

Abstract
A novel method was developed to detect molecular associations of dystrophin with actin in cryostat muscle tissue sections by combining resonance energy transfer technology with immunohistochemical techniques. This method takes advantage of the long phosphorescent lifetime of terbium chelates, a property that enables the accurate determination of energy transfer in biological tissues by lifetime measurements of sensitized emission. After a brief excitation pulse, terbium chelates emit for milliseconds after the intrinsically high autofluorescence of biological specimens has decayed to negligible levels. Rat skeletal muscle tissue sections were labeled with both anti-dystrophin monoclonal antibody conjugated to a terbium-based resonance energy transfer donor and anti-actin tetramethylrhodamine phalloidin as an acceptor. Resonance energy transfer between the two probes indicated that the distance separating the probes is within 10 nm (about the size of an IgG2b antibody molecule). The fraction of antibodies that participated in resonance energy transfer was estimated to be 80–90% because of the close agreement between the quenching of donor phosphorescence and the efficiency of resonance energy transfer revealed by lifetime measurements of sensitized emission by tetramethyl-rhodamine phalloidin. Sensitized emission was detectable only when both anti-dystrophin antibody and tetramethyl-rhodamine phalloidin were present. These results indicate that actin and dystrophin are closely associated within the cell. This method is potentially applicable to the investigation of many types of intracellular associations.
Dystrophin is the gene product of the Duchenne muscular dystrophy gene, the absence of which leads to progressive atrophy of muscle (1, 2). Accordingly, the precise cellular functions of dystrophin and its role in Duchenne muscular dystrophy are tightly linked and remain to be elucidated. Immunofluorescence, electron microscopy, and subcellular fractionation techniques localize dystrophin to the inner surface of the muscle plasmalemma (4–7). Dystrophin is a significant component of the plasmalemma, comprising nearly 5% of the membrane-bound proteins in skeletal muscle (6, 7). Current models of dystrophin function propose that it has a role in linking actin to the dystrophin–glycoprotein complex in the plasmalemma that is ultimately connected to the extracellular matrix (7–9). This model implies that dystrophin may aid in stabilizing muscle fibers by linking actin filament networks to external connective elements.
The association of actin with dystrophin comes from several lines of evidence. Initially, the homology of the N-terminal portion of the dystrophin gene sequence with the actin-binding proteins α-actinin and spectrin suggested that dystrophin might likewise be an actin-binding protein (1, 9–11). Perhaps the strongest supporting evidence for this hypothesis came from in vitro binding studies of actin with bacterially expressed fragments of dystrophin (12–15). These studies indicated that a specific sequence on the N terminus of dystrophin bound to actin. Interestingly, transgenic mice that express only dystrophin lacking the presumptive actin-binding site display symptoms of muscular dystrophy (16).
Numerous proteins bind actin in vitro; however, some of these proteins do not bind to actin in the cell. The best characterized example is that of DNAse I binding to actin, a process that was used to generate crystals for the first crystallographic determination of actin structure (17). Although DNAse I forms a very specific complex with actin, the distinctly different distributions of actin and DNAse I are not consistent with a physiologic role for this interaction. Additionally, other apparent actin-binding proteins might be segregated from actin in the cell. Therefore, it is important to determine whether actin-binding proteins such as dystrophin actually associate with actin in the muscle fiber. Few methods are currently available to probe such interactions with molecular resolution in cells.
To investigate interactions of proteins in cells at molecular resolution, new resonance energy transfer donors are conjugated to monoclonal antibodies that label specifically dystrophin in skeletal muscle cryostat sections. The resonance energy transfer from antibody-labeled dystrophin to tetramethylrhodamine phalloidin-labeled actin can only occur if the resonance energy transfer probes are within about 10 nm of each other. Therefore, this sensitized emission immuno-resonance energy transfer (SEIRET) method can detect associations between two macromolecules at the molecular level using a combination of spectroscopy, light microscopy, and immunohistochemistry. The data show strong resonance energy transfer between probes bound specifically to actin and dystrophin. These results indicate a close association between actin and dystrophin within muscle fibers and illustrate the use of a new technology for studying intracellular interactions at the molecular level.
MATERIALS AND METHODS
Reagents and Chemicals.
Mouse monoclonal anti-dystrophin IgG2b MANDYS8 and alkaline phosphatase-conjugated rabbit anti-mouse IgG, A-4312, antibodies were procured (Sigma). Luminescent chemicals included tetramethylrhodamine phalloidin (ɛA552 = 85,000 M−1⋅cm−1; Sigma), terbium chloride hexahydrate (Aldrich), and fluorescein-5-maleimide (ɛA490 = 83,000 M−1⋅cm−1; Molecular Probes). The following chemicals were obtained from Sigma: diethylenetriaminepentaacetic acid anhydride (DTPA anhydride); N,N-dimethylformamide; carbostryl-124; and 2-mercaptoethanol. Posterior back skeletal muscle actin was purified from New Zealand White rabbits by the method of Spudich and Watt (18). Deionized water passed through a Milli-Q filtration system (Millipore) was used in all experiments. CY3-ATP was prepared as described by Funatsu et al. (19) and purified by preparative TLC.
