

Abstract
As a long-term goal we want to use cryoelectron tomography to understand how non-enveloped viruses, such as picornaviruses, enter cells and translocate their genomes across membranes. To this end, we developed new image-processing tools using an in vitro system to model viral interactions with membranes. The complex of poliovirus with its membrane-bound receptors was reconstructed by averaging multiple sub-tomograms, thereby producing three-dimensional maps of surprisingly high resolution (30Å). Recognizable images of the complex could be produced by averaging as few as 20 copies. Additionally, model-free reconstructions of free poliovirus particles, clearly showing the major surface features, could be calculated from 60 virions. All calculations were designed to avoid artifacts caused by missing information typical for tomographic data (“missing wedge”). To investigate structural and conformational variability we applied a principal component analysis classification to specific regions. We show that the missing wedge causes a bias in classification, and that this bias can be minimized by supplementation with data from the Fourier transform of the averaged structure. After classifying images of the receptor into groups with high similarity, we were able to see differences in receptor density consistent with the known variability in receptor glycosylation.
Introduction
Electron tomography is an emerging methodology that is capable of providing three-dimensional (3D) information of individual large cellular specimens (Baumeister, 2005; Frank, 2006; McIntosh et al., 2005). Traditionally, electron tomographic studies were done on biological samples that had been fixed, stained to improve contrast, and plastic-embedded for sectioning. Such studies have provided invaluable insights concerning the structure of cellular organelles and supramolecular complexes with irregular structures. Within the past few years there has been an increased focus on the use of electron tomography of samples embedded in vitreous ice. These low-dose cryoelectron tomography studies (as reviwed in (Baumeister, 2005; Grunewald and Cyrklaff, 2006; Lucic et al., 2005; McIntosh et al., 2005; Subramaniam, 2005) originally emerged as a way to investigate different sorts of problems from those addressed by the more common single-particle cryo-EM reconstruction approaches. In conventional single particle methods, reconstructions from two-dimensional projections focus on systems where averaging over multiple copies of conformationally identical particles can produce images at relatively high (sub-nanometer) resolution (Cheng et al., 2004; Fotin et al., 2004; Ludtke et al., 2004). In contrast, early cryoelectron tomographic studies focused on rather thick, unique or conformationally diverse structures, and asked questions where much lower resolution information (>10 nm) was sufficient.
Recently a new class of problem has emerged that is well-suited to a combination of the two approaches: it combines the power of tomographic methods to derive three-dimensional structures of individual molecules with the improvement in signal-to-noise (and consequently resolution) provided by averaging over multiple similar structures (Walz et al., 1997). Initially, this approach was applied to determine the structures of macromolecular complexes in vitro (Beck et al., 2004; Cardone et al., 2007; Chang et al., 2007; Cheng et al., 2007; Deng et al., 2007; Grunewald et al., 2003; Harris et al., 2006; Nicastro et al., 2005; Nicastro et al., 2006; Nickell et al., 2007; Zhu et al., 2006). Perhaps more excitingly, the approach has begun to be used to study macromolecular complexes within the context of intact cells. The ultimate goal is to be able to interpret tomographic reconstructions of crowded cell interiors in terms of their smaller components by matching a library of known higher-resolution structures to features in the low-resolution landscape. Several recent papers have demonstrated substantial progress in this direction, showing that is possible to map individual particles in cells (Bohm et al., 2000; Frangakis et al., 2002; Garvalov et al., 2006; Medalia et al., 2002; Murphy et al., 2006; Ortiz et al., 2006), and in favorable cases to derive reconstructions of macromolecular complexes such as the ribosome, to a few nanometers resolution (Ortiz et al., 2006).
Studying the process of viral infection in cells is an exciting potential use of this technique, both because there has already been extensive work characterizing the structures of viruses and viral components in isolation, and because there is often internal symmetry in the component structures. This aids in the identification of entry intermediates within cells and assists in the production of reconstructions at significantly higher resolution than would be possible with asymmetric structures.
The process of viral entry by non-enveloped viruses is poorly understood. In contrast to enveloped viruses, where the fusion of an outer viral membrane with a cellular membrane provides a conceptually simple mechanism for delivering the viral genome into the cytoplasm, non-enveloped viruses face a more difficult topological problem: they must pass a large, highly charged nucleic acid genome or nucleoprotein complex across a lipid bilayer to gain access to the appropriate intracellular compartment for replication. Different models have been suggested for this entry process, such as disrupting the membrane, creating a pore, or co-opting the translocation machinery of its targeted host cells (Hogle, 2002).
We have focused on poliovirus as a particularly attractive model for understanding how non-enveloped viruses solve this topological problem. Poliovirus, a well-studied prototype member of the picornavirus family (Hogle, 2002), is a small, non-enveloped virus consisting primarily of a T=1 icosahedral protein capsid, 30 nm in diameter, enclosing a 7500-nucleotide positive-sense RNA genome (Hogle et al., 1985). Poliovirus infection is initiated though binding of its receptor, called Pvr or CD155, a CAM-like molecule with three extracellular Ig-like domains, a transmembrane domain and a C-terminal cytoplasmic domain (Mendelsohn et al., 1989). At physiological temperature, receptor binding catalyzes a dramatic conformational rearrangement to form an altered particle called the 135S particle or A particle (Joklik and Darnell, 1961). This conformational rearrangement results in the externalization of a myristoylated capsid protein, VP4, and of the N-terminal extension of capsid protein VP1, both of which are located on the inner surface of the mature virion capsid (Fricks and Hogle, 1990). These two externalized peptides then insert into membranes. Electrophysiological experiments in vitro show that the externalized peptides can form channels and pores (Tosteson and Chow, 1997; Tosteson et al., 2004). Genetic experiments demonstrate that certain mutations can alter both the ability to form channels and the ability to release the viral genome during infection (Danthi et al., 2003). These results have led to the suggestion that the channels, or some related perturbation of the membrane, play an essential role, allowing the viral genome to cross cell membranes and to be delivered into the cytoplasm.
To better understand this process, we and others have used a combination of x-ray crystallography and cryoelectron microscopy to study the structures of soluble forms of several key cell entry intermediates of poliovirus, including the mature virions (160S particles), virions decorated with the ectodomains of host-cell-specific receptors (Belnap et al., 2000b), the 135S particles (Belnap et al., 2000a; Bubeck et al., 2005a), and the empty particles (80S particles) that result after loss of the viral genome (Belnap et al., 2000a). More recently, we have developed a simple model system for biochemical and structural characterization of membrane-associated cell entry intermediates. In this model membrane system, liposomes containing low levels of lipids with NTA head-groups are used to capture C-terminally His-tagged ectodomains of Pvr. These receptor-decorated liposomes have been shown to bind virus, to induce the transition to form 135S particles, and to result in the insertion of VP4 and the N-terminus of VP1 into membranes (Tuthill et al., 2006). Structural studies of the virus in complex with a membrane-anchored receptor (Bubeck et al., 2005b) demonstrate that poliovirus approaches the membrane along its five-fold axis and binds five copies of Pvr. The density in the reconstruction also suggests that receptor binding produces a distortion of the outer leaflet of the membrane, which is seen as an outward projection of the bilayer, occurring just below the five-fold peak on the viral surface (Bubeck et al., 2005b).
