

SUMMARY
Ferrochelatase (protoheme ferrolyase, E.C. 4.99.1.1) is the terminal enzyme in heme biosynthesis and catalyzes the insertion of ferrous iron into protoporphyrin IX to form protoheme IX (heme). Due to the many critical roles of heme, synthesis of heme is required by the vast majority of organisms. Despite significant investigation of both the microbial and eucaryotic enzyme, details of metal chelation remain unidentified. In this work we present the first structure of the wild-type human enzyme, a lead-inhibited intermediate of the wild-type enzyme with bound metallated porphyrin macrocycle, the product bound form of the enzyme, and a higher resolution model for the substrate-bound form of the E343K variant. These data paint a picture of an enzyme that undergoes significant changes in secondary structure during the catalytic cycle. The role that these structural alterations play in overall catalysis and potential protein-protein interactions with other proteins, as well as the possible molecular basis for these changes, is discussed. The atomic details and structural rearrangements presented herein significantly advance our understanding of the substrate binding mode of ferrochelatase and reveal new conformational changes in a structurally conserved π helix that is predicted to have a central role in product release.
Keywords: Heme synthesis, Ferrochelatase, Protoporphyrin IX, X-ray crystallography
INTRODUCTION
Heme is a cofactor found in essentially all aerobic organisms and a majority of anaerobes and facultative organisms. Most organisms that possess heme synthesize it themselves1. With the exception of Caenorhabditis elegans and related helminthes2, heme acquired via dietary sources is generally degraded to release free iron and is not utilized as a source of cellular heme3. While the traditional textbook roles for heme as a cofactor include hemo- and myoglobins, cytochromes and a handful of enzymes, considerable evidence has emerged that demonstrates a central role for heme in regulation of gene transcription4; 5, as a gas sensor6, in the regulation of circadian rhythm7, during development8 and in RNAi processing9. Disordered heme metabolism can have profound developmental and health consequences10; 11.
The terminal step in heme biosynthesis is the insertion of ferrous iron into protoporphyrin IX to make protoheme IX12. This step, catalyzed by the enzyme ferrochelatase, thus represents the convergence of two cellular pathways: the synthesis of the organic macrocycle protoporphyrin and the supply of ferrous iron13. These two pathways are tightly regulated since their substrates are both chemically reactive and potentially damaging to the cell14; 15. Ferrochelatase was the first enzyme activity to be identified with a recognized function as a biological metal chelator16; 17. While there was considerable questioning early on about the physiological need for such an enzyme, it is now clear that chelatases and metal chaperones are key elements of biological systems18; 19; 20; 21; 22; 23.
Ferrochelatases among all organisms are highly conserved at the level of tertiary structure although there is less than ten percent conservation at the level of amino acid sequence13. Crystal structures for the enzyme from Bacillus subtilis24; 25; 26; 27; 28, Saccharomyces cerevisiae29, and human30; 31; 32 have been published along with a number of structure/function studies33; 34; 35; 36; 37. Based upon enzymatic studies that demonstrated strong competitive inhibition of ferrochelatase by N-alkyl porphyrins38, Lavalee39 proposed that N-alkyl porphyrins, because of their distorted macrocycle, may represent a transition state analog for the ferrochelatase reaction13; 40. This proposal has been reviewed, discussed and refined, but has been generally accepted. Putative experimental validation for such a model came from studies with catalytic antibodies produced with N-methylmesoporphyrin (N-MeMP) as antigen. These catalytic antibodies bind porphyrin and in doing so distort the macrocycle so that divalent cations of the appropriate size are nonspecifically chelated into the porphyrin41; 42; 43. A similar mode of action is envisioned for ferrochelation catalyzed by DNA- and RNAzymes44; 45. It was, however, the series of structural studies on ferrochelatase from B. subtilis with bound N-MeMP that seemed to solidify this argument26; 28.
The most recently reviewed ferrochelatase catalytic model proposes that the enzyme undergoes relatively minor structural motion as the porphyrin molecule binds in a distorted (ruffled or saddled) conformation46. Metallation is then facilitated by macrocycle distortion and product release from the active site is due to the strain created by metal insertion28. However, recent structural data from human ferrochelatase with bound porphyrin substrate challenges the view that the enzyme exhibits only minor backbone movement and opens slightly upon porphyrin binding26. Specifically, a substrate bound form of human ferrochelatase revealed that the active site mouth closes around the porphyrin substrate with a select group of active site residues becoming reoriented so as to substantially remodel the shape of the active site pocket30; 31. The resulting question then becomes is there protein molecular motion involved in product release, or does it occur simply due to planarization of the macrocycle after metal insertion? Given that protoporphyrin macrocycle distortion is only about 12° in the human ferrochelatase structure31 as opposed to the 35° distortion of N-MeMP in the B. subtilis ferrochelatase structure26; 46, it would seem that planarization alone would not be a driving factor in product release.
Ferrochelatase binds porphyrins with micromolar affinity47, which is similar to what is found for heme binding by many hemoproteins. In hemoproteins the binding of heme is frequently stabilized in part by a coordinating histidine ligand to the heme iron. Interestingly, the available crystal structures of ferrochelatases show the presence of a conserved histidine residue (H263 in human) in the active site that is appropriately positioned for ligation with the heme iron. Thus, with an active site that binds the porphyrin macrocycle with avidity and the presence of a possible ligating histidine residue, it is not clear what would destabilize heme binding at the active site to sufficiently cause heme release. The answer to this question becomes even more intriguing if the conserved histidine residue is involved in insertion of the ferrous iron since this represents a very stable heme binding mode in biology. This question is further complicated for eucaryotes in that another acceptable heme ligand, M76 (human numbering), is positioned near the center of the porphyrin macrocycle on the opposite side of the active site from H26331. While it can be argued that in vivo a heme chaperone system may exist to directly acquire heme from ferrochelatase and transport it to the site of utilization, the fact that in vitro assays of the enzyme have reasonable product release rates demonstrates that ferrochelatase can “eject” the protoheme product34; 48.
Herein we present structural data for human ferrochelatase that we propose represent snapshots of several stages of the turnover mechanism. Some of these data demonstrate that following metallation the conserved π helix on one side of the active site pocket is unwound resulting in the presence of an extended “tongue and chin” of the active site mouth. We propose that this conformational alteration and the reorientation of H263 and one porphyrin propionate substituent are involved in product ejection from the active site. In addition, it may be envisioned that this structural alteration plays a role in making and breaking contacts with other proteins such as protoporphyrinogen oxidase49; 50; 51 or presenting a new binding surface for a heme transport protein.
RESULTS
Structures of the wild-type human ferrochelatase enzyme and the initial steps in catalysis
After extensive screening, crystals of wild type human ferrochelatase were obtained in the current study under a number of conditions. The crystals for the two structures of the wild-type enzyme reported herein were obtained in the presence of the salts ammonium chloride (WT1) or manganese chloride (WT2). Consistent with the structure of the R115L variant of human ferrochelatase that was originally reported32, the enzyme forms a homodimeric structure with each monomer consisting of two similar domains composed of β-α-β motifs and each subunit possessing a single [2Fe-2S] cluster. The wild type enzyme also adopts a predominantly open conformation that is similar to what has been observed for the R115L variant31; 32. However, a comparison of the electron density observed in the active site for the two data sets reveals some significant differences.