Antibody Conjugations.
The synthesis of terbium chelates followed the protocol of Selvin et al. (20) and was applied to conjugation of IgG by a modification of the method of Hnatowich et al. (21). One part of a 281 mM solution of carbostryl-124 in N,N-dimethylformamide was mixed with 7.5 parts of a 37.5 mM solution of DTPA anhydride to form an equimolar mixture. After 3 h at 22°C, the formation of a 1:1 linkage of carbostryl-124 with DTPA anhydride is greater than 50% (20). Because DTPA anhydride is a dianhydride, one anhydride can react with carbostryl-124 to form CS-DTPA while leaving the second anhydride free for coupling to amine groups on IgG (21). This reagent (1 μl) was added directly to the antibody (5 μl, containing 0.14 mg of protein) and incubated with mixing at 22°C for 5 min. Any anhydride that does not couple to the protein will hydrolyze rapidly in water, so the reaction is self-terminating. Free reagent was removed by either gel filtration chromatography or dialysis; however, control experiments with unpurified antibody indicated that unreacted reagent did not affect background in muscle sections. After adding a stoichiometric amount of terbium chloride hexahydrate to the chelate (1 μl of a 33 mM solution in water), the labeled antibody was highly luminescent with an intensity nearly 10,000 times higher than free terbium. The decay lifetime of the chelate (approximately 1.7 ms) was also much higher than that of free terbium chloride (approximately 0.4 ms), although the shapes of the emission spectra were identical. Analysis of the elution profiles of gel filtration chromatography on a G-100 Sephadex column indicated that approximately 5% of the phosphorescent reagent was incorporated into the protein for either anti-dystrophin IgG or control IgG.
The fluorescein-conjugated anti-dystrophin antibody was prepared by addition of 1 μl of fluorescein-5-maleimide (2 mM in N,N-dimethylformamide) to 5 μl of antibody (0.14 mg of protein) and incubation for 30 min at 4°C. The chemical modification was terminated by the addition of a 10-fold molar excess of 2-mercaptoethanol over the fluorescein-5-maleimide. This reaction provided efficient labeling of the antibody by the fluorescein-5-maleimide, resulting in insignificant amounts of unreacted reagent as judged by gel filtration chromatography on a G-100 sephadex column.
Tissue Preparation and Antibody Labeling.
Tissues were prepared by standard protocols (3, 22). The posterior back muscle of a laboratory rat was excised immediately after the rat was killed. The muscle tissue was either first fixed in 4% paraformaldehyde (as indicated) or directly frozen at −35°C in 2-methylbutane, and mounted in a microtome-cryostat (Frigocut 2800, Reichert-Jung). Sequential, 20-μm-thick sections were thaw-mounted on gelatin-coated microscope slides and stored at −70°C until use. After thawing at room temperature for 30 min, the tissue section was blocked with 3% BSA in 150 mM KCl/10 mM imidazole (pH 7.0) for 1 h at room temperature. The tissue section was immersed in 0.1 ml tetramethylrhodamine phalloidin (1 μM) and/or the antibody conjugate was diluted 1:400 in 3% BSA/150 mM KCl/10 mM imidazole (pH 7.0) for 16 h at 4°C, and then washed to remove unbound reagent. Controls labeled with CY3-ATP were incubated initially with anti-dystrophin antibody conjugate as described above and then with 1 μM CY3-ATP for 5 min. The sample was then washed briefly and examined in the spectrometer and the fluorescence microscope within 30 min.
Fluorescence Microscopy.
Microsections labeled with fluorescein-conjugated anti-dystrophin antibody were immediately coverslipped and observed by epifluorescence microscopy. Fluorescein was excited at a 490-nm wavelength from a 150-W xenon arc lamp light source passed through a monochromator set at 8 nm bandpass and coupled to the epifluorescence port of a modified Zeiss IM-35 microscope through a randomized quartz fiber optic bundle. Fluorescence was imaged with a gated GEN III intensified charge-coupled device camera (Videoscope, Sterling, VA) and grabbed to nih image 1.60 (version by Wayne Rasband, National Institutes of Health, Bethesda) through a Scion frame grabber (Frederick, MD) on a Power Macintosh computer (Apple Computer, Cupertino, CA). Images were printed with a dye-sublimation printer (Supermatch Proofpositive, SuperMac, Sunnyvale, CA).