Although both optical microscopy and electron microscopy have been used extensively in characterizing viral infection, the inefficiency of establishing infections with poliovirus (and indeed most animal viruses) has raised questions concerning the relevance of the observations. Thus, even with very high quality preparation, it takes, on average, 100 (for poliovirus) and often thousands or even tens of thousands of particles to establish infection. It is therefore impossible to determine, by visualization methods alone, whether a given particle is on a productive or nonproductive pathway. For this reason we have previously developed a dual-fluorescent label approach that allowed us to follow the viral capsid and the viral genome independently at the single particle level in live cells and to identify pathways leading to release of the RNA genome (Brandenburg et al., 2007). In that study, we coupled this visualization assay with a biological infectivity-based assay for RNA release, to ensure that the genome release that was observed in the visualization assay was productive. Using the combined assays we demonstrated that productive RNA release is surprisingly efficient. This observation implies that the inefficiency of poliovirus infection arises at some post-entry step (e.g., the initiation of translation). We have also shown that viral entry occurs via a non-canonical endocytic pathway, that productive entry occurs at both the apical and attached surface of HeLa cells with equal efficiency, and that RNA release occurs within 100–200nm from the cell surface.
The combination of structural, biochemical and optical microscopy studies have provided significant insights into the cell entry process. However, much remains to be learned, including the endocytic pathway, the compartment from which RNA is released, and the mechanism used to transfer RNA from the interior of the virus across the vesicle membrane and into the cytoplasm. We believe that cryoelectron tomography of virus-infected cells could provide a powerful tool to address these questions. Moreover, the fact that productive RNA release is efficient, and that it takes place in regions of cells that are sufficiently thin to be visualized without the need for cryo-sectioning, make poliovirus entry an ideal candidate for developing the tools needed to characterize low abundance macromolecular complexes in intact cells.
As a first step in establishing the feasibility of this approach, we have used cryoelectron tomography to probe the structure of poliovirus attached to receptor-decorated liposomes. We show that by orienting and averaging large numbers (~1500) of sub-tomograms of the virus-receptor-liposome complex, it is possible to produce accurate maps at surprisingly high resolution (3 nm), that free virus and virus-receptor complexes can easily be distinguished by visual inspection of the sub-tomograms, and that clearly recognizable structures of the virus and virus receptor complex can already be obtained by averaging only 20–100 sub-tomograms. Principal component analysis (PCA), coupled with a compensation for the missing wedge, allowed us to determine the conformational variability of different regions of the complex, including differences consistent with the presence of variable levels of glycosylation of receptor molecules. These studies lay the foundation for probing the structures of cell-entry intermediates during the course of an actual infection in situ, wherein the cellular environment is crowded, and where infection may be due to a minority of the viral particles that are present. Understanding viral infection may require the ability to correlate the structures of individual virus particles with their locations and activities within the cell – information that can only be obtained by combining cryoelectron tomography with other techniques.
Materials and methods
Sample Preparation
Liposomes were prepared using a mixture of phosphatidylethanolamine, phosphatidylcholine, sphingomyelin, cholesterol and phosphatidic acid in a molar ratio of 1:1:1.5:0.3 with 10% DOGS-NTA and 1% rhodamine-PE (Tuthill et al., 2006). The lipids were dried, re-hydrated and extruded through 100 nm pores to produce unilaminar vesicles 100–150 nm in diameter. A quantity of 33 μl of 1.5 mg/ml liposomes was incubated with 1.5 μl of 0.67 mg/ml Poliovirus receptor (Pvr) for 10 minutes followed by another 2 μl of Pvr for 10 minutes. Subsequently, 8.5 μl of re-hydration buffer was added to the mixture, followed by 8.5 μl of 0.5 mg/ml virus (type 1 Mahoney strain). An additional 1 μl of 10–15 nm colloidal gold was mixed into the 4μl drop of sample before being applied to glow-discharged quantifoil holey carbon grids and plunge-frozen in liquid ethane (Dubochet et al., 1988).
Electron microscopy
Images were recorded using a Tecnai F-30 microscope (FEI, Eindhoven, Netherlands) equipped with a post-column energy filter (GIF), and a 2Kx2K CCD camera (Gatan, Pleasanton, CA). Tilt series were recorded using the automated acquisition software SerialEM (Mastronarde, 2005). The stage was tilted over the angular range of +66º to −66º in 1.5º increments. The electron dosage during the series was set to ~1 electron/Å2 at 0º tilt (<100 electron/Å2 total dose per tilt series). The beam intensity was kept constant. Pictures were taken at a defocus of 4 μm and at magnifications that corresponds to a pixel size of 0.99 nm or 0.71 nm, respectively.
Image processing
The eTomo graphical user interface of the IMOD tomography package (Kremer et al., 1996; Mastronarde, 2005) was used to calculate the 3-D reconstruction (Fig. 1A and B). The images in the tilt series were aligned using the gold fiducial markers and the final tomograms were calculated by weighted backprojection.
Figure 1.

Cryoelectron tomography of poliovirus-receptor-liposome complexes. (A) A projection along the z-axis (beam direction), showing a portion of a 3D tomogram. This rectangle (~2 μm wide) includes about a quarter of the original tomogram. Liposomes (narrow arrowheads), virus particles (wide arrowheads), and gold fiducials are present. Scale bar: 10 nm. (B) Surface rendering, cropped for clarity, showing a few virus particles and liposomes. A representative virus particle (wide arrowhead), and liposome (narrow arrowhead) are indicated (C) Fourier shell correlation (FSC) indicates that the resolution of the reconstruction is around 3.0 nm, and is limited by the pixel size. (D) Reconstruction of the complex by single particle cryo-EM (Bubeck et al., 2005b). (E) Averaged tomographic structure: ~1500 copies of the complex were identified in the tomograms, boxed, and aligned to the reference by comparing Fourier terms. Terms in the missing wedge were not considered. Aligned particles were summed and five-fold averaged. The resulting reconstruction clearly shows the expected peaks and valleys on the viral surface, and prominent glycosylation sites on the middle domain of the receptor. As in (D), the membrane has a pronounced distortion in the vicinity of the five-fold axis. Panels (D) and (E) show the complex in the same orientation, and are calculated using the same pixel size (1 nm). Scale: the diameter of the virus is 30 nm.
