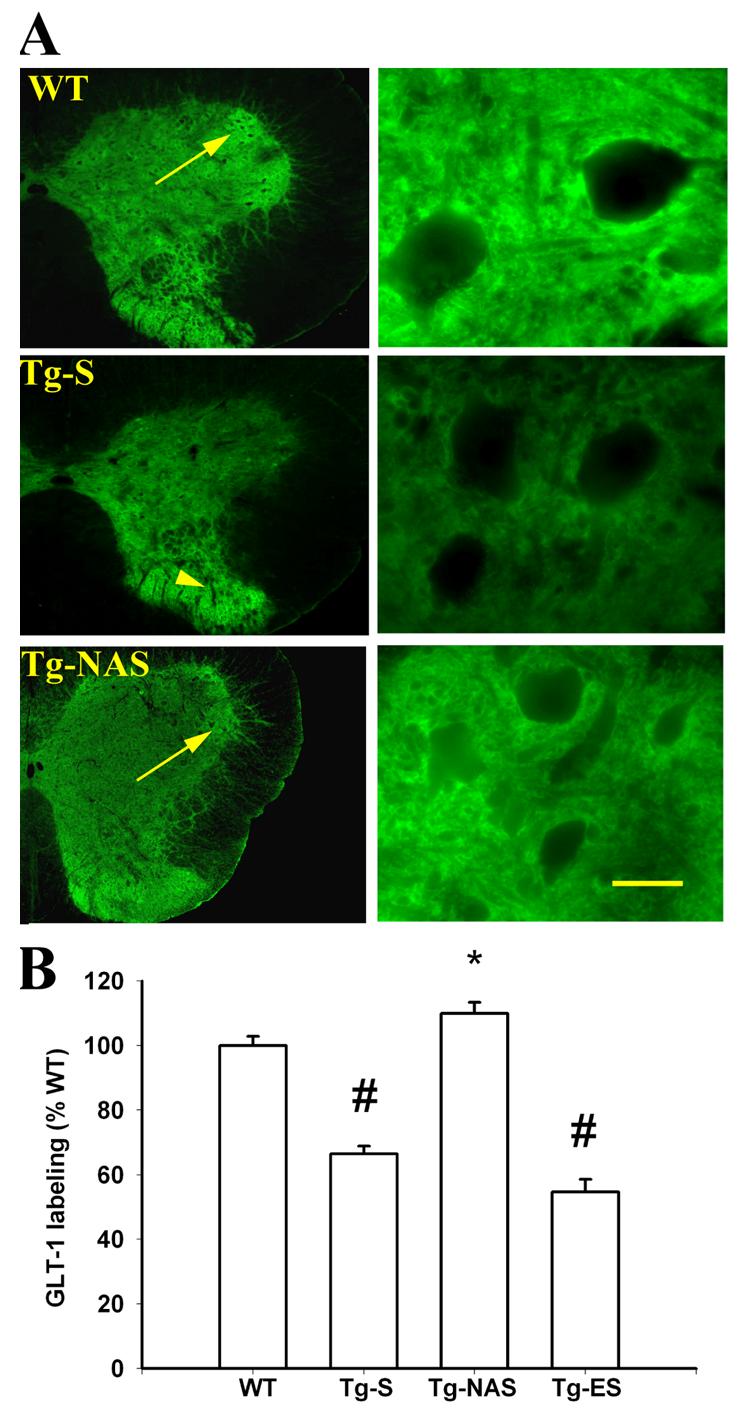
A. GLT-1 immunofluorescence: Photomicrographs show the ventral horn region of lumbar spinal cord sections derived from 97±2 day old rats (upon termination of the 30 day intrathecal infusions), stained for GLT-1 (40, 400x). Note the strong GLT-1 labeling surrounding MNs and throughout ventral horn MN clusters in wild type (WT) animals, in contrast to the preferential loss of ventral horn labeling, with preservation of dorsal horn labeling in transgenic animals (Tg-S). Also note the marked preservation of the ventral horn labeling with NAS treatment (Tg-NAS). Arrows show clusters of ventral horn motor neurons with strong astrocytic GLT-1 labeling; arrowhead shows persistent strong GLT-1 labeling in dorsal horn of untreated transgenic animals. Bar = 500 μm (left column) or 50 μm (right column).