Spectroscopy.
To minimize light scattering, the slides were mounted at 30° to the excitation beam path and 60° to the emission path in a SLM-Aminco Bowman II luminescence spectrometer (Spectronic Instruments, Rochester, NY) with dual continuous-wavelength xenon arc and flashlamp light sources. Delayed luminescence and lifetime measurements were collected after 200 μs following the excitation pulse of the flashlamp at 342 nm. Delayed emission spectra were accumulated over a gate period of 4 ms. Luminescence decay curves were acquired at a resolution of 100–200 μs and averaged for 100 repetitions. The UV-visible absorbance spectra were measured at 1-nm resolution on a diode array spectrometer (Hewlett–Packard). All emission spectra were corrected in software for the wavelength-dependent instrument response.
RESULTS
Resonance Energy Transfer Probes.
The donor of the resonance energy transfer pair is terbium, protected largely from quenching by water in a chelate complex. Although free terbium is weakly luminescent due primarily to its very low absorbance, the reagent used in this study employs a chromophore, carbostryl-124, which participates with DTPA in the chelation of lanthanides and transfers energy to them, thereby increasing greatly the brightness of terbium phosphorescence (23, 24). The chemical reaction of amino groups with DTPA allows the attachment of these chelates to lysine residues. The emission spectrum of terbium is unique in that it has four sharp peaks in the visible region at 489, 547, 589, and 622 nm, with no emissions between the peaks (Fig. 1). These properties are particularly useful for resonance energy transfer coupling to a variety of fluorophores that absorb and emit in the visible spectrum.
Figure 1.
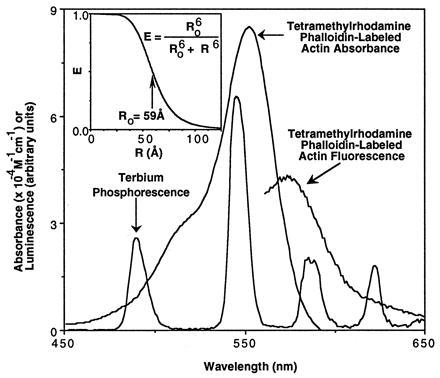
Spectral characteristics of resonance energy transfer probes. The emission spectrum of terbium phosphorescence of the unconjugated terbium chelate in distilled water was collected at a 2-nm bandwidth with the excitation monochromator set at 342 nm. Excitation at 540 nm of a stained rat skeletal muscle cryostat section yielded the tetramethylrhodamine phalloidin-labeled actin fluorescence emission curve. The absorption spectrum of tetramethylrhodamine phalloidin-labeled actin (purified rabbit skeletal muscle actin) was measured on a diode array spectrometer with a bandwidth of 1 nm in 2 mM MgCl2/0.2 mM CaCl2/0.2 mM ATP/0.5 mM 2-mercaptoethanol/2 mM Tris (pH 7.0). Unlabeled actin did not absorb in the part of the spectrum shown. (Inset) Dependence of efficiency of energy transfer (E) on separation distance (R) between a donor and acceptor probe pair. Using the inset equation, the theoretical relationship between E and R was calculated for critical transfer distance (Ro) of 59 Å. The critical transfer distance was calculated from the overlap between the donor’s emission spectrum and the acceptor’s absorbance spectrum. (See text for calculations.)
For resonance energy transfer to occur, the absorbance spectrum of an acceptor fluorophore must overlap with the emission spectrum of a donor probe. The absorbance spectrum of tetramethylrhodamine phalloidin-labeled actin overlaps very well with the emission spectrum of terbium (Fig. 1). From these data, an overlap integral, J, is calculated by the equation J = ∫fDɛAλ4dλ/∫fDdλ, where fD is the fluorescence emission of the donor and ɛA is the extinction coefficient of the acceptor at wavelength λ. The value of J, 3.6 × 1015 nm4⋅M−1, agrees well with that reported for free tetramethylrhodamine; therefore, the critical transfer distance (Ro) is also a similar value of 59 Å in water (23). This large critical transfer distance means that significant amounts of resonance energy transfer will occur between donor and acceptor probes out to a separation distance of just over 100 Å (Fig. 1 Inset). This large critical transfer distance enables the detection of resonance energy transfer even between two large macromolecules such as antibodies.
These resonance energy transfer probes permit the discrete detection of sensitized emission of the acceptor, which fluoresces brightly between the sharp terbium emission peaks where there is no background (Figs. 1 and 2). The measurement of sensitized emission eliminates background fluorescence, such as cellular autofluorescence, which decay on the nanosecond time scale by introducing a delay of 200 μs between the excitation flash and the acquisition of data by the photomultiplier tube (23). The sensitized emission from the tetramethylrhodamine acceptor will decay at the same rate as the quenched terbium donor because tetramethylrhodamine fluorescence decays almost instantaneously relative to terbium phosphorescence. Therefore, tetramethylrhodamine fluorescence after a delay of 200 μs can occur only if terbium is providing the energy by resonance energy transfer. For example, tetramethylrhodamine phalloidin labeling of muscle sections in the absence of terbium produces no significant signal after the 200-μs delay (Fig. 2).