3D single particle averaging of the virus-receptor-liposome complex
Particle selection
A data set of 1500 sub-tomograms containing virion-receptor-liposome complexes were selected from seven tomograms with a pixel size of 0.99 nm, as follows. First, each tomogram was projected along the z-direction (beam), where visible distortion is minimal, and the approximate x and y position for each complex was determined interactively using the program WEB (Frank et al., 1996). Each particle was cropped from the tomogram in a column extended over the thickness of the tomogram stack. Then, in order to determine the x, y, and z coordinates of the virus accurately, each slice of the sub-tomogram was cross-correlated with a strongly low-pass-filtered central slice through the reference virus structure (Bubeck et al., 2005b). Finally, the particles were extracted, each centered in a cubic volume with 128-pixels on each edge.
Alignment
The alignment of the complex in each centered sub-tomogram was determined using SPIDER (Frank et al., 1996). References for the alignment process were derived either from previously determined structures of the complex (Bubeck et al., 2005b) or as detailed below. All alignments were calculated taking into account only the experimentally recorded Fourier terms as described in (Forster et al., 2005). Thus, a missing-wedge corresponding to that of the raw tomogram was applied to the Fourier transform of the reference for each possible orientation in the alignment process and the cross correlation function (cc n) was normalized accordingly in SPIDER (see below). For each sub-tomogram, either two or three of the Eulerian angles that define the orientation of the reference particle were determined by systematic search over some specified range of values, with the range and the coarseness of the search being reduced in successive iterations of the search. At each point in the search grid, improvements in the three Cartesian shift coordinates that define the position of a particle were determined automatically.
Averaging of the complex
We tried a number of different approaches for obtaining averages of the complex, based on the aligned particles. The initial crude approach method was a simple summation of the oriented particles. A second tactic was to weight each particle according to the cross-correlation coefficient. Finally, a more elaborate method was tried, in which the average at each point in Fourier space was weighted according to the number of contributing particles, following the approach of Nicastro et al (Nicastro et al., 2006). Such a scheme can be shown to minimize in a least-squares manner the difference between the resulting map and each contributing particle. Probably due to the relatively large data set employed, to the five-fold symmetry of the complex, and to the uniformity of the distribution of orientations (see below) all three methods gave nearly identical results. By excluding the 20% of the particles having the poorest cross-correlation coefficients with the average of all the particles, the resulting map had a better-defined structure and better resolution statistics.
Maps were visualized and figures were prepared using CHIMERA (Pettersen et al., 2004).
Model-independent single particle reconstruction of the mature virion
From a tomogram recorded at pixel size 0.71 nm a total of 80 particles of mature virus (160S particles) were selected as described above and extracted in an 80 pixel cube. An average of the centered particles was icosahedrally symmetrized and used as a starting model for an icosahedral search. Several alignment iterations were performed using the missing wedge correction. The coarse initial search grid was made smaller at each step, and the process was stopped when no further improvement in resolution was obtained.
Classification
To detect and account for structural variability in certain portions of each particle image, specific masked subsets of the voxels were extracted from individual tomograms, after first aligning each complex to the frame-of-reference of the averaged standard. In particular, we looked for variability in density regions associated with receptor molecules and membranes. For classifying the particles we used principal component analysis, followed by Diday method of classification with moving centers (Diday, 1971), and finally hierarchical ascendant classification as implemented in SPIDER (Frank et al., 1996). This procedure produces a dendrogram, resulting in a number of classes that depends on a choice of a cutoff value. Typically we chose to create ten classes, but the interpretation of the results was not sensitive to that choice.
Classification, intended to detect structural heterogeneity, was applied to small regions of the map. Only pixels inside a mask delineating the area of interest were used. In the case of the receptor region, each of the five copies of the receptor for every individual particle was oriented in the same frame of reference, giving a total of 7500 individual receptor molecules. For analyzing the degree of variability of the membrane shape, each five-fold-related view was included in the classification procedure as a separate asymmetric object.
We were concerned that the systematic distortion of particle images that is due to the missing wedge might bias the classification, creating an artifactual tendency for similarly oriented particles to cluster together. In order to assess such a bias in classification, and to correct for it, the classification procedure was repeated twice: once with the original aligned particle images, and second, with particles corrected for the missing Fourier terms. In order to fill the missing wedge for each particle, replacement Fourier terms were selected from the scaled Fourier transform of the reference, where the reference was scaled to each individual image by the ratio of their power spectra.
Results and Discussion
Reconstruction of poliovirus-receptor-liposome complex
An undistorted tomographic reconstruction could only be calculated if a tilt series were recorded over the full angular range of +/−90. However, in practice, the stage cannot be tilted higher than 60–70º, which causes a range of views to be missing. This problem is commonly known in electron tomography as the “missing wedge”, describing the incomplete coverage of Fourier space. The existence of the missing wedge must be taken into account, and compensated for, in every stage of the process of creating a reconstruction by averaging multiple sub-tomograms. For example, in choosing and isolating individual particles, projections along the z-axis (beam direction) are much less distorted than views perpendicular to the beam. Consequently, we relied entirely on z-axis projections (Fig. 1A,B) in both stages of a two-step process for isolating and centering individual complexes in sub-tomograms (see Methods). A total of ~1500 sub-tomograms were isolated from tomographic reconstructions.
Similarly, the alignment of each individual complex was determined using a procedure that compensates for the missing wedge (Forster et al., 2005). This procedure takes advantage of the fact that every particle chosen from a tomographic reconstruction has its missing wedge in the same position, relative to the microscope’s frame-of-reference. Thus, a Fourier mask that sets all missing terms to zero was applied to each possible trial orientation of the reference before estimating the normalized cross-correlation function between the particle and the reference. The reference used (Fig. 1D) was a low-pass-filtered copy of the 3.2 nm resolution structure of the poliovirus-receptor-membrane complex obtained earlier by projection-based “single-particle” methods (Bubeck et al., 2005b). In order to reduce the large computational cost, the task was divided into two stages. In a first step, the position of the membrane (and of the associated five-fold axis) were determined using a cylindrically averaged reference. This was carried out as a complete search over two of the three Euler angles on a 9-degree grid. Once the direction of the membrane was found, a finer grid was used in a search for all three Euler angles, taking advantage of the five-fold symmetry to reduce the searchable volume. This restricted search around the unique five-fold axis permitted the receptor positions to become apparent in the averaged structure. Once all the particles were roughly aligned we performed several additional refinement steps on increasingly finer grids, using as a reference the averaged structure obtained in the previous cycle. In the final stages, the search grid used 0.5-degree steps.