Most significant is the nonequivalence of the density in the active site of the A versus the B monomer for the WT1 data set (Fig. 1a). In the A monomer the density is best represented by the presence of just cholate molecules which have been reported previously for the R115L variant of human ferrochelatase31; 32. In contrast, modeling of cholate molecules alone in the active site of the B monomer resulted in the presence of a 10 sigma peak in the difference (1Fo-Fc) map (Fig. 1b). Modeling of a heme molecule into the B monomer in the same orientation as is found for the macrocycle in the E343K variant31 is consistent with an iron in the position of the 10 sigma peak, but shows less than ideal density for the complete tetrapyrrole macrocycle. This may simply indicate a partial occupancy of heme and detergent. Despite the differences in occupancies observed between the active site of the A and B monomers, a superposition of the A and B subunits reveals very few differences in the overall secondary structure, which may be attributable to the partial occupancy of heme in the B subunit. The main differences between chain A and chain B are found in the side chain and main chain atoms of residues 90 to 115. This region corresponds to the upper lip region, which shows significant movement in the protoporphyrin IX bound structure31.
Figure 1.

Occupancy of the active site for WT1 and R115L. (a) Overlay of cholate residues and the side chains of H263, R115 and M76 of the WT1 model with the model previously reported for the R115L variant of human ferrochelatase. (b) Heme molecule modeled into the density present in the active site of monomer B of the WT1 data with side chains of M76 below and H263 above the macrocycle ring. All atoms in the WT1 model are represented as sticks with nitrogen, oxygen, carbon, sulfur, and iron atoms colored blue, red, tan, cyan, and black, respectively. All atoms in the model previously reported for R115L variant are colored green in (a). In all cases the 2Fo-Fc composite omit map was generated using the simulated annealing protocol and is contoured at 1σ (purple and green cage in panels a and b, respectively).
For the data set collected on crystals with manganese chloride added to the purified protein (WT2) there exist three chloride binding sites that were not observed in the WT1 crystals. Two of these sites are in addition to the previously reported chloride site seen in the higher resolution data of the R115L variant31. The positions of these sites and a composite omit map generated with the simulated annealing protocol are shown in Supplemental Figure 1. The recently identified chloride binding sites are found at either end of the α helix (α16) formed by residues 395–400. The role of these chloride atoms is currently unknown other than as counterions to residue side chains.
Porphyrin binding
The initial structure reported for human ferrochelatase was an R115L variant of the enzyme. Crystallization of the wild type protein was not accomplished under the conditions attempted at that time. Recent structural data on ferrochelatase that has the wild type R115 has revealed that the R115 side chain is an important player in substrate binding31. Consistent with this observation are kinetic data (Table II) showing that the Kcat for the R115L variant is slightly lower than that of the wild type enzyme, but the Km for porphyrin is slightly higher. Because of the resolution of the data previously reported for the E343K variant and the pseudo-symmetrical nature of the protoporphyrin IX, it was possible to model the substrate in two orientations with a negligible impact on the R factor31. Since kinetic data clearly demonstrate that the two propionate groups are not equivalent52, it is crucial to properly orient the substrate in order to further advance our understanding of the mechanism of human ferrochelatase. The higher resolution data reported here for the E343K variant unequivocally place the 6 position propionate group at the N-terminal end of a π helix in human ferrochelatase (Fig. 2). The higher resolution data also provide a better orientation of the additional substrate molecule bound just outside of the active site (Supplemental Fig. 2). The orientation of the second protoporphyrin IX molecule is consistent with the binding mode of the substrate molecule bound within the active site. A pair of arginine residues (R114 and R115 in the human enzyme) are found in the upper lip region and interact directly with the substrate on the outside and inside of the active site.
Table II.
Kinetic parameters for wild-type and variant ferrochelatases
| kcat (min−1) | KmFe (μM) | KmMeso (μM) | |
|---|---|---|---|
| Wild-type | 4.2 ± 0.2 | 18 ± 1.2 | 28 ± 2.8 |
| R115L | 3.2 ± 0.1 | 21 ± 1.0 | 34 ± 1.0 |
| F110A | 3.2 ± 0.2 | 20 ± 3.2 | 24 ± 4.7 |
Figure 2.

Substrate binding to the E343K variant of human ferrochelatase. (a) Schematic representation of protoporphyrin IX. (b) Cartoon representation of the overall fold of the protoporphyrin-bound structure of the E343K variant showing the position of the substrate relative to the π helix (colored dark blue) in human ferrochelatase. (c) Wall-eyed stereo view of the composite omit map for the substrate and residues near the substrate in the π helix. The atoms are represented as sticks and are colored blue, red and tan for nitrogen, oxygen, and carbon, respectively. The 2Fo-Fc composite omit map is contoured at 1σ (green cage) and was generated using the simulated annealing protocol with 7 % of the model being omitted per cycle.
Within the active site several amino acid side chains are close enough to have van der Waals contacts with the substrate (Fig. 3). These include residues L92, L98, T198, V305 (which are well conserved in most ferrochelatases), and Y276 and W310 (which are identical in all known ferrochelatases). These highly conserved hydrophobic residues may support the closure of the active site and/or exclusion of water as well as participate in porphyrin substrate orientation.
Figure 3.

Model of protoporphyrin IX in the active site and composite omit map of monomer A for the E343K data highlighting residues within van der Waals contact of the substrate. (a) View of the porphyrin macrocycle and nearby residues on the H263 side of the pocket. (b) View of the porphyrin in the active site looking at the substrate from the same side of the substrate as residue M76. The atoms are represented as sticks and are colored blue, red, yellow and tan for nitrogen, oxygen, sulfur and carbon, respectively. For clarity the 2Fo-Fc omit map is contoured at 1σ around the substrate only and was generated as described in the legend to Figure 2.
Metallation and product release
Ferrochelatase is inhibited by a number of divalent cations including lead53. Lead is a weak competitive inhibitor with respect to iron and because of this it was initially thought that a lead-inhibited enzyme would have lead bound by the same set of residues that bind iron. With an ionic radius of 0.98 Å vs 0.64Å for ferrous iron it was not known if lead could access the active site or be inserted into protoporphyrin IX. To further investigate lead as a potential inhibitor of human ferrochelatase the enzyme was incubated with protoporphyrin IX and lead during crystallization. Data collection and phasing by molecular replacement revealed a 20 σ peak (well above the 9 σ peak for each iron in the [2Fe-2S] clusters) in the active site of each monomer as well as density consistent with protoporphyrin IX. Given that the data was collected at 0.98 Ǻ, the lead atom should have an anomalous signal that is at least two times larger than that of iron.