Figure 2.
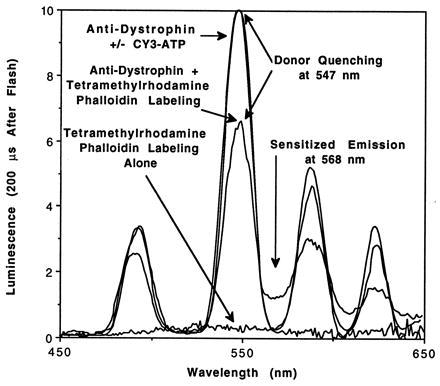
Delayed emission spectra of labeled skeletal muscle fiber tissue sections. Rat skeletal muscle fixed tissue sections were labeled as indicated and mounted in a luminescence spectrometer. Flashlamp excitation at 342 nm was followed by a 200-μs delay, and then data were collected by the photomultiplier tube for 5 ms at an emission bandwidth of 8 nm. Resonance energy transfer was observed during labeling of actin with an acceptor, tetramethylrhodamine phalloidin, but not during labeling of nucleotide-binding proteins with the acceptor, CY3-ATP. Tetramethylrhodamine phalloidin labeling alone did not generate measurable emission peaks. Clear terbium quenching at 547 nm concurrent with sensitized emission of tetramethylrhodamine at 568 nm verify that resonance energy transfer is occurring. Data were normalized as described in Appendix.
SEIRET in Muscle Tissue Sections.
When paraformaldehyde-fixed muscle sections were labeled with both terbium chelate-conjugated anti-dystrophin antibodies and tetramethylrhodamine phalloidin, both donor quenching and sensitized emission showed clearly resonance energy transfer between the probes (Fig. 2). Direct inspection of the delayed emission at 547 nm in the presence versus the absence of tetramethylrhodamine phalloidin indicates a decrease in intensity by approximately 40% in fixed tissue and 60% in unfixed tissue (Table 1). This large decrease indicates quenching of most of the terbium chelate-labeled antibodies in the muscle section, although the absolute amount of quenching may be affected by the labeling efficiency and the difference in decay lifetimes between quenched and unquenched donors.
Table 1.
SEIRET on paraformaldehyde-fixed and unfixed tissue sections
| Muscle section | τD547, ms | τDA547, ms | τDA568, ms | E | EavgDQ | EavgL | FDQ | FL |
|---|---|---|---|---|---|---|---|---|
| Fixed | 2.0 ± 0.3 | 1.2 ± 0.1 | 0.7 ± 0.1 | 0.65 | 0.4 | 0.4 | 0.6 | 0.6 |
| Unfixed | 1.8 ± 0.1 | 0.8 ± 0.1 | 0.59 ± 0.04 | 0.67 | 0.6 | 0.56 | 0.9 | 0.83 |
τD547, donor only lifetime at 547 mm; τDA547, donor plus acceptor lifetime at 547 nm; τDA568, donor plus acceptor lifetime at 568 nm; E, efficiency of energy transfer; EavgDQ, average energy transfer by donor quenching; EavgL, average energy transfer by lifetime method; FDQ, fraction of coupled donors by donor quenching; FL, fraction of coupled donors by lifetime method.
The proximity between a pair of resonance energy transfer probes is related to the decrease in decay lifetimes between an unquenched and quenched donor probe. The decay of sensitized emission after an excitation flash reports the lifetime of the quenched donor, and the lifetime of the donor in the absence of acceptor probe yields the lifetime of the unquenched donor. These measurements on labeled muscle tissue sections are illustrated in Fig. 3. The lifetime of the muscle section labeled with anti-dystrophin antibody alone was 1.80 ± 0.07 ms (Fig. 3), which is only slightly different from that of the chelate before conjugation to antibody (1.68 ± 0.02 ms; data not shown). The lifetime of the muscle section, which was labeled with both donor and acceptor probes, was 0.59 ± 0.03 ms as fit by a single exponential decay (Fig. 3). These numbers correspond to an efficiency of resonance energy transfer (E) of 1 − (0.59 ms/1.8 ms) = 0.67. For a single donor and acceptor pair, this efficiency of resonance energy transfer would correspond to a separation distance of 52 Å according to the Inset of Fig. 1; however, the physical size an IgG molecule is larger than this distance, so the resolution of colocalization of dystrophin with actin is limited by the molecular size of the antibody (about 100 Å). Similar results were obtained for rabbit skeletal muscle fibers that were dual-labeled after mechanical disruption by a blender and suspended in buffer (2 mM MgCl2/150 mM KCl/10 mM imidazole, pH 7.0) for measurement of lifetimes in a quartz cuvette. In this case, the efficiency of resonance energy transfer between the antibody-labeled dystrophin and the tetramethylrhodamine phalloidin-labeled actin was 0.66 (data not shown).