Several approaches were tried for obtaining the reconstruction of the complex based on the aligned particles (see Methods), and all produced nearly identical results. The final map (Fig. 1E) is a simple average of the 80% of the particles having the best cross-correlation coefficients. Its resolution was calculated by Fourier shell correlation of two averages (Saxton and Baumeister, 1982), each derived from a randomly chosen half of the selected particles (Fig. 1C). Using the conservative 0.5 criterion, the resolution of the complex was estimated to be 3.0 nm. One alternative criterion, which infers the correlation between a perfect reference and a reconstruction from the full data set, as proposed in (Grunewald et al., 2003; Rosenthal and Henderson, 2003) gives a resolution of 2.6 nm. Another alternative, accepting the 1/2 bit criterion formulated in (van Heel and Schatz, 2005), gives an estimate of 2.7 nm. Based on these estimates, the map we report has been low-pass filtered to 3.0 nm, which corresponds to the first zero of the contrast transfer function. Taking into account that the pixel size for the data set was 0.99 nm, the resolution obtained is probably the highest achievable value, i.e. we are at as close as possible to a compromise between the theoretical Shannon limit and the interpolation errors made in the process of aligning the particles. Consistent with this hypothesis, imposing 60-fold symmetry on the capsid region of the complex did not improve the resolution. By comparison, in a lower-resolution study of Herpes Simplex nucleocapsids, where neither the pixel size nor the first zero of the CTF were limiting, the authors reported obtaining higher resolution reconstructions by averaging sub-tomograms than they were able to obtain by single-particle methods (Cardone et al., 2007).
The structure obtained for the poliovirus-receptor-membrane complex is in good agreement with the previously determined map of the complex (Bubeck et al., 2005b) (Fig. 1D) and with the map of poliovirus fully decorated by receptor in solution (Belnap et al., 2000b). The capsid shows strong icosahedral features even though in its alignment only the five-fold symmetry was assumed. Density is present for the three domains of the receptor; and its connections with the virus and with the membrane are well resolved. A broadening of the middle domain is caused by the two large glycosylation sites known to be present there (Belnap et al., 2000b). These two large glycosylation sites were first seen in receptor-decorated virus in solution (Belnap et al., 2000b; He et al., 2003), and can be eliminated by mutations (He et al., 2003) or enzymatically (P. Kwong and D. Belnap, personal communication). The membrane bilayer is seen as a well-ordered continuous density, about 5 nm wide, despite the relatively wide range of liposome sizes and curvatures. As might be expected given the 1nm pixel size, the leaflets of the bilayer are not as well resolved as they were in the previously published single particle reconstruction (Bubeck et al., 2005b), where a band of lower density was readily detectable between them. However, as in the single particle reconstruction, a strong distortion of the membrane in the vicinity of the five-fold axis protrudes towards the virus. Thus, the tomographic reconstruction helps to confirm that the membrane distortion was not an artifact of the previous reconstruction process, and supports the idea of its biological relevance. Moreover, we have also determined that the membrane distortion is not simply an artifact of the application of five-fold symmetry: reconstructions without symmetry applied, showing similar distortions, are presented and discussed below.
Although sub-tomograms derived from 1200 particles were averaged to obtain the 3.0 nm resolution reconstruction, the number of particles necessary to produce a reconstruction with a recognizable morphology is much lower. Thus, with only 20 particles (Fig 2) it is possible to recognize the viral features and to distinguish the middle domain of the receptor, and by averaging 100 particles it is possible to obtain a medium resolution image of the particle having all of the major features clearly defined. Based on FSC comparisons with the final reconstruction, the resolutions of the 20-particle and 100-particle reconstructions is estimated to be 5 nm and 4.2 nm, respectively.) The ability to clearly identify a structure using only a limited number of particles could prove to be key to future applications looking at cells infected at a low multiplicity-of-infection.
Figure 2.
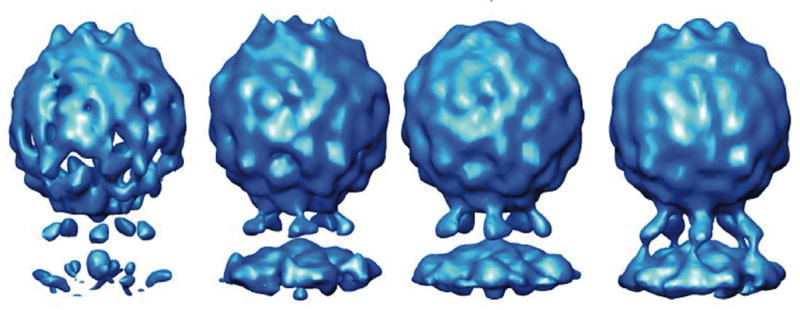
Recognizable reconstructions can be produced by combining a surprisingly small number of individual sub-tomograms. Left-to-right: randomly chosen subsets of the data (consisting of 20, 40, 80 or 100 sub-tomograms) were summed and five-fold averaged. Densities for receptors and membrane are visible after averaging as few as 20 particles. Fourier shell correlation (vs. the ~1500-particle reconstruction) indicates resolutions of 5.0, 4.7, 4.4. and 4.2 nm for these four maps respectively.
The 160S structure
A total of 80 virus particles that were not attached to liposomes were selected from a single tomogram with a pixel size of 0.71 nm. Starting with a 60-fold-symmetrized average of the particles as an initial reference, we repeatedly conducted orientation searches in an icosahedral frame-of-reference, and then reconstructed the reference map from the oriented particle images. This process converged quickly to a stable structure, estimated by the FSC 0.5 criterion to be at 3.6 nm resolution. The structure obtained using this ab initio approach is shown in Fig 3. Major features of the viral surface are defined, such as the canyon surrounding the five-fold mesa, and the propeller pattern at the three-fold axis. As in previous reconstructions (Belnap et al., 2000a) and crystal structures (Hogle et al., 1985), the internal structure of the virus also shows a large cavity under each five-fold vertex. Due to the geometrical constraints imposed by the icosahedral symmetry, the orientation search was much easier to perform in the case of the virus particle. In addition, the 60-fold symmetry simplifies the problem of the missing wedge, as much of the information that is missing is redundant. This compensation makes misalignment an infrequent event.
Figure 3.