While an anomalous signal consistent with this prediction was observed, the potential lead position could not be refined suggesting a partially occupied site or poorly defined site. All of the structures that are currently available for lead-porphyrin compounds show that the lead is situated out of the mean plane of the porphyrin with an approximate N-lead bond distances of 2.37 to 2.4 Ǻ54; 55. Similar to what was found with ferrochelatase, in these inorganic complexes the lead atom appears to be highly disordered. Consistent with these observations, the best R-factors for our lead-porphyrin ferrochelatase model and the structures of known lead-porphyrin compounds are obtained if the lead atom is modeled with partial occupancy on either side of the protoporphyrin IX as is shown in Figure 4a. A significant amount of additional density was observed on either side of the protoporphyrin IX. As lead acetate was used during preparation of the crystals, it is not surprising that the placement of an acetate moiety coordinated to the lead could accommodate the observed density nicely, if given a partial occupancy similar to the lead atoms (Supplemental Fig. 3a). A minor exception to this occurs on one side of the lead porphyrin moiety in the B monomer where the density was best fit by modeling in molecular oxygen (Supplemental Fig. 3b). Considering the resolution of the data (2.3 angstroms) and the observation that lead-porphyrin species are meta-stable, further work will be needed to accurately describe the atomic details of the lead inhibition of ferrochelatase.
Figure 4.
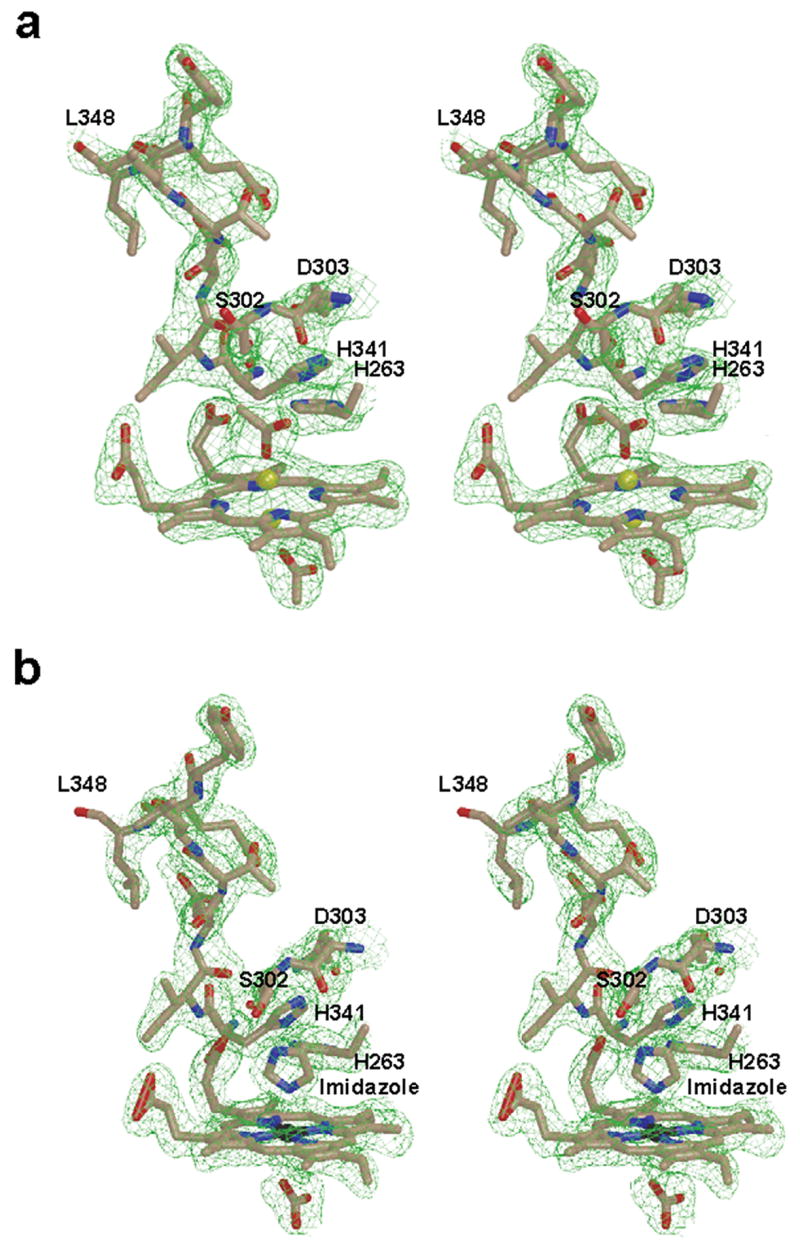
Wall-eyed stereo diagram for the model and 2Fo-Fc composite omit map (green cage) showing porphyrin macrocycle and the unwound π helix observed in (a) the lead-inhibited ferrochelatase and (b) the protoheme-bound F110A variant. Also visible in are the axial ligands for the porphyrin-bound metal: acetates in (a) and imidazole (labeled) and bicarbonate in (b). The models are shown in ball and stick format with the nitrogen, oxygen, carbon, lead, and iron atoms colored blue, red, tan, yellow, and black, respectively. The 2Fo-Fc composite omit map is contoured at 1σ and was generated as described in the legend to Figure 2.
In the lead-porphyrin bound enzyme several additional observations were made. First, in comparison to the binding mode of protoporphyrin IX alone in the E343K variant, the propionate at position 6 of the lead porphyrin is oriented such that it folds back towards the center of the macrocycle. Second, and most strikingly, the conserved π helix56 is “unwound” from residues 340 to 349. In addition, the imidazole of H263 is no longer located 3.0 Å from the center of the macrocycle but is now approximately 5 Å distant (depending upon the orientation of the imidazole either 4.5 Å or 5.3 Å from the closest pyrrole nitrogen). Other residues that are found in a different spatial orientation in the lead-inhibited structure relative to the wild-type enzyme include M76, R164, F337, E343 and H341. The lead-inhibited enzyme also has a slightly more “open” conformation in comparison to the protoporphyrin IX bound structure. These observations are shown in Figure 4a and underscore the dynamic nature of ferrochelatase.
During a series of kinetic studies on human ferrochelatase variants an F110A variant was noted to possess significant amounts of bound heme as isolated (data not shown). Kinetic characterization of this variant revealed an active enzyme with unaltered Kms, but with a diminished Kcat with respect to the wild-type enzyme (Table II). Crystallization of this enzyme yielded data consistent with enzyme that had product bound. These data are presented in Figure 4b and clearly reveal the presence of a heme group within the active site. More importantly, the F110A variant data reveal that the propionate at position 6 is bent back towards the metallated porphyrin, the side chain of H263 is moved and the π helix is unwound, as we observed for the lead inhibited wild-type enzyme. Additional density on either side of the heme group in the F110A variant data set was also observed. The slightly higher resolution clearly indicated that the heme group had two non-protein ligands and that imidazole, which was present in the elution buffer, was one of the ligands while bicarbonate could be modeled as the other. With respect to the bound imidazole in the F110A variant active site, it is of interest that it occupies the same position as the imidazole side chain of H263 in the E343K variant and the wild-type ferrochelatase. In the heme-bound F110A variant the imidazole is spatially oriented to be a ligand to the heme iron and we believe that this stabilizes product binding in the enzyme active site. In support of this view is the observation that it was not possible to obtain crystals of enzyme-bound heme when efforts were made to remove imidazole from the purified enzyme. Many of the same residues that were observed to have altered orientation in the lead inhibited wild-type data relative to the lead-free wild-type structure occupy similar spatial orientations in the heme bound F110A variant structure.