Figure 3.
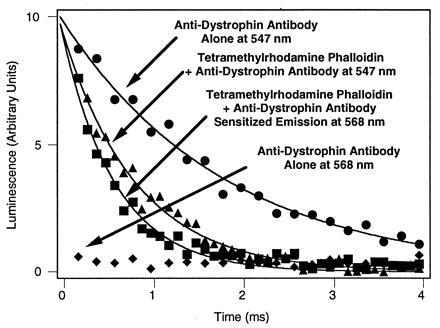
Determination of efficiency of resonance energy transfer by luminescence decay on unfixed muscle sections. Unquenched donor phosphorescence was detected at 547 nm in the presence or absence of acceptor. No donor phosphorescence was detectable at 568 nm, permitting measurement of sensitized emission at 568 nm. Measurements made on large cross-sections of muscle mounted in the luminescence spectrometer. Decay curves were measured in a luminescence spectrometer after flashlamp excitation at 342 nm. Lifetimes were estimated by single exponential curve fitting that yielded 1.80 ± 0.07 ms (r2 = 0.984) for unquenched phosphorescence and 0.59 ± 0.03 ms (r2 = 0.964), indicating an efficiency of energy transfer, E, of 0.67.
Lifetime measurements were also made on 90 μm diameter regions of the muscle tissue sections (data not shown). To attain the sensitivity necessary for these measurements, a pulsed nitrogen laser (VSL-337ND-S, Laser Science, Newton, MA) was used a light source for epiillumination in a microscope and focused with a Nikon UV-F ×100 objective for high UV light transmittance. Emissions were routed back to the luminescence spectrometer by a light guide and analyzed spectrally and temporally. Individual fields on dual-labeled samples yielded similar values for the efficiency of energy transfer (0.7 ± 0.1) based on lifetime measurements at 568 nm. However, the lifetime measurements at 547 nm varied from field to field (0.7 ms to 1.4 ms), indicating that the fraction of donors transferring energy was not identical in all parts of the tissue section. This microscopic heterogeneity might suggest that dystrophin in different cells or parts of cells may differ in their associations with actin.
Controls for Specificity of Anti-Dystrophin Labeling.
The anti-dystrophin monoclonal antibody was raised against peptides synthesized based on the sequence of dystrophin gene and has been well characterized (22). As a positive control, the antibody was directly conjugated with fluorescein and used to label muscle sections. Consistent with the specific reaction of the antibody with dystrophin, the sarcolemma was preferentially labeled as judged by the bright fluorescence along the perimeter of sectioned fibers visualized with immunofluorescence microscopy (Fig. 4).
Figure 4.

Labeling of muscle tissue sections. Frozen tissue sections (20 μm) of rat skeletal muscle on a gelatin-coated microscope slide were labeled and examined under an epifluorescence microscope. (A and B) Sections were labeled with fluorescein-conjugated anti-dystrophin monoclonal antibody and observed through a Zeiss ×63 Plan-Apo, 1.4 n.a. objective. Immunofluorescence labeling of the sarcolemma was well defined. Immunofluorescence (A) images correlated with the cell morphology observed by bright field (B) microscopy of the same field. This correlation of cell morphology with immunofluorescence is consistent with specific labeling of dystrophin on the sarcolemma by the anti-dystrophin antibody. (C) Muscle sections were labeled briefly with CY3-ATP and illuminated by a green helium-neon laser (Uniphase, Manteca, CA). Images illustrate labeling at intracellular sites and strong signals at the cell periphery which might erroneously be interpreted as colocalization with dystrophin by immunofluorescence. In contrast, Fig. 2 shows no resonance energy transfer between CY3-ATP and terbium chelate-conjugated anti-dystrophin antibody in dual-labeled sections. (D) Muscle sections were dual-labeled with chelate-conjugated anti-dystrophin monoclonal antibody and excited by a pulsed nitrogen laser and viewed through an emission filter which allows transmission from 565 to 585 nm. Cellular autofluorescence dominates the image of three adjacent cells. (E) This field is identical to that in D but obtained after applying a delay to the charge-coupled device. This delayed luminescence image illustrates resonance energy transfer at the periphery of the adjacent cells. The intensity of this delayed emission is considerably less than that in D.