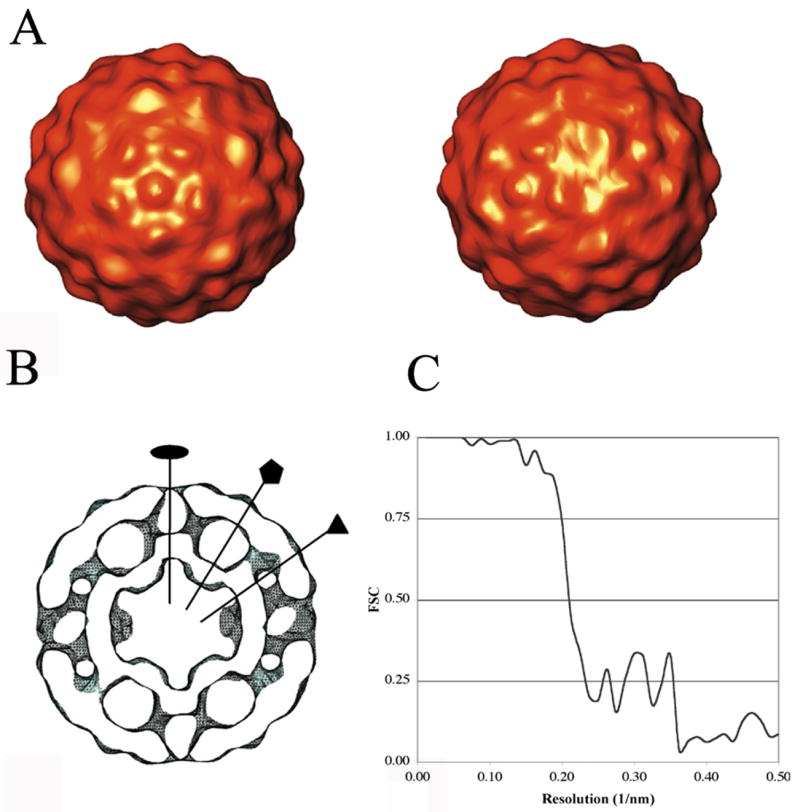
Model-free icosahedral reconstruction of 160S poliovirions from tomographic images. (A) Views along its five-fold (left) and two-fold (right) axes. (B) Central slice through the reconstruction, shown as an iso-contour surface. Two-fold, five-fold, and three-fold axes are marked. Scale: the diameter of the virus is 30 nm. (C) Fourier shell correlation (FSC) indicates that a resolution of 3.6 nm was obtained by adding 60 virus particles.
Classification
In electron micrographs of biological samples, it is not uncommon for large conformational variations to occur that are easy to detect by visual inspection. Often in single-particle reconstructions, classification must be employed (followed by averaging within a class) to raise the signal-to-noise ratio so that differences either in conformation or orientation become discernible. The level of resolution at which the differences become meaningful, and the stage of structure solution that classification should be undertaken, depends both on the magnitude of the differences and the extent of variability. Since poliovirus forms a compact and stable structure, and the structural variations in the complex are subtle, classification was undertaken after an averaged structure was available, as a way to study localized differences, rather than in the early stages of the calculation, as a way to eliminate large numbers of conformational outliers.
Reducing Bias in Classification of tomograms
A major concern in single particle electron microscopy regards the homogeneity of the sample. Electron tomography can cope with this problem, as the data, despite its low signal-to-noise ratio and the distortion caused by the missing data, contain three-dimensional information. Classification can be done, in this case, on pixel-by-pixel basis. Nevertheless, we suspected that the missing wedge could introduce significant bias in this procedure, as particles being distorted in similar ways might have a tendency to cluster in the same class. We hypothesized that one way to compensate for the lack of data would be to replace the null terms in each sub-tomogram with the corresponding Fourier coefficients from the averaged structure.
The search models leading to the 3 nm structure assumed five-fold symmetry. In order to break that assumed symmetry, prior to classification each sub-tomogram was reoriented in all five of its symmetry-equivalent orientations, and each symmetry-related copy was then treated as a separate (asymmetric) sample. We then scaled the transform of the current average to each individual sub-tomogram, and then replaced the missing coefficients, hoping to obtain a better, less distorted approximation to the original particle. Chosen subsets of the pixels from these modified tomographic reconstructions were then classified as described. Finally, the distribution of tomograms into classes, both with and without compensation for missing terms, were assessed and compared.
The existence of a bias in classification (and of a method to alleviate it) would be demonstrated if particles with similar derived orientations (Euler angles) had a greater tendency to cluster together in the unmodified samples. One way to check for bias in the case of the ‘uncorrected’ sub-tomograms is to assess the uniformity of Fourier space coverage within individual large classes. For a single particle image (or symmetry copy), a weighting mask having a value of ‘one’ for each recorded coefficient in Fourier space, and ‘zero’ for each missing coefficient, covers 72% of the space. Thus, within any particular class, if all of the corresponding masks are rotated into the frame of reference of the reconstructed average and added together, the sum-of-the-masks will give a description of the uniformity of reciprocal space coverage. Under-sampled regions will have lower sums, which will appear as cavities when visualized at a chosen iso-contour threshold. Thus, in Fig 4A we show how angular coverage within the largest class differs between the corrected (blue) and uncorrected (red) data sets. At the chosen threshold, 1.1 standard deviations above the mean, it appears that reciprocal space is sampled less uniformly when uncorrected images (shown in red) are classified (though the appearance of the cavities would clearly depend on the choice of contour).
Figure 4.
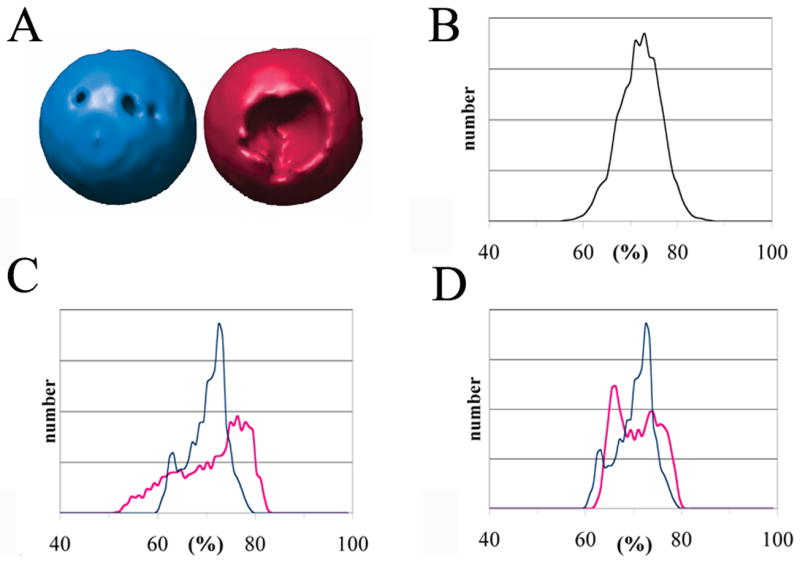
The missing wedge causes a bias in classification: particles with similarly oriented missing wedges become distorted in similar ways, and tend to be grouped together. Filling in the missing wedge with Fourier coefficients from the averaged structure reduces the bias. For the largest classes, filled (blue) and unfilled (red), respectively, this figure illustrates how uniformly reciprocal space is sampled. (A) Angular coverage: the cavities in the surfaces correspond to under-sampled regions of reciprocal space after the members of each class are added together. The contour level is 1.1 standard deviations above the mean. (B) Simulated histogram showing how often points in reciprocal space are sampled for a random distribution of Euler angles. As in the tomograms, 72% of reciprocal space is occupied; a class with 100 members was assumed. (C, D) Actual histograms calculated from the two largest classes. Observe that leaving the missing wedge unfilled (red) causes a non-uniform sampling, reflecting a bias in classification. Restoring the missing wedge (blue) significantly reduces the bias. In panels B, C and D the horizontal axis represents the percentage (%) of boxed particles whose contribution to a reciprocal space direction lies outside of the missing wedge. The verical axis represents the number of reciprocal space directions having the indicated percentage; this value is on an arbitrary scale, depending on the finess of sampling of reciprocal space.