B. Quantification of labeling: Graph shown quantification of GLT-1 labeling in 5 μm zones surrounding each MN (compiled from 4 independent experiments, > 100 surround regions for WT, Tg-S, and Tg-NAS conditions; 3 animals, >60 regions for Tg-ES condition). # indicates difference from WT, * indicates difference from Tg-S by 2-tailed t test (p<0.01).
Discussion
Summary of principal findings
In agreement with prior studies of these animals (Howland et al., 2002) we observe substantial loss of the GLT-1 glutamate transporter, most evident in the ventral horn in close proximity to MNs, in late presymptomatic animals (∼97 days), shortly before an acceleration in the rate of MN death with development of symptoms. The loss of transporter was paralleled by the appearance of nitrotyrosine labeling of the neuropil surrounding many ventral horn MNs in transgenic but not in wild type animals. The primary new findings of present studies are that animals treated with the Ca-AMPA channel blocker, NAS, demonstrate a marked preservation not only of ventral horn MNs, but also of GLT-1 labeling near ventral horn MNs. While we cannot rule out the possibility that NAS has direct effects on astrocytes, observations that MNs are strong ROS generators in response to Ca-AMPA channel activation taken together with present observations of strong oxidative damage in the vicinity of MNs, and protection by NAS of MNs themselves as well as of GLT-1 levels in the adjacent astrocytes is most consistent with the primary relevant effect of NAS being MN Ca-AMPA channel blockade.
Clues to MN loss in G93A model of ALS
As discussed in the introduction, observations of impaired astrocytic glutamate transport in ALS support an excitotoxic contribution to MN damage in the disease (Rothstein et al., 1992). Indeed, observations that substantial loss of GLT-1 preceded much of the MN loss (Howland et al., 2002), and that increasing the level of this transporter slows disease onset (Guo et al., 2003) whereas decreasing its levels accelerates disease in G93A transgenic mice (Pardo et al., 2006) support the idea that transporter loss contributes directly to MN damage.
However, the reasons for this loss of astrocytic glutamate transport have remained largely obscure. In our prior culture studies, we found that the ROS generated in MNs in response to excitotoxic activation appeared able to cause a rapid disruption of glutamate transport in adjacent astrocytes, leading us to suggest that similar MN ROS generation might contribute to astrocyte dysfunction in ALS. Consistent with this possibility, oxidative tissue damage is prominent in ALS, and progressive nitrotyrosine labeling of astrocytes as well as MNs is well documented in both sporadic human and SOD1 linked forms of the disease (Beal et al., 1997; Ferrante et al., 1997). Perhaps, in addition to suggesting a general role of tissue oxidation in the disease, our observations of distinct nitrotyrosine labeling surrounding ventral horn MNs in transgenic animals reflect tissue damage resulting in part from ROS production in MNs.
Present studies provide new clues to mechanisms of transport loss. Specifically, we now find that a Ca-AMPA channel blocker slows the loss of astrocytic transporter. Thus, whereas transport loss appears to contribute to excitotoxic MN damage, present observations support the converse: that activation of MNs by glutamate contributes to the dysfunction of nearby astrocytes (likely at least in part through oxidative mechanisms), resulting in further elevations in extracellular glutamate. If these reciprocal interactions between MNs and astrocytes occur in concert, the stage could be set for a feed forward vicious cycle, which once established could be self propagating (Rao and Weiss, 2004). Such a mechanism could help to explain features of ALS including the rapid progression of the disease after onset, as well as observations that the disease often seems to spread contiguously through the spinal cord. Of note, while SOD1 mutations only account for a small percentage of human ALS cases, these mutations cause a disease which is virtually indistinguishable from sporadic ALS (which itself likely has multiple causes), with both manifesting similar pathological hallmarks including oxidative tissue damage, mitochondrial dysfunction, protein aggregates and loss of astrocytic glutamate transport. Furthermore, there is no reason to assume that the reciprocal interactions between MNs and glia which we suggest are unique to SOD1 linked forms of disease; transporter loss and oxidative tissue damage are equally prevalent in sporadic disease. Thus, it seems likely that in ALS, a spectrum of inciting insults can converge into a final common self propagating disease pathway, accounting for the clinicopathological manifestations of the disease.