Additional controls included comparing different muscle sections incubated in parallel with terbium chelate-conjugated anti-dystrophin IgG or with IgG that was not raised against dystrophin. After washing away unbound IgG, only the anti-dystrophin IgG-labeled muscle tissue sections had measurable terbium phosphorescence in the luminescence spectrometer (data not shown). Similarly low background was observed with tetramethylrhodamine phalloidin-labeled tissue muscle sections in the absence of anti-dystrophin labeling, which exhibited both strong fluorescence in the luminescence spectrometer (Fig. 1) and I band labeling under the fluorescence microscope (data not shown), but no significant phosphorescence in the spectral range of interest (Fig. 2). Therefore, background interference with the spectroscopic measurements can be ruled out.
To illustrate the higher resolution of SEIRET over conventional immunofluorescence colocalization, muscle sections were dual-labeled with CY3-ATP and chelate-labeled IgG. Immunofluorescence images of the CY3-ATP-labeled sections showed strong staining at the cell periphery in addition to intracellular sites. The peripheral staining might suggest that part of the CY3-ATP colocalized with dystrophin (Fig. 4 A and C); however, no resonance energy transfer was detectable (Fig. 2) even though CY3-ATP is a more efficient acceptor for terbium phosphorescence than tetramethylrhodamine phalloidin. (The calculated critical transfer distances are 65 Å and 59 Å for CY3-ATP and tetramethylrhodamine phalloidin, respectively.)
Preliminary evidence from delayed luminescence imaging is also consistent with the spectroscopic finding reported. Muscle tissue sections labeled with both chelate-conjugated anti-dystrophin antibody and tetramethylrhodamine phalloidin were observed with a gated intensified charge-coupled device after illumination by a nitrogen laser light pulse. Cell autofluorescence dominated ungated images (Fig. 4D), but when a delay after excitation was imposed before imaging, the sensitized emission signal at 565–585 nm became clear (Fig. 4E). This delayed luminescence image shows that most of the energy transfer occurs at the periphery of the muscle cells, particularly where adjacent cells overlap in the tissue section.
DISCUSSION
Intracellular Resonance Energy Transfer.
A technique that can provide information on the proximity between two sites with molecular resolution is resonance energy transfer. In resonance energy transfer, an excited luminescent donor couples through space to an acceptor probe which, if also luminescent, may emit that energy even when the acceptor has not been excited directly by light. This sensitized emission resonance energy transfer occurs only if the donor and acceptor are very close to each other and the absorbance spectrum of the acceptor overlaps with the emission spectrum of the acceptor. This physical property is useful for measuring intramolecular distances (see Fig. 1 Inset) and may be modified to study intermolecular interactions on purified proteins as well (23).
This technology holds great promise for measuring intracellular interactions between proteins. To apply this method within the cell, several requirements must be fulfilled. First, a specific method of labeling the desired proteins with acceptor or donor probes must be developed. Second, the probes must be capable of transferring energy over distances large enough that intramolecular associations can be detected. In addition, the transfer of energy between the probes must be readily measurable over the intrinsic autofluorescence of cells. Pioneering studies by Selvin and Hearst (24) have produced resonance energy transfer probes that can measure separation distances over 100 Å even in the presence of background from fluorescence and light scattering (25). In addition, antibodies can be used to label proteins for resonance energy transfer (26). The present study combines these new probes with immunohistochemistry to produce a method that can detect at molecular resolution the association between any two molecules in the cell for which antibodies or other specific labels are available. This method is illustrated schematically in Fig. 5.
Figure 5.
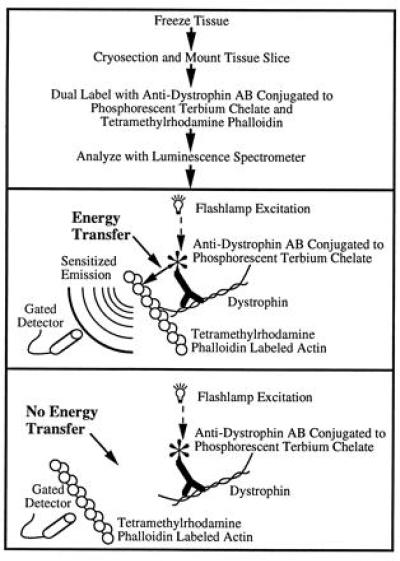
Schematic illustration of sensitized emission resonance energy transfer between antibody-labeled dystrophin and tetramethylrhodamine phalloidin-labeled actin. The steps in the protocol (Top) and the two possible results of the experiment and their interpretations (Middle and Bottom) are shown. Sensitized emission of the tetramethylrhodamine phalloidin-labeled actin occurs only if the labeled dystrophin is within about 10 nm from the labeled actin. Protein sizes are not drawn to scale.
Association of Dystrophin with Actin in Skeletal Muscle.