Another way to visualize the uniformity of coverage, independent of the contour choice, is to construct a histogram of the values occurring in each class-specific sum-of-the-masks. Perfect uniform coverage would give every (non-origin) pixel the same value. In the case of a random distribution of Euler angles, a simulated histogram for 100 particles (Fig 4B) has a narrow peak centered at 72%, which is the fraction of non-zero terms within each mask. Less uniform coverage of reciprocal space would cause a broader distribution of values to occur. Histograms are shown for the two largest classes (Fig 4C). Leaving the missing wedge unfilled (red) causes a less uniform sampling of reciprocal space, reflecting a bias in classification. On the other hand, restoring the missing wedge (blue) significantly reduces the bias.
In the foregoing discussion, we used the theoretical distribution (a symmetric peak at 72%) as the standard, to assess whether or not bias was present. However, perhaps a better (and more widely-applicable) choice would have been to use the sum-of-the-masks histogram distribution for the population as a whole as a standard. Then the distribution within an individual class could be compared with that standard, to see how closely it agrees. Clearly, the skewness of the distribution within a class cannot decrease below the skewness of the population as a whole, whatever that happens to be. In the case of the poliovirus-receptor-liposome complexes, it made no difference which standard was used because these complexes happen to adopt a very uniform distribution of orientations (data not shown). For other biological samples, it might have made a difference if, for example, the particles adopt a preferred orientation, as has been observed in many single particle studies and in other tomographic studies (Cardone et al., 2007; Schmid et al., 2006).
Classification of receptors and membranes
Our current and prior experience with the poliovirus-receptor-liposome complex leads us to expect that conformational diversity would be more of an issue in the receptors and membranes than it is in the capsid region. Indeed, the receptor density in the averaged reconstructions is weaker; and the averaged membrane density disappears beyond the footprint of the receptors. Furthermore, the FSC goes to higher resolution when only the capsid region is considered, as shown in (Bubeck et al., 2005b). Like many other complexes of biological interest, the structure contains conformationally consistent regions that can form the basis of an averaged structure, and less-consistent regions that may be resolved by classification after-the-fact, hoping to gain additional biological insights from the structures of the individual class averages. This method would be the only way to elucidate the structures of possible minority populations of particles (perhaps previously unseen ones) during the process of viral infection.
To study receptors and membranes, we adopted the strategy of filling in the missing wedge prior to classification, and of treating the five-fold symmetry-related views as separate asymmetric samples. First, we investigated the membrane morphology. The classification confirmed the existence of a membrane protuberance located at the five-fold axis in all the classes. Interestingly, some of the classes differ markedly in the shape and inclination of the membrane, and thus in the orientation of the protuberance. For example, in Fig 5A, two central sections are compared, one derived from the largest class, comprising 36% of the views (blue), and the other derived from one of the smaller classes (5%, in red); these two classes show distinctly different tilts in the membrane. This indicates that virus can attach to the membrane obliquely, possibly due to incomplete occupancy of the receptor binding sites, to the flexibility of the receptor, and/or to the fluidity of the membrane.
Figure 5.
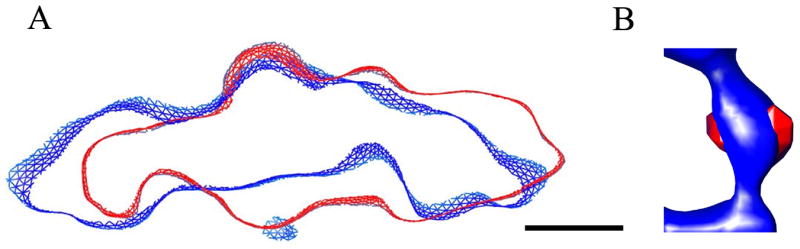
Conformational diversity, as revealed by classification. (A) The classification was based on pixels sampled from the membrane regions of 7500 views. Some of the classes differ markedly in the shape and inclination of the membrane. This example shows corresponding sections from a large class (2700 members, blue) and a small class (400 members, red). (B) Classification in the receptor region. The average receptor is shown in blue; the density in red represents the variability between the largest class averages. The large classes included in this calculation collectively represent 62% of the receptor population. The major differences coincide with the positions of the two large known glycosylation sites on the middle domain of the receptor. Scale bar: 5 nm.
To examine whether some of the individual receptor molecules were either bent or missing, pixels in the vicinity of the receptor were sampled for classification. Each of the ~1500 sub-tomograms was oriented in the frame-of-reference of the reconstruction, and then rotated about the particle five-fold axis by various multiples of 72 degrees, thus yielding ~7500 independent asymmetric density samples. After classification, average structures were calculated within each of the ten largest classes, and inspected visually. There were two important findings. First, none of the classes showed a detectable level of non-occupancy of the receptor site. This is not entirely unexpected because the early receptor-binding events help to promote subsequent binding events. This happens both because the virus is oriented correctly and because the effective local concentration of receptor is inherently much higher in a two-dimensional search of the membrane surface than it would have been in a three-dimensional search, free in solution. Second, there were qualitative differences in the shape of the density seen in different class averages. Consistent with the fact that both ends of each receptor are tethered, the differences are small, corresponding to a shift in the domain positions, rather than to a radical rearrangement. It would be difficult to know whether such differences were caused by noise or by genuine structural variability, except for one very interesting finding, seen in the variance map for the 4 or 5 largest classes, which collectively account for 62% of the receptors (Fig 5B). (This variance map shows which pixels exhibit the greatest variability, once structural differences in the minor classes are ignored.) In this variance map the main structural variability (in red) is seen at the sides of the middle domain, where two very large glycosylation sites are known to be present (Belnap et al., 2000b). This suggests that the largest classes (whose exact number depends on the random seed used to initiate classification) all share similar, strong, structurally consistent protein domains; and that once a conformationally consistent set of receptors has been classed together, variability in the extent and disposition of glycosylation can be detected. Further subdivision of this class failed to divide the population cleanly into groups with and without the putative glycosylation, suggesting that a continuous range of sizes is likely to be present.
Classification of virus particles
It is clear from the present work and from previous studies (Belnap et al., 2000a) that reconstructions of native poliovirions can be visually distinguished from 135S particles on structural grounds alone, even at low resolution, once a population of similar particles has been classified together and then averaged, and poorly correlating particles are excluded. This explains the rationale for our experiment: tomography may be the only way to locate particles in relatively thick samples, such as the interior of cells. The averaging of tomograms then represents the only unbiased way to verify the identity of a group of similar particles. It still remains to be proven that previously uncharacterized conformational states of the virus can be reliably identified in a similar way.