Implications for therapy
It has become increasingly clear that MN degeneration in SOD1 linked models of ALS is non-cell-autonomous, with the genotype of glia importantly impacting the fate of nearby MNs (Boillee et al., 2006; Clement et al., 2003; Gong et al., 2000; Pramatarova et al., 2001). There has been much interest in ways in which glial cells may contribute to MN injury; implicated mechanisms include the loss of glutamate transport, as well as the production of toxic compounds that can damage MNs (Nagai et al., 2007). Conversely, present observations lend preliminary in vivo support to the idea that ROS produced in MNs in response to excitotoxic activation and mitochondrial Ca2+ loading constitutes one MN-derived signal contributing to glial dysfunction.
However, there is likely to be more than one type of interaction between dysfunctional MNs and glia contributing to disease pathogenesis. For instance, another potential mechanism that could underlie a positive feedback is inflammation, which could enhance MN damage and/or impair astrocyte function, with consequent tissue damage resulting in further inflammation (Darman et al., 2004; Zhao et al., 2004). Since it may generally be impossible to identify initiating causes early enough to intervene and prevent the evolution of disease, our best chance for effective treatment of most sporadic ALS may be to target the mechanisms underlying disease propagation and progression. Indeed, present studies only demonstrate effects on the evolution of pathology during presymptomatic stages of disease, and further studies will be necessary to determine whether Ca-AMPA channel blockers alone will provide substantial clinical benefit. However, if we can further characterize late stage events in ALS, particularly those that are components of positive feedback cycles between MN and glia, it is likely that combined interventions targeting multiple limbs of the cycles will prove most effective at slowing disease while minimizing disruption of normal function.
Acknowledgements
This work was supported by NIH grant NS36548 (JHW) and a grant from the Muscular Dystrophy Association (JHW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander GM, Deitch JS, Seeburger JL, Del Valle L, Heiman-Patterson TD. Elevated cortical extracellular fluid glutamate in transgenic mice expressing human mutant (G93A) Cu/Zn superoxide dismutase. J. Neurochem. 2000;74:1666–1673. doi: 10.1046/j.1471-4159.2000.0741666.x. [DOI] [PubMed] [Google Scholar]
- Beal MF, Ferrante RJ, Browne SE, Matthews RT, Kowall NW, Brown RH., Jr. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann. Neurol. 1997;42:644–654. doi: 10.1002/ana.410420416. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Carriedo SG, Sensi SL, Yin HZ, Weiss JH. AMPA exposures induce mitochondrial Ca(2+) overload and ROS generation in spinal motor neurons in vitro. J. Neurosci. 2000;20:240–250. doi: 10.1523/JNEUROSCI.20-01-00240.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriedo SG, Yin HZ, Lamberta R, Weiss JH. In vitro kainate injury to large, SMI-32(+) spinal neurons is Ca2+ dependent. Neuroreport. 1995;6:945–948. doi: 10.1097/00001756-199504190-00030. [DOI] [PubMed] [Google Scholar]
- Carriedo SG, Yin HZ, Weiss JH. Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J. Neurosci. 1996;16:4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH, Jr., Julien JP, Goldstein LS, Cleveland DW. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Darman J, Backovic S, Dike S, Maragakis NJ, Krishnan C, Rothstein JD, Irani DN, Kerr DA. Viral-induced spinal motor neuron death is non-cell-autonomous and involves glutamate excitotoxicity. J. Neurosci. 2004;24:7566–7575. doi: 10.1523/JNEUROSCI.2002-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Shinobu LA, Schulz JB, Matthews RT, Thomas CE, Kowall NW, Gurney ME, Beal MF. Increased 3-nitrotyrosine and oxidative damage in mice with a human copper/zinc superoxide dismutase mutation. Ann. Neurol. 1997;42:326–334. doi: 10.1002/ana.410420309. [DOI] [PubMed] [Google Scholar]
- Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J. Neurosci. 2000;20:660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Lai L, Butchbach ME, Stockinger MP, Shan X, Bishop GA, Lin CL. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum. Mol. Genet. 2003;12:2519–2532. doi: 10.1093/hmg/ddg267. [DOI] [PubMed] [Google Scholar]