In both the lead-bound and F110A variant data sets an extensive number of residues associated with the π helix are clearly repositioned which results in a significant change in the overall shape of ferrochelatase with the extension of a “chin,” as well as a substantial change in surface potential encompassing this region of the active site (Fig. 5).
Figure 5.

Electrostatic surface potential showing the active site region for the (a) wild type, (b) the substrate bound, and (c) heme bound human ferrochelatase. For clarity, the upper lip and π helix regions are highlighted. The figure was generated with PYMOL.
DISCUSSION
Redox active metals play central roles in a wide variety of metabolic and regulatory pathways. In spite of this, the molecular mechanism by which metal is inserted into either a cofactor or protein has received minimal attention until relatively recently. Although ferrochelatase was the first enzyme activity identified as being obligately involved in iron chelation almost fifty years ago16; 17, little real progress has been made in understanding the molecular mechanism. This is in contrast to copper metabolism and iron sulfur cluster assembly about which entire research fields have evolved over the past twenty years18; 19; 20.
Ferrochelatase, like many of the enzymes of the heme biosynthetic pathway, is an evolutionarily old protein12. While primary sequences for ferrochelatase are poorly conserved, the overall fold of the ferrochelatase enzyme from human32, yeast29, and bacteria24 is highly conserved13. In fact, the human enzyme structure aligns quite nicely with the B. subtilis ferrochelatase, having a root mean square deviation of 2.4 Ǻ for the Cα atoms of residues 130–39132. In addition to substrate metal selectivity, ferrochelatases have the challenge of binding and differentiating between two similar molecules: the substrate, protoporphyrin IX, and the product, protoheme IX. The active site pocket must bind transiently to both of these macrocycles in a fashion so as not to create a thermodynamic barrier to product release.
Previously we have demonstrated that porphyrin binding to human ferrochelatase involves backbone structural movement that closes the active site mouth around the porphyrin and results in the reorientation of a set of side chains that reshape the pocket (Supplemental Movie 1). The combination of these events causes a 12° distortion of the porphyrin macrocycle which facilitates iron insertion30; 31. The data presented above show that following iron insertion additional secondary structural changes occur that may facilitate heme release. The key player in all of these structural rearrangements appears to be H263, the conserved histidine that is located centrally in the active site. Several observations related to H263 are of note. First, in the resting state enzyme H263, by hydrogen bonding with E343, anchors one end of a complex hydrogen bond network within the active site30. Upon binding of porphyrin substrate this network reorients as H263 switches from a hydrogen bond with E343 to bond with a pyrrole nitrogen31. Mutations of H263 which prevent hydrogen bonding with E343 also cause similar reorientations to occur30. Secondly is the observation that in the post metallation structures the side chain of H263 is located several angstroms from its initial position and is no longer spatially close to the center of the porphyrin macrocycle, but now sits outside the meso carbon between rings B and C. In this position it no longer participates in any hydrogen bond network. This is consistent with the side chain of H263 being fully protonated. Thirdly, no H263 variant ferrochelatase has any in vivo or in vitro activity and the activity of the H263A variant is not rescued in vitro by added imidazole35. If H263 served simply as a metal donor or proton acceptor, then one might expect to see some activity with either the H263C variant or with assays spiked with imidazole. Neither is observed, suggesting that the role of H263 is more substantial.
Based upon these data we propose that the catalytic cycle for human ferrochelatase involves a series of reactions that revolve around H263 and an active site hydrogen bond network. In this model initial binding of porphyrin substrate allows H263 to release its hydrogen bond with E343 and form one with a protonated pyrrole ring (most likely the B ring). This sets in motion the reorientation of several key active site residues, which along with mouth closing, results in macrocycle distortion. Mouth closing may be independent of the hydrogen bond network realignment30 and dependent upon hydrophobic interactions between the fully conjugated macrocycle and select hydrophobic active site residues such as L92, L98, V305, and W310. Interactions between the porphyrin propionate 7 with R115 and propionate 6 with the conserved residues S130 and Y123 effectively orient the macrocycle in proper position for metallation31. Upon metallation two significant events occur: interactions of the 6 propionate substituent and S130 and Y123 are broken, and H263 swings away from the metallated porphyrin. Also occurring after metallation is movement of the guanido side chain of R114. This is of interest since it rotates into the position occupied by F110 in the porphyrin-bound, closed mouth enzyme molecule. We suggest that metal insertion displaces the proton from the pyrrole nitrogen to H263, thereby creating the imidazole cation which would be subject to charge repulsion from the incoming divalent metal cation. This particular feature would seem essential to ensure that a nonionized imidazole not be left near the heme iron where it would be expected to form a stable interaction that would make subsequent removal of the heme product problematic. This same issue argues against H263 as a metal donor in the reaction since there appears to be no obvious mechanism in place to protonate a metal binding imidazole. Clearly the 12° distortion of the macrocycle that has been identified31 would not provide sufficient out of plane strain following metal insertion to pull the newly positioned iron away from a donating imidazole group57.
Completing the catalytic cycle requires product release. This is facilitated by significant secondary structure reorganization of the previously identified conserved π helix56 that extends from D340 to C360 in human ferrochelatase (Supplemental Movie 1). We propose that this is triggered by the movement of the H263 imidazole side chain and the 6 position heme propionate. The structures of the F110A variant with bound heme and the wild type enzyme with bound lead-protoporphyrin show that these enzymes have unwound half of the π helix with the resultant change in the position of the conserved H341 and E343 side chains. The side chain of D340 is moved only slightly while the backbone and side chains of H341 to G361 occupy significantly different spatial areas. The Cα of E343 is 4.4 Å distant from its site in the resting state and the side chain is rotated about 180°. Movement of the H263 imidazole side chain is into the area occupied by the carboxylate side chain of E343 in the resting state and porphyrin bound structures. Movement of E343 results in disruption of the π helix and the secondary structural perturbation extends to E351. The remaining π helix is extended, canted, and turned relative to that segment of the π helix in the resting state enzyme (Fig. 6). It should be noted that this entire region (340–360) has a very high B value in the crystal structures of F110A and wild type with lead bound, indicating the dynamic nature of this region. The reorientation of side chains results in an altered electrostatic surface contour in this region. The conserved acid patch is eliminated, the active site mouth opens, and a relatively nonpolar “slide” is created for the heme to exit (Fig. 5). The considerable spatial and temporally orchestrated molecular movement that must occur during catalysis was not anticipated, but does explain the kinetic data which demonstrated the essentially irreversible nature of the enzymatic metallation48.
Figure 6.