Summary
The question of poliovirus entry provides an unprecedented opportunity to combine a variety of technologies to study the function of a biologically relevant machine from the atomic to the cellular level. Technical issues to be considered include the sparseness and diversity of virus particles, and the relative thickness of eukaryotic cells. Fortunately, the recent observation that poliovirus entry occurs close to the cell periphery (Brandenburg et al., 2007) suggests that tomograms of vitreous cell sections would provide pertinent information. Several key observations of the present study suggest that cryoelectron tomography could make important contributions to understanding poliovirus entry, including: 1) the demonstration that it is possible to obtain recognizable structures of membrane-associated particles at useful resolution from experimentally tractable numbers of sub-tomograms, obtained from membrane-crowded environments; 2) the demonstration that known structures can be used to identify specific structural states in mixed samples; and 3) the demonstration that with simple modifications to common image-processing methods, images in mixed samples can be classified in such a way that subtle conformational differences in large and complex structures can be detected. Finally, with foreseeable technological advances in electron microscope optics, improved electron detectors, and the development of correlative cryo-light microscopy and cryoelectron tomography, this approach may become the preferred method for the more general problem of characterizing the function of complex biological machines in the context of whole, unperturbed cells.
Acknowledgments
This work was supported by NIH grants AI20566 (to J.M.H.) and an NSF pre-doctoral fellowship (to D.B.). The Boulder Laboratory for 3-Dimensional Electron Microscopy of Cells was supported by NCRR grant RR000592 (to Andreas Hoenger).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baumeister W. From proteomic inventory to architecture. FEBS Lett. 2005;579:933–937. doi: 10.1016/j.febslet.2004.10.102. [DOI] [PubMed] [Google Scholar]
- Beck M, Forster F, Ecke M, Plitzko JM, Melchior F, Gerisch G, Baumeister W, Medalia O. Nuclear pore complex structure and dynamics revealed by cryoelectron tomography. Science. 2004;306:1387–1390. doi: 10.1126/science.1104808. [DOI] [PubMed] [Google Scholar]
- Belnap DM, Filman DJ, Trus BL, Cheng N, Booy FP, Conway JF, Curry S, Hiremath CN, Tsang SK, Steven AC, Hogle JM. Molecular tectonic model of virus structural transitions: the putative cell entry states of poliovirus. J Virol. 2000a;74:1342–1354. doi: 10.1128/jvi.74.3.1342-1354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belnap DM, McDermott BM, Jr, Filman DJ, Cheng N, Trus BL, Zuccola HJ, Racaniello VR, Hogle JM, Steven AC. Three-dimensional structure of poliovirus receptor bound to poliovirus. Proc Natl Acad Sci U S A. 2000b;97:73–78. doi: 10.1073/pnas.97.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm J, Frangakis AS, Hegerl R, Nickell S, Typke D, Baumeister W. Toward detecting and identifying macromolecules in a cellular context: template matching applied to electron tomograms. Proc Natl Acad Sci U S A. 2000;97:14245–14250. doi: 10.1073/pnas.230282097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandenburg B, Lee LY, Lakadamyali M, Rust MJ, Zhuang X, Hogle JM. Imaging poliovirus entry in live cells. PLoS Biology. 2007 doi: 10.1371/journal.pbio.0050183. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck D, Filman DJ, Cheng N, Steven AC, Hogle JM, Belnap DM. The structure of the poliovirus 135S cell entry intermediate at 10-angstrom resolution reveals the location of an externalized polypeptide that binds to membranes. J Virol. 2005a;79:7745–7755. doi: 10.1128/JVI.79.12.7745-7755.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck D, Filman DJ, Hogle JM. Cryo-electron microscopy reconstruction of a poliovirus-receptor-membrane complex. Nat Struct Mol Biol. 2005b;12:615–618. doi: 10.1038/nsmb955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone G, Winkler DC, Trus BL, Cheng N, Heuser JE, Newcomb WW, Brown JC, Steven AC. Visualization of the herpes simplex virus portal in situ by cryo-electron tomography. Virology. 2007;361:426–434. doi: 10.1016/j.virol.2006.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JT, Schmid MF, Rixon FJ, Chiu W. Electron cryotomography reveals the portal in the herpesvirus capsid. J Virol. 2007;81:2065–2068. doi: 10.1128/JVI.02053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Boll W, Kirchhausen T, Harrison SC, Walz T. Cryo-electron Tomography of Clathrin-coated Vesicles: Structural Implications for Coat Assembly. J Mol Biol. 2007;365:892–899. doi: 10.1016/j.jmb.2006.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. Structure of the human transferrin receptor-transferrin complex. Cell. 2004;116:565–576. doi: 10.1016/s0092-8674(04)00130-8. [DOI] [PubMed] [Google Scholar]
- Danthi P, Tosteson M, Li QH, Chow M. Genome delivery and ion channel properties are altered in VP4 mutants of poliovirus. J Virol. 2003;77:5266–5274. doi: 10.1128/JVI.77.9.5266-5274.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng B, O’Connor CM, Kedes DH, Zhou ZH. Direct visualization of the putative portal in the Kaposi’s sarcoma-associated herpesvirus capsid by cryoelectron tomography. J Virol. 2007;81:3640–3644. doi: 10.1128/JVI.02254-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diday E. La methode de nuees dynamiques. Revue Statistique Appliquée. 1971;19:19–34. [Google Scholar]
- Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P. Cryo-electron microscopy of vitrified specimens. Q Rev Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- Forster F, Medalia O, Zauberman N, Baumeister W, Fass D. Retrovirus envelope protein complex structure in situ studied by cryo-electron tomography. Proc Natl Acad Sci U S A. 2005;102:4729–4734. doi: 10.1073/pnas.0409178102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotin A, Cheng Y, Sliz P, Grigorieff N, Harrison SC, Kirchhausen T, Walz T. Molecular model for a complete clathrin lattice from electron cryomicroscopy. Nature. 2004;432:573–579. doi: 10.1038/nature03079. [DOI] [PubMed] [Google Scholar]