- Hayes CS, Mulkmus SA, Cizkova D, Yaksh TL, Hua XY. A double-lumen intrathecal catheter for studies of modulation of spinal opiate tolerance. J. Neurosci. Methods. 2003;126:165–173. doi: 10.1016/s0165-0270(03)00078-5. [DOI] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc. Natl. Acad. Sci. U.S.A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugon J, Vallat JM, Spencer PS, Leboutet MJ, Barthe D. Kainic acid induces early and delayed degenerative neuronal changes in rat spinal cord. Neurosci. Lett. 1989;104:258–262. doi: 10.1016/0304-3940(89)90585-5. [DOI] [PubMed] [Google Scholar]
- Kuner R, Groom AJ, Bresink I, Kornau HC, Stefovska V, Muller G, Hartmann B, Tschauner K, Waibel S, Ludolph AC, Ikonomidou C, Seeburg PH, Turski L. Late-onset motoneuron disease caused by a functionally modified AMPA receptor subunit. Proc. Natl. Acad. Sci. U.S.A. 2005;102:5826–5831. doi: 10.1073/pnas.0501316102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C, Xie C, McCormack SG, Chiang HC, Michalak MK, Lin X, Chandran J, Shim H, Shimoji M, Cookson MR, Huganir RL, Rothstein JD, Price DL, Wong PC, Martin LJ, Zhu JJ, Cai H. Amyotrophic lateral sclerosis 2-deficiency leads to neuronal degeneration in amyotrophic lateral sclerosis through altered AMPA receptor trafficking. J. Neurosci. 2006;26:11798–11806. doi: 10.1523/JNEUROSCI.2084-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips MB, Keller BU. Endogenous calcium buffering in motoneurones of the nucleus hypoglossus from mouse. J. Physiol. 1998;511(Pt 1):105–117. doi: 10.1111/j.1469-7793.1998.105bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo AC, Wong V, Benson LM, Dykes M, Tanaka K, Rothstein JD, Maragakis NJ. Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1(G93A) mice. Exp. Neurol. 2006;201:120–130. doi: 10.1016/j.expneurol.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Pramatarova A, Laganiere J, Roussel J, Brisebois K, Rouleau GA. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci. 2001;21:3369–3374. doi: 10.1523/JNEUROSCI.21-10-03369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SD, Weiss JH. Excitotoxic and oxidative cross-talk between motor neurons and glia in ALS pathogenesis. Trends Neurosci. 2004;27:17–23. doi: 10.1016/j.tins.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Rao SD, Yin HZ, Weiss JH. Disruption of glial glutamate transport by reactive oxygen species produced in motor neurons. J. Neurosci. 2003;23:2627–2633. doi: 10.1523/JNEUROSCI.23-07-02627.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc. Natl. Acad. Sci. USA. 1993;90:6591–6595. doi: 10.1073/pnas.90.14.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Kuncl RW. Neuroprotective strategies in a model of chronic glutamate-mediated motor neuron toxicity. J. Neurochem. 1995;65:643–651. doi: 10.1046/j.1471-4159.1995.65020643.x. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Tateno M, Sadakata H, Tanaka M, Itohara S, Shin RM, Miura M, Masuda M, Aosaki T, Urushitani M, Misawa H, Takahashi R. Calcium-permeable AMPA receptors promote misfolding of mutant SOD1 protein and development of amyotrophic lateral sclerosis in a transgenic mouse model. Hum. Mol. Genet. 2004;13:2183–2196. doi: 10.1093/hmg/ddh246. [DOI] [PubMed] [Google Scholar]
- Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VM. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc. Natl. Acad. Sci. U.S.A. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Braeken D, Callewaert G, Robberecht W, Van Den Bosch L. GluR2 deficiency accelerates motor neuron degeneration in a mouse model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2005;64:605–612. doi: 10.1097/01.jnen.0000171647.09589.07. [DOI] [PubMed] [Google Scholar]
- Van Den Bosch L, Vandenberghe W, Klaassen H, Van Houtte E, Robberecht W. Ca(2+)-permeable AMPA receptors and selective vulnerability of motor neurons. J. Neurol. Sci. 2000;180:29–34. doi: 10.1016/s0022-510x(00)00414-7. [DOI] [PubMed] [Google Scholar]
- Vandenberghe W, Robberecht W, Brorson JR. AMPA receptor calcium permeability, GluR2 expression, and selective motoneuron vulnerability. J. Neurosci. 2000;20:123–132. doi: 10.1523/JNEUROSCI.20-01-00123.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Xie W, Le W, Beers DR, He Y, Henkel JS, Simpson EP, Yen AA, Xiao Q, Appel SH. Activated microglia initiate motor neuron injury by a nitric oxide and glutamate-mediated mechanism. J. Neuropathol. Exp. Neurol. 2004;63:964–977. doi: 10.1093/jnen/63.9.964. [DOI] [PubMed] [Google Scholar]