Cartoon representation of the structural alignment of the free, substrate-bound and product-bound forms of human ferrochelatase. (a) Cartoon overlay of the wild-type, substrate-free structure in cream color. Areas of significant movement are highlighted in color. The upper lip of the active site that is altered in spatial orientation in the E343K substrate-bound variant relative to the wild-type enzyme structure is shown in green. The unwound π helix segment of the F110A product-bound variant enzyme is shown in red. Bound protoporphyrin IX is shown in brick and highlighted using dots to denote the electron density. The [2Fe-2S] cluster is shown as yellow and blue spheres. (b) Enlarged view of the bound protoporphyrin IX (brick) and bound heme (purple) in the same spatial orientation as shown in the cartoon model. The movement of the protoporphyrin/protoheme IX propionate 6, which is on the corner of the macrocycle in this orientation, is highlighted by the grey arrow.
The altered configuration of the π helix as seen in the product bound structures has multiple possible implications. First, because it alters the electrostatic nature of the putative heme exit pathway, it may facilitate product release as discussed above. Second, the change in the surface shape and charge that occurs when the π helix is unwound may alter the dynamics of possible protein-protein interactions (Fig. 7). It has been suggested both experimentally and from a structural basis that ferrochelatase may physically interact with protoporphyrinogen oxidase, the previous enzyme in the pathway49; 50; 51. If this interaction takes place, then alteration of the surface geometry or charge of ferrochelatase upon the completion of metal insertion may alter the thermodynamics of this putative protein-protein interaction so as to make it unfavorable and also facilitate interactions with a heme-accepting chaperone protein (currently unidentified) that may the transport heme to its site of use. Thus, the structurally conserved π helix may play a role significantly greater than just as a proton conduit during catalysis as was previously proposed13; 35; 58. The π helix may also function in heme release and as a modulator of protein-protein interactions. Interestingly, the surface electrostatics on the back side of ferrochelatase are also altered in the product bound stage, which may have an impact on possible interactions with the putative iron transporter/donor mitoferrin59.
Figure 7.

Model for the possible role of structural changes in putative protein-protein interactions. Cartoon representation of how human ferrochelatase (Fc) might interact with protoporphyrinogen oxidase (PPO) during binding of protoporphyrin IX. Following iron insertion the π helix is unwound and a new binding surface is presented for the release of protoheme to a currently unidentified heme carrier protein (HCP). The inner mitochondrial membrane is shown as solid lines.
Neither the ultimate source of iron for the ferrochelatase reaction nor path of iron transport to the active site is identified by the current studies. Previously it has been suggested that metal enters via the acidic patch (E343, D340) on the π helix to ultimately be passed to H263 for metallation25. That proposal requires that metallation and pyrrole deprotonation occur on one surface of the macrocycle and that it employs only the single histidine residue for both functions. It does not explain how the iron-imidazole bond that would be formed following metallation is broken once metal insertion is accomplished. This proposal is also inconsistent with the observed differences in the metal selectivity observed for various ferrochelatases. Recent pre-steady state kinetic analyses have shown that the conserved residue E343 may serve multiple roles, but that its primary function occurs after metal insertion48. Another suggestion based largely upon kinetic analysis of site-directed mutant variants is that the iron enters via a channel that extends from the active site pocket to the back side of the enzyme35(unpublished data). Such a proposal is consistent with iron delivery via a matrix-located iron donor such as the mitochondrial iron transporter mitoferrin59.
METHODS
Mutagenesis, enzyme expression and purification
Wild-type ferrochelatase was expressed and purified as previously described60; 61. The F110A variant was created using the QuikChange® Site-Directed Mutagenesis protocol (Stratagene, Lo Jolla, CA). Both the E343K and F110A variants were expressed and purified as previously described35; 60.
Enzyme assay and crystallization
Wild-type and variant ferrochelatases were assayed using the previously described continuous direct spectroscopic method62.
Wild-type and variant enzymes were concentrated to between 500 and 800 μM in concentration buffer31. Approximately 50 μmoles of lead acetate and protoporphyrin IX were added to wild-type enzyme prior to concentration. Manganese chloride was added following concentration in two-fold excess of the enzyme concentration. Mother liquor for wild-type (WT1 and WT2), wild-type with lead and protoporphyrin IX and the F110A variant was composed of 0.05M ammonium acetate, 0.05M Bis-Tris, pH 6.5 and 40% (v/v) pentaerythritol ethoxylate (15/4 EO/OH). E343K was crystallized as previously described31.
Data collection, structure determination and refinement
Diffraction data for the WT1 and WT2 data sets were collected remotely using the SER-CAT remote user participation program63 on beamline SER-C. All other data were collected at the Advanced Light Source and were processed using the program HKL200064. Phases were obtained by molecular replacement using a poly alanine model of monomer A taken from the R115L structure published previously (PDB ID 1HRK). Iterative rounds of model building and refinement were performed using the programs COOT65 and CNS66.
Coordinates
Coordinates for the E343K, F110A, WT1, WT2, and lead-inhibited wild type ferrochelatase models have been deposited in the protein data bank under the accession codes 2QD1, 2QD2, 2QD3, 2QD4, and 2QD5, respectively.
Supplementary Material
Supplementary Figure 1. Wall-eyed stereo view of the model and composite omit map for two chloride binding sites at the (a) N-terminal and (b) C-terminal end of helix α16 in the WT2 data set for human ferrochelatase. Atoms in the WT2 model are represented as balls and sticks with nitrogen, oxygen, carbon, and chloride atoms colored blue, red, tan, and yellow, respectively. The 2Fo-Fc composite omit map is contoured at 1σ (green cage) and was generated using the simulated annealing protocol with 7 % of the model being omitted per cycle.
Supplementary Figure 2. Wall-eyed stereo cartoon representation showing the entrance to the active site in the E343K model and the relative positions of the protoporphyrin IX molecules, detergent, and some basic residues in the upper lip of the active site “mouth”. For clarity the 2Fo-Fc composite omit map is contoured at 1σ around the substrate only (green cage) and was generated as described in the legend to Supplementary Figure 1.
Supplemental Figure 3. Wall-eyed stereoview for the model and 2Fo-Fc composite omit map (green cage) of the lead-porphyrin complex observed in the active site of the lead and protoporphyrin-treated human ferrochelatase crystals. Data for both monomer A (a) and monomer B (b) are shown with nitrogen, oxygen, carbon, and lead atoms represented as ball and sticks and colored blue, red, tan, and orange, respectively. The 2Fo-Fc composite omit map is contoured at 1σ and was generated as described in the legend to Figure 2.
Supplemental Movie 1. Animation showing the proposed movement of the upper lip upon binding of protoporphyrin IX and unwinding of π helix following heme production. Movie created using PDB ID files 2QD4, 2QD1, 2QD5, and 2QD3. The animation and rendering were created using the Yale Morph Server and PyMol.
Table I.