- Frangakis AS, Bohm J, Forster F, Nickell S, Nicastro D, Typke D, Hegerl R, Baumeister W. Identification of macromolecular complexes in cryoelectron tomograms of phantom cells. Proc Natl Acad Sci U S A. 2002;99:14153–14158. doi: 10.1073/pnas.172520299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, editor. Electron Tomography: Methods for Three-dimensional Visualization of Structures in the Cell. 2. Springer; 2006. [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Fricks CE, Hogle JM. Cell-induced conformational change in poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. J Virol. 1990;64:1934–1945. doi: 10.1128/jvi.64.5.1934-1945.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvalov BK, Zuber B, Bouchet-Marquis C, Kudryashev M, Gruska M, Beck M, Leis A, Frischknecht F, Bradke F, Baumeister W, et al. Luminal particles within cellular microtubules. J Cell Biol. 2006;174:759–765. doi: 10.1083/jcb.200606074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunewald K, Cyrklaff M. Structure of complex viruses and virus-infected cells by electron cryo tomography. Curr Opin Microbiol. 2006;9:437–442. doi: 10.1016/j.mib.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Grunewald K, Desai P, Winkler DC, Heymann JB, Belnap DM, Baumeister W, Steven AC. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science. 2003;302:1396–1398. doi: 10.1126/science.1090284. [DOI] [PubMed] [Google Scholar]
- Harris A, Cardone G, Winkler DC, Heymann JB, Brecher M, White JM, Steven AC. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc Natl Acad Sci U S A. 2006;103:19123–19127. doi: 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Mueller S, Chipman PR, Bator CM, Peng X, Bowman VD, Mukhopadhyay S, Wimmer E, Kuhn RJ, Rossmann MG. Complexes of poliovirus serotypes with their common cellular receptor, CD155. J Virol. 2003;77:4827–4835. doi: 10.1128/JVI.77.8.4827-4835.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogle JM. Poliovirus cell entry: common structural themes in viral cell entry pathways. Annu Rev Microbiol. 2002;56:677–702. doi: 10.1146/annurev.micro.56.012302.160757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogle JM, Chow M, Filman DJ. Three-dimensional structure of poliovirus at 2.9 A resolution. Science. 1985;229:1358–1365. doi: 10.1126/science.2994218. [DOI] [PubMed] [Google Scholar]
- Joklik WK, Darnell JE., Jr The adsorption and early fate of purified poliovirus in HeLa cells. Virology. 1961;13:439–447. doi: 10.1016/0042-6822(61)90275-6. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Lucic V, Forster F, Baumeister W. Structural studies by electron tomography: from cells to molecules. Annu Rev Biochem. 2005;74:833–865. doi: 10.1146/annurev.biochem.73.011303.074112. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Chen DH, Song JL, Chuang DT, Chiu W. Seeing GroEL at 6 A resolution by single particle electron cryomicroscopy. Structure. 2004;12:1129–1136. doi: 10.1016/j.str.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- McIntosh R, Nicastro D, Mastronarde D. New views of cells in 3D: an introduction to electron tomography. Trends Cell Biol. 2005;15:43–51. doi: 10.1016/j.tcb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Medalia O, Weber I, Frangakis AS, Nicastro D, Gerisch G, Baumeister W. Macromolecular architecture in eukaryotic cells visualized by cryoelectron tomography. Science. 2002;298:1209–1213. doi: 10.1126/science.1076184. [DOI] [PubMed] [Google Scholar]
- Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56:855–865. doi: 10.1016/0092-8674(89)90690-9. [DOI] [PubMed] [Google Scholar]
- Murphy GE, Leadbetter JR, Jensen GJ. In situ structure of the complete Treponema primitia flagellar motor. Nature. 2006;442:1062–1064. doi: 10.1038/nature05015. [DOI] [PubMed] [Google Scholar]
- Nicastro D, McIntosh JR, Baumeister W. 3D structure of eukaryotic flagella in a quiescent state revealed by cryo-electron tomography. Proc Natl Acad Sci U S A. 2005;102:15889–15894. doi: 10.1073/pnas.0508274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicastro D, Schwartz C, Pierson J, Gaudette R, Porter ME, McIntosh JR. The molecular architecture of axonemes revealed by cryoelectron tomography. Science. 2006;313:944–948. doi: 10.1126/science.1128618. [DOI] [PubMed] [Google Scholar]
- Nickell S, Mihalache O, Beck F, Hegerl R, Korinek A, Baumeister W. Structural analysis of the 26S proteasome by cryoelectron tomography. Biochem Biophys Res Commun. 2007;353:115–120. doi: 10.1016/j.bbrc.2006.11.141. [DOI] [PubMed] [Google Scholar]
- Ortiz JO, Forster F, Kurner J, Linaroudis AA, Baumeister W. Mapping 70S ribosomes in intact cells by cryoelectron tomography and pattern recognition. J Struct Biol. 2006;156:334–341. doi: 10.1016/j.jsb.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Saxton WO, Baumeister W. The correlation averaging of a regularly arranged bacterial cell envelope protein. J Microsc. 1982;127:127–138. doi: 10.1111/j.1365-2818.1982.tb00405.x. [DOI] [PubMed] [Google Scholar]
- Schmid MF, Paredes AM, Khant HA, Soyer F, Aldrich HC, Chiu W, Shively JM. Structure of Halothiobacillus neapolitanus Carboxysomes by Cryo-electron Tomography. J Mol Biol. 2006;364:526–535. doi: 10.1016/j.jmb.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S. Bridging the imaging gap: visualizing subcellular architecture with electron tomography. Curr Opin Microbiol. 2005;8:316–322. doi: 10.1016/j.mib.2005.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosteson MT, Chow M. Characterization of the ion channels formed by poliovirus in planar lipid membranes. J Virol. 1997;71:507–511. doi: 10.1128/jvi.71.1.507-511.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosteson MT, Wang H, Naumov A, Chow M. Poliovirus binding to its receptor in lipid bilayers results in particle-specific, temperature-sensitive channels. J Gen Virol. 2004;85:1581–1589. doi: 10.1099/vir.0.19745-0. [DOI] [PubMed] [Google Scholar]
- Tuthill TJ, Bubeck D, Rowlands DJ, Hogle JM. Characterization of early steps in the poliovirus infection process: receptor-decorated liposomes induce conversion of the virus to membrane-anchored entry-intermediate particles. J Virol. 2006;80:172–180. doi: 10.1128/JVI.80.1.172-180.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heel M, Schatz M. Fourier shell correlation threshold criteria. J Struct Biol. 2005;151:250–262. doi: 10.1016/j.jsb.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Walz J, Typke D, Nitsch M, Koster AJ, Hegerl R, Baumeister W. Electron Tomography of Single Ice-Embedded Macromolecules: Three-Dimensional Alignment and Classification. J Struct Biol. 1997;120:387–395. doi: 10.1006/jsbi.1997.3934. [DOI] [PubMed] [Google Scholar]
- Zanetti G, Briggs JA, Grunewald K, Sattentau QJ, Fuller SD. Cryo-Electron Tomographic Structure of an Immunodeficiency Virus Envelope Complex In Situ. PLoS Pathog. 2006;2 doi: 10.1371/journal.ppat.0020083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Liu J, Bess J, Jr, Chertova E, Lifson JD, Grise H, Ofek GA, Taylor KA, Roux KH. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441:847–852. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]