This new methodology, SEIRET, is used here to determine whether dystrophin interacts with actin in rat skeletal muscle fibers. The association between actin and dystrophin was examined by dual-labeling tissue sections of frozen muscle tissue with terbium chelate-conjugated anti-dystrophin monoclonal antibody and actin-specific tetramethylrhodamine phalloidin. The resonance energy transfer between donor-labeled dystrophin and acceptor-labeled actin indicate a close association between these proteins in situ. The resolution of this method is limited by the size of the IgG molecule used as a probe (less than 100 Å, depending on the modification site). This resolution is therefore 10–100 times higher than achieved by traditional immunofluorescence colocalization studies. Together with biochemical demonstrations that purified dystrophin binds actin in solution (12–16), such a close association virtually guarantees that direct binding between actin and dystrophin occurs in the muscle fiber.
The primary limitation of SEIRET is its dependence on the specificity of actin and dystrophin labeling. However, the specificity of labeling does not appear to be a problem in this study, based on the correlation between the efficiency of energy transfer (0.67) calculated from sensitized emission decay curves in Fig. 3 with the average efficiency of energy transfer (0.6 for unfixed tissue), determined independently by donor quenching or lifetime measurements (see Table 1 and Appendix). This close correspondence indicates that only 10–20% of the antibodies (resonance energy transfer donors) did not colocalize with actin because donor quenching depends on both the fraction of donors that are quenched and the efficiency of resonance energy transfer, whereas the sensitized emission decay lifetimes depend only on the efficiency of resonance energy transfer (25). Based on the strong specificity of the monoclonal antibody for dystrophin, it is highly improbable that 80–90% of the antibodies cross-reacted with other proteins that are bound to actin. This internal control in the data provides evidence that any small amounts of cross-reactivities of the antibody will not cloud the interpretations of the results. Measurements on paraformaldehyde-fixed tissue sections also indicate a high fraction (60%) of the anti-dystrophin antibodies colocalized with actin (Table 1), even though fixed sections did not stain with as strong an intensity as unfixed muscle. The correspondence between results on unfixed and paraformaldehyde fixed muscle indicate that the observed association of dystrophin with actin is not related to the tissue preparation.
The data presented here support models in which dystrophin binds directly to actin in skeletal muscle (refs. 7, 27, and 28 and references therein). This finding is consistent with proposals that dystrophin aids in the stabilization of muscle fibers by attaching actin to the dystrophin–glycoprotein complex within the sarcolemma which is then linked to the extracellular matrix. The absence of dystrophin in Duchenne muscular dystrophy might result in greater stress on the sarcolemma or other subcellular structures that can be damaged during fiber shortening. This interpretation might explain why muscular atrophy occurs in patients who lack dystrophin.
Acknowledgments
I am indebted to Dr. Jannon Fuchs for enthusiastically providing comments and encouragement on this manuscript. In addition, her assistance and expertise in preparing cryostat tissue sections were instrumental to this work.
ABBREVIATIONS
- DTPA
diethylenetriaminepentaacetic acid
- SEIRET
sensitized emission immuno-resonance energy transfer
Appendix
The equation used to normalize emission spectra from samples labeled with donor only to those labeled with both donor and acceptor was
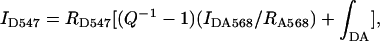 |
in which ID547 is the intensity at 547 nm of the donor only emission, RD547 is the ratio of ID547 to the integral of the donor only emission spectrum and has a value of 0.021 nm−1, Q is the quantum yield of tetramethylrhodamine for which a value of 0.28 was used (29), IDA568 is the intensity at 568 nm of the dual-labeled donor and acceptor sample, RA568 is the ratio of the intensity at 568 nm of the acceptor fluorescence to the integral of its emission spectrum which has a measured value of 0.015 nm−1, and ∫DA is the integral of the emission spectrum of the dual-labeled donor and acceptor sample. This equation is easily derived from the relation that the emission of donor only sample is the sum of the emission of the dual-labeled donor and acceptor sample plus a correction for the energy loss due to the quantum yield of the acceptor. It is similar in principle to other methods based on sensitized emission (24, 30). After normalization, the average efficiency of energy transfer, Eavg, may be calculated by the donor quenching at 547 nm.
An independent method for determining the average efficiency of energy transfer, Eavg, is by luminescence decay measurements using the equation, Eavg = 1 − (τDA547/τD547). In this equation, average lifetimes of the dual-labeled donor and acceptor (τDA547) and the donor only (τD547) samples are measured at 547 nm. The empirical agreement between this lifetime method and the donor quenching method above is shown in Table 1.