Data collection and refinement statistics.
| Data | WT1 | WT2 | E343K | Pb-inhibited | F110A |
| Space Group | P212121 | P212121 | P1 | P212121 | P212121 |
| Wavelength | 0.98 | 0.98 | 0.98 | 0.98 | 0.98 |
| Resolution Range (Ǻ) | 50.0–2.2 | 50.0–2.0 | 50.0–2.2 | 50.0–2.3 | 50.0–2.2 |
| Outer Shell | 2.33–2.2 | 2.07–2.0 | 2.30–2.2 | 2.38–2.3 | 2.35–2.2 |
| Unique Observations | 89,803 | 117,326 | 86,145 | 71,861 | 44,989 |
| Completeness (%) | 100.0(99.8)a | 99.9(98.3) | 95.4(78.8) | 94.7(80.0) | 99.9(99.8) |
| Rsym (%)b | 0.08(0.23) | 0.06(0.28) | 0.08(0.32) | 0.06(0.26) | 0.07(0.25) |
| I/σ | 32.1(5.3) | 27.2(4.7) | 28.9(4.2) | 24.3(2.4) | 31.6(5.1) |
|
| |||||
| Model | WT1 | WT2 | E343K | Pb-inhibited | F110A |
| Unit Cell (a, b, c) in Ǻ | 88,93,111 | 87,93,109 | 61,88,93c | 86,92,109 | 86,92,109 |
| Protein Atoms | 5,790 | 5,790 | 11,580 | 5,790 | 5,790 |
| Solvent Atoms | 427 | 500 | 686 | 182 | 385 |
| Resolution Limits (Ǻ) | 50.0–2.2 | 50.0–2.0 | 50.0–2.2 | 50.0–2.3 | 50.0–2.2 |
| Rcryst (%) | 20.9 | 20.6 | 22.2 | 23.6 | 22.0 |
| Rfree (%) | 24.2 | 23.0 | 26.1 | 28.0 | 26.0 |
| rmsd bonds (Ǻ) | 0.007 | 0.006 | 0.008 | 0.009 | 0.008 |
| rmsd angles ( ° ) | 1.29 | 1.27 | 1.47 | 1.69 | 1.55 |
| average B factor (Ǻ2) | 30.2 | 27.1 | 32.7 | 54.1 | 32.9 |
Numbers in parentheses denote values for the outermost resolution shell.
Rsym = Σhkl[ΣI(|Ihkl,I - <Ihkl>|)]/Σhkl,I<Ihkl>, where Ihkl is the intensity of an individual measurement of the reflection with indices hkl and <Ihkl> is the mean intensity of that reflection.
Angles for the triclinic data were α=102.49 β=108.99 and γ=105.55.
Acknowledgments
This work was supported by a grant from the American Heart Association Grant AHA0465228B to W.N.L. and the National Institutes of Health Grant DK32303 to H.A.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dailey HA. Ferrochelatase. In: Hausinger RP, Eichhorn GL, Marzilli LG, editors. Mechanisms of Metallocenter Assembly. VCH Publisher, Inc; New York: 1996. pp. 77–98. [Google Scholar]
- 2.Rao AU, Carta LK, Lesuisse E, Hamza I. Lack of heme synthesis in a free-living eukaryote. Proc Natl Acad Sci U S A. 2005;102:4270–5. doi: 10.1073/pnas.0500877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunn LL, Rahmanto YS, Richardson DR. Iron uptake and metabolism in the new millennium. Trends Cell Biol. 2007;17:93–100. doi: 10.1016/j.tcb.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Hou S, Reynolds MF, Horrigan FT, Heinemann SH, Hoshi T. Reversible binding of heme to proteins in cellular signal transduction. Acc Chem Res. 2006;39:918–24. doi: 10.1021/ar040020w. [DOI] [PubMed] [Google Scholar]
- 5.Mense SM, Zhang L. Heme: a versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 2006;16:681–92. doi: 10.1038/sj.cr.7310086. [DOI] [PubMed] [Google Scholar]
- 6.Gilles-Gonzalez MA. Oxygen signal transduction. IUBMB Life. 2001;51:165–73. doi: 10.1080/152165401753544232. [DOI] [PubMed] [Google Scholar]
- 7.Dioum EM, Rutter J, Tuckerman JR, Gonzalez G, Gilles-Gonzalez MA, McKnight SL. NPAS2: a gas-responsive transcription factor. Science. 2002;298:2385–7. doi: 10.1126/science.1078456. [DOI] [PubMed] [Google Scholar]
- 8.Tsiftsoglou AS, Tsamadou AI, Papadopoulou LC. Heme as key regulator of major mammalian cellular functions: molecular, cellular, and pharmacological aspects. Pharmacol Ther. 2006;111:327–45. doi: 10.1016/j.pharmthera.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 9.Faller M, Matsunaga M, Yin S, Loo JA, Guo F. Heme is involved in microRNA processing. Nat Struct Mol Biol. 2007;14:23–9. doi: 10.1038/nsmb1182. [DOI] [PubMed] [Google Scholar]
- 10.Kappas A, Sassa S, Galbraith RA, Nordmann Y. The Porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecuar Bases of Inherited Disease. The Mc-Graw-Hill Companies Inc; New York: 1995. pp. 2103–2160. [Google Scholar]
- 11.Kauppinen R. Porphyrias. Lancet. 2005;365:241–52. doi: 10.1016/S0140-6736(05)17744-7. [DOI] [PubMed] [Google Scholar]
- 12.Dailey HA. Terminal steps of haem biosynthesis. Biochem Soc Trans. 2002;30:590–5. doi: 10.1042/bst0300590. [DOI] [PubMed] [Google Scholar]
- 13.Dailey HA, Dailey TA. Ferrochelatase. In: Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. Vol. 12. Academic Press; New York: 2003. pp. 93–121. 20 vols. [Google Scholar]
- 14.Medlock AE, Dailey HA. Regulation of Mammalian Heme Biosynthesis. In: Warren M, Smith AG, editors. Tetrapyrroles. Landes Bioscience and Springer Science +; Business Media, Austin: in press. [Google Scholar]
- 15.Napier I, Ponka P, Richardson DR. Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood. 2005;105:1867–74. doi: 10.1182/blood-2004-10-3856. [DOI] [PubMed] [Google Scholar]
- 16.Goldberg A, Ashenbrucker H, Cartwright GE, Wintrobe MM. Studies on the Biosynthesis of Heme In Vitro by Avian Erythrocytes. Blood. 1956;11:821–833. [PubMed] [Google Scholar]
- 17.Klein JR, Krueger RC, Melnick I. Formation of heme by broken-cell preparations of duck erythrocytes. Arch Biochem Biophys. 1956;64:302–10. doi: 10.1016/0003-9861(56)90273-9. [DOI] [PubMed] [Google Scholar]
- 18.Hamza I, Gitlin JD. Copper chaperones for cytochrome c oxidase and human disease. J Bioenerg Biomembr. 2002;34:381–8. doi: 10.1023/a:1021254104012. [DOI] [PubMed] [Google Scholar]
- 19.Johnson DC, Dean DR, Smith AD, Johnson MK. Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem. 2005;74:247–81. doi: 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- 20.Lill R, Dutkiewicz R, Elsasser HP, Hausmann A, Netz DJ, Pierik AJ, Stehling O, Urzica E, Muhlenhoff U. Mechanisms of iron-sulfur protein maturation in mitochondria, cytosol and nucleus of eukaryotes. Biochim Biophys Acta. 2006;1763:652–67. doi: 10.1016/j.bbamcr.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 21.Dailey HA, Dailey TA, Wu CK, Medlock AE, Wang KF, Rose JP, Wang BC. Ferrochelatase at the millennium: structures, mechanisms and [2Fe-2S] clusters. Cell Mol Life Sci. 2000;57:1909–26. doi: 10.1007/PL00000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reid JD, Hunter CN. Current understanding of the function of magnesium chelatase. Biochem Soc Trans. 2002;30:643–5. doi: 10.1042/bst0300643. [DOI] [PubMed] [Google Scholar]