References
- 1.Koenig M, Monaco A P, Kunkel L M. Cell. 1988;53:219–228. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- 2.Worton R. Science. 1995;270:755–756. doi: 10.1126/science.270.5237.755. [DOI] [PubMed] [Google Scholar]
- 3.Byers T J, Kunkel L M, Watkins S C. J Cell Biol. 1991;115:411–421. doi: 10.1083/jcb.115.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dickson G, Azad A, Morris G E, Simon H, Noursadeghi M, Walsh F S. J Cell Sci. 1992;103:1223–1233. doi: 10.1242/jcs.103.4.1223. [DOI] [PubMed] [Google Scholar]
- 5.Wakayama Y, Shibuya S, Jim T, Takeda A, Oniki H. Acta Neuropathol. 1993;86:567–577. doi: 10.1007/BF00294294. [DOI] [PubMed] [Google Scholar]
- 6.Love D R, Byth B C, Tinsley J M, Blake D J, Davies K E. Neuromuscular Disorders. 1993;3:5–21. doi: 10.1016/0960-8966(93)90037-k. [DOI] [PubMed] [Google Scholar]
- 7.Ervasti J M, Campbell K P. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown S C, Lucy J A. BioEssays. 1993;15:413–419. doi: 10.1002/bies.950150608. [DOI] [PubMed] [Google Scholar]
- 9.Hammonds R G H. Cell. 1987;51:1. doi: 10.1016/0092-8674(87)90002-x. [DOI] [PubMed] [Google Scholar]
- 10.Byers T J, Husain-Chishti A, Dubreuil R R, Branton D, Goldstein L S B. J Cell Biol. 1989;111:1849–1858. doi: 10.1083/jcb.109.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ibraghimov-Beskrovnaya O, Ervasti J M, Leveille C J, Slaughter C A, Sernett S W, Campbell K P. Nature (London) 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 12.Levine B A, Moir A J G, Patchell V B, Perry S V. FEBS Lett. 1990;263:159–162. doi: 10.1016/0014-5793(90)80728-2. [DOI] [PubMed] [Google Scholar]
- 13.Fabbrizio E, Bonet-Kerrache A, Leger J J, Mornet D. Biochemistry. 1993;32:10457–10463. doi: 10.1021/bi00090a023. [DOI] [PubMed] [Google Scholar]
- 14.Corrado K, Mills P L, Chamberlain J S. FEBS Lett. 1994;344:255–260. doi: 10.1016/0014-5793(94)00397-1. [DOI] [PubMed] [Google Scholar]
- 15.Senter L, Ceoldo S, Meznaric Petrusa M, Salviati G. Biochem Biophys Res Commun. 1995;206:57–63. doi: 10.1006/bbrc.1995.1009. [DOI] [PubMed] [Google Scholar]
- 16.Corrado K, Rafael J A, Mills P L, Cole N M, Faulkner J A, Wang K, Chamberlain J S. J Cell Biol. 1996;134:873–884. doi: 10.1083/jcb.134.4.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kabsch W, Mannherz H G, Suck D, Pai E F, Holmes K C. Nature (London) 1990;347:37–44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- 18.Spudich J A, Watt S. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 19.Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida T. Nature (London) 1995;374:555–559. doi: 10.1038/374555a0. [DOI] [PubMed] [Google Scholar]
- 20.Selvin P R, Jancarik J, Li M, Hung L. Inorg Chem. 1996;35:700–705. [Google Scholar]
- 21.Hnatowich D J, Layne W W, Childs R L, Lanteigne D, Davis M A, Griffin T W, Doherty P W. Science. 1983;220:613–615. doi: 10.1126/science.6836304. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen T M, Ginjaar I B, van Ommen G, Morris G E. Biochem J. 1992;288:663–668. doi: 10.1042/bj2880663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dos Remedios C G, Moens P D J. J Struct Biol. 1995;115:175–185. doi: 10.1006/jsbi.1995.1042. [DOI] [PubMed] [Google Scholar]
- 24.Selvin P R, Hearst J E. Proc Natl Acad Sci USA. 1994;91:10024–10028. doi: 10.1073/pnas.91.21.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selvin P R. Methods Enzymol. 1995;246:300–334. doi: 10.1016/0076-6879(95)46015-2. [DOI] [PubMed] [Google Scholar]
- 26.Mathis G. Clin Chem. 1995;41:1391–1397. [PubMed] [Google Scholar]
- 27.Matsumura K, Campbell K P. Muscle Nerve. 1994;17:2–15. doi: 10.1002/mus.880170103. [DOI] [PubMed] [Google Scholar]
- 28.Ohlendiek K. Eur J Cell Biol. 1996;69:1–10. [PubMed] [Google Scholar]
- 29.Haughland R P. In: Excited States of Biopolymers. Steiner R F, editor. New York: Plenum; 1983. pp. 29–58. [Google Scholar]
- 30.Clegg R M. Methods Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]