- 23.Schubert HL, Raux E, Matthews MA, Phillips JD, Wilson KS, Hill CP, Warren MJ. Structural diversity in metal ion chelation and the structure of uroporphyrinogen III synthase. Biochem Soc Trans. 2002;30:595–600. doi: 10.1042/bst0300595. [DOI] [PubMed] [Google Scholar]
- 24.Al-Karadaghi S, Hansson M, Nikonov S, Jonsson B, Hederstedt L. Crystal structure of ferrochelatase: the terminal enzyme in heme biosynthesis. Structure. 1997;5:1501–10. doi: 10.1016/s0969-2126(97)00299-2. [DOI] [PubMed] [Google Scholar]
- 25.Hansson MD, Karlberg T, Rahardja MA, Al-Karadaghi S, Hansson M. Amino acid residues His183 and Glu264 in Bacillus subtilis ferrochelatase direct and facilitate the insertion of metal ion into protoporphyrin IX. Biochemistry. 2007;46:87–94. doi: 10.1021/bi061760a. [DOI] [PubMed] [Google Scholar]
- 26.Lecerof D, Fodje M, Hansson A, Hansson M, Al-Karadaghi S. Structural and mechanistic basis of porphyrin metallation by ferrochelatase. J Mol Biol. 2000;297:221–32. doi: 10.1006/jmbi.2000.3569. [DOI] [PubMed] [Google Scholar]
- 27.Lecerof D, Fodje MN, Alvarez Leon R, Olsson U, Hansson A, Sigfridsson E, Ryde U, Hansson M, Al-Karadaghi S. Metal binding to Bacillus subtilis ferrochelatase and interaction between metal sites. J Biol Inorg Chem. 2003;8:452–8. doi: 10.1007/s00775-002-0436-1. [DOI] [PubMed] [Google Scholar]
- 28.Shipovskov S, Karlberg T, Fodje M, Hansson MD, Ferreira GC, Hansson M, Reimann CT, Al-Karadaghi S. Metallation of the transition-state inhibitor N-methyl mesoporphyrin by ferrochelatase: implications for the catalytic reaction mechanism. J Mol Biol. 2005;352:1081–90. doi: 10.1016/j.jmb.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Karlberg T, Lecerof D, Gora M, Silvegren G, Labbe-Bois R, Hansson M, Al-Karadaghi S. Metal binding to Saccharomyces cerevisiae ferrochelatase. Biochemistry. 2002;41:13499–506. doi: 10.1021/bi0260785. [DOI] [PubMed] [Google Scholar]
- 30.Dailey HA, Wu CK, Horanyi P, Medlock AE, Najahi-Missaoui W, Burden AE, Dailey TA, Rose J. Altered orientation of active site residues in variants of human ferrochelatase. Evidence for a hydrogen bond network involved in catalysis. Biochemistry. 2007;46:7973–9. doi: 10.1021/bi700151f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Medlock A, Swartz L, Dailey TA, Dailey HA, Lanzilotta WN. Substrate interactions with human ferrochelatase. Proc Natl Acad Sci U S A. 2007;104:1789–93. doi: 10.1073/pnas.0606144104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu CK, Dailey HA, Rose JP, Burden A, Sellers VM, Wang BC. The 2.0 A structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat Struct Biol. 2001;8:156–60. doi: 10.1038/84152. [DOI] [PubMed] [Google Scholar]
- 33.Franco R, Ma JG, Lu Y, Ferreira GC, Shelnutt JA. Porphyrin interactions with wild-type and mutant mouse ferrochelatase. Biochemistry. 2000;39:2517–29. doi: 10.1021/bi991346t. [DOI] [PubMed] [Google Scholar]
- 34.Franco R, Pereira AS, Tavares P, Mangravita A, Barber MJ, Moura I, Ferreira GC. Substitution of murine ferrochelatase glutamate-287 with glutamine or alanine leads to porphyrin substrate-bound variants. Biochem J. 2001;356:217–22. doi: 10.1042/0264-6021:3560217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sellers VM, Wu CK, Dailey TA, Dailey HA. Human ferrochelatase: characterization of substrate-iron binding and proton-abstracting residues. Biochemistry. 2001;40:9821–7. doi: 10.1021/bi010012c. [DOI] [PubMed] [Google Scholar]
- 36.Shi Z, Ferreira GC. Probing the active site loop motif of murine ferrochelatase by random mutagenesis. J Biol Chem. 2004;279:19977–86. doi: 10.1074/jbc.M313821200. [DOI] [PubMed] [Google Scholar]
- 37.Hansson MD, Lindstam M, Hansson M. Crosstalk between metal ions in Bacillus subtilis ferrochelatase. J Biol Inorg Chem. 2006;11:325–33. doi: 10.1007/s00775-006-0080-2. [DOI] [PubMed] [Google Scholar]
- 38.Dailey HA, Fleming JE. Bovine ferrochelatase. Kinetic analysis of inhibition by N-methylprotoporphyrin, manganese, and heme. J Biol Chem. 1983;258:11453–9. [PubMed] [Google Scholar]
- 39.Lavallee DK. In: Mechanistic Principles of Enzyme Activity. Liebman JF, Greenberg A, editors. VCH; New York: 1988. pp. 279–314. [Google Scholar]
- 40.Dailey HA. Conversion of Coproporphyrinogen to Protoheme in Higher Eukaryotes and Bacterial: Terminal Three Enzymes. In: Dailey HA, editor. Biosynthesis of Heme and Chlorophylls. McGraw-Hill Publishing Company; New York: 1990. pp. 123–161. [Google Scholar]
- 41.Blackwood ME, Jr, Rush TS, 3rd, Romesberg F, Schultz PG, Spiro TG. Alternative modes of substrate distortion in enzyme and antibody catalyzed ferrochelation reactions. Biochemistry. 1998;37:779–82. doi: 10.1021/bi972616f. [DOI] [PubMed] [Google Scholar]
- 42.Cochran AG, Schultz PG. Antibody-catalyzed porphyrin metallation. Science. 1990;249:781–3. doi: 10.1126/science.2389144. [DOI] [PubMed] [Google Scholar]
- 43.Venkateshrao S, Yin J, Jarzecki AA, Schultz PG, Spiro TG. Porphyrin distortion during affinity maturation of a ferrochelatase antibody, monitored by Resonance Raman spectroscopy. J Am Chem Soc. 2004;126:16361–7. doi: 10.1021/ja0465395. [DOI] [PubMed] [Google Scholar]
- 44.Conn MM, Prudent JR, Schultz PG. Porphyrin Metalation Catalyzed by a Small RNA Molecule. J Am Chem Soc. 1996;118:7012–7013. [Google Scholar]
- 45.Li Y, Sen D. Toward an efficient DNAzyme. Biochemistry. 1997;36:5589–99. doi: 10.1021/bi962694n. [DOI] [PubMed] [Google Scholar]
- 46.Al-Karadaghi S, Franco R, Hansson M, Shelnutt JA, Isaya G, Ferreira GC. Chelatases: distort to select? Trends Biochem Sci. 2006;31:135–42. doi: 10.1016/j.tibs.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dailey HA, Jones CS, Karr SW. Interaction of free porphyrins and metalloporphyrins with mouse ferrochelatase. A model for the active site of ferrochelatase. Biochim Biophys Acta. 1989;999:7–11. doi: 10.1016/0167-4838(89)90021-6. [DOI] [PubMed] [Google Scholar]
- 48.Hoggins M, Dailey HA, Hunter CN, Reid JD. Direct measurement of metal ion chelation in the active site of human ferrochelatase. Biochemistry. 2007;46:8121–7. doi: 10.1021/bi602418e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferreira GC, Andrew TL, Karr SW, Dailey HA. Organization of the terminal two enzymes of the heme biosynthetic pathway. Orientation of protoporphyrinogen oxidase and evidence for a membrane complex. J Biol Chem. 1988;263:3835–9. [PubMed] [Google Scholar]
- 50.Koch M, Breithaupt C, Kiefersauer R, Freigang J, Huber R, Messerschmidt A. Crystal structure of protoporphyrinogen IX oxidase: a key enzyme in haem and chlorophyll biosynthesis. Embo J. 2004;23:1720–8. doi: 10.1038/sj.emboj.7600189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Proulx KL, Woodard SI, Dailey HA. In situ conversion of coproporphyrinogen to heme by murine mitochondria: terminal steps of the heme biosynthetic pathway. Protein Sci. 1993;2:1092–8. doi: 10.1002/pro.5560020703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Honeybourne CL, Jackson JT, Jones OT. The interaction of mitochondrial ferrochelatase with a range of porphyrin substrates. FEBS Lett. 1979;98:207–10. doi: 10.1016/0014-5793(79)80185-4. [DOI] [PubMed] [Google Scholar]
- 53.Dailey HA. Metal inhibition of ferrochelatase. Ann N Y Acad Sci. 1987;514:81–6. doi: 10.1111/j.1749-6632.1987.tb48763.x. [DOI] [PubMed] [Google Scholar]
- 54.Barkigia KM, Fajer J, Adler AD. Crystal and Molecular Structure of (5,10,15,20-Tetra-N-propylporphinato)lead(II): A "Roof" Porphyrin. Inorg Chem. 1980;19:2057–2061. [Google Scholar]
- 55.Plater JM, Aiken S, Gelbrich T, Hursthouse MB, Bourhill G. Structures of Pb(II) porphyrins. Polyhedron. 2001;20:3219–3224. [Google Scholar]
- 56.Fodje MN, Al-Karadaghi S. Occurrence, conformational features and amino acid propensities for the pi-helix. Protein Eng. 2002;15:353–8. doi: 10.1093/protein/15.5.353. [DOI] [PubMed] [Google Scholar]
- 57.Sigfridsson E, Ryde U. The importance of porphyrin distortions for the ferrochelatase reaction. J Biol Inorg Chem. 2003;8:273–82. doi: 10.1007/s00775-002-0413-8. [DOI] [PubMed] [Google Scholar]
- 58.Gora M, Grzybowska E, Rytka J, Labbe-Bois R. Probing the active-site residues in Saccharomyces cerevisiae ferrochelatase by directed mutagenesis. In vivo and in vitro analyses. J Biol Chem. 1996;271:11810–6. doi: 10.1074/jbc.271.20.11810. [DOI] [PubMed] [Google Scholar]
- 59.Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, Gwynn B, Lambert AJ, Wingert RA, Traver D, Trede NS, Barut BA, Zhou Y, Minet E, Donovan A, Brownlie A, Balzan R, Weiss MJ, Peters LL, Kaplan J, Zon LI, Paw BH. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440:96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]
- 60.Burden AE, Wu C, Dailey TA, Busch JL, Dhawan IK, Rose JP, Wang B, Dailey HA. Human ferrochelatase: crystallization, characterization of the [2Fe-2S] cluster and determination that the enzyme is a homodimer. Biochim Biophys Acta. 1999;1435:191–7. doi: 10.1016/s0167-4838(99)00196-x. [DOI] [PubMed] [Google Scholar]
- 61.Dailey HA, Sellers VM, Dailey TA. Mammalian ferrochelatase. Expression and characterization of normal and two human protoporphyric ferrochelatases. J Biol Chem. 1994;269:390–5. [PubMed] [Google Scholar]
- 62.Najahi-Missaoui W, Dailey HA. Production and characterization of erythropoietic protoporphyric heterodimeric ferrochelatases. Blood. 2005;106:1098–104. doi: 10.1182/blood-2004-12-4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rose JP, Chzras J, Jin Z, Fait J, Babson V, Wang BC. Annual Meeting of the American Crystallographic Association; Honolulu HI. 2006. [Google Scholar]
- 64.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 65.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica, Section D: Biological Crystallography. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 66.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Wall-eyed stereo view of the model and composite omit map for two chloride binding sites at the (a) N-terminal and (b) C-terminal end of helix α16 in the WT2 data set for human ferrochelatase. Atoms in the WT2 model are represented as balls and sticks with nitrogen, oxygen, carbon, and chloride atoms colored blue, red, tan, and yellow, respectively. The 2Fo-Fc composite omit map is contoured at 1σ (green cage) and was generated using the simulated annealing protocol with 7 % of the model being omitted per cycle.
Supplementary Figure 2. Wall-eyed stereo cartoon representation showing the entrance to the active site in the E343K model and the relative positions of the protoporphyrin IX molecules, detergent, and some basic residues in the upper lip of the active site “mouth”. For clarity the 2Fo-Fc composite omit map is contoured at 1σ around the substrate only (green cage) and was generated as described in the legend to Supplementary Figure 1.
Supplemental Figure 3. Wall-eyed stereoview for the model and 2Fo-Fc composite omit map (green cage) of the lead-porphyrin complex observed in the active site of the lead and protoporphyrin-treated human ferrochelatase crystals. Data for both monomer A (a) and monomer B (b) are shown with nitrogen, oxygen, carbon, and lead atoms represented as ball and sticks and colored blue, red, tan, and orange, respectively. The 2Fo-Fc composite omit map is contoured at 1σ and was generated as described in the legend to Figure 2.
Supplemental Movie 1. Animation showing the proposed movement of the upper lip upon binding of protoporphyrin IX and unwinding of π helix following heme production. Movie created using PDB ID files 2QD4, 2QD1, 2QD5, and 2QD3. The animation and rendering were created using the Yale Morph Server and PyMol.