

Abstract
Apoptotic cell death in wing imaginal discs takes place in single cells or small clusters of neighboring cells. These cells are distributed throughout the anlage at early stages and in recognizable territories at late larval and pupal stages. Apoptotic cells remain in the epithelium 2–4 h, prior to being engulfed in place by hemolymph cells. Experimentally induced apoptosis in single cells or territories is accompanied by nonautonomous death of adjacent cells and of cells further away in adjacent territories. These effects are followed by changes in cell proliferation in both territories. Apogenetic mosaics in mutant discs show cell death throughout the anlage. Apoptosis provides a mechanism, in addition to cell proliferation control, for matching territories with different positional values or different genetic specifications.
Keywords: apoptosis, regeneration, cell proliferation, morphogenesis
Morphogenesis in multicellular organisms is associated with the control of cell proliferation. However, apoptosis has also been proposed to play an important part in the process (1). Extra cells in a complete pattern or misplaced cells in different tissues are eliminated through cell death (2). Thus, in normal Drosophila development, extra cells in the eye imaginal disc undergo apoptosis (3). Correct matching of different cell types may also operate through cell death (2). Thus, in the developing vertebrate nervous system, neurons and glia cells are overproduced, their final functional numbers being attained by apoptosis (2). Lethality results from reception of apoptotic signals or deprivation of survival signals from the neighboring cells. In fact, addition of growth factors to those cells rescues them from apoptosis (2). Upstream activators of the cell death machinery have been identified in Drosophila (4–6), and the morphogenetic role of apoptosis in some differentiating tissues (i.e., in ommatidia patterning and in the central nervous system) (3, 7) is known. However, the role of apoptosis in morphogenesis associated to cell proliferation is poorly explored. Many morphogenetic mutants (e.g., vestigial, nubbin, and many others) (8, 9) show increased apoptosis in the imaginal discs but its causal mechanism, which is relevant to understanding the function of the normal gene, is unclear. It is a requisite for that understanding to know the cellular and developmental parameters of cell death in the wild-type imaginal disc. In this report we show that apoptosis takes place through local cell-cell interactions. It provides a mechanism, in addition to cell proliferation, for generating cell territories with continuous positional values, relevant to size and shape developmental control.
MATERIALS AND METHODS
Drosophila Strains.
The following stocks are used: Vallecas wild-type D. melanogaster; hsp70-head involution defective (HS-hid) (5); the GAL4 expressing lines 734, 766, MS209, Scalloped-GAL4 (Sd-GAL4), engrailed-GAL4 (en-GAL4), apterous-GAL4 (ap-GAL4) (10, 11); ContrabithoraxM1 (CbxM1) (12), UAS-lacZ; UAS-cold-sensitive ricin A (UAS-RAcs) (13), and hsp70-flp; PUFWTRA; UAS-lacZ (14) (UAS, upstream activation sequence). Other stocks are described in ref. 15. Larvae were staged in hours after egg laying, and pupae in hours after puparium formation (APF).
Imaginal Disc Staining.
In situ hybridization with a digoxigenin labeled string (stg) cDNA probe, 5-bromo-4-chloro-3-indolyl β-d-galactoside staining, β-galactosidase detection, in vitro 5-bromo-2′-deoxyuridine (BrdUrd) incorporation, and acridine orange and Hoechst 33258 staining were performed as described (16). Terminal deoxynucleotidyltransferase-mediated UTP end labeling (TUNEL) was adapted from ref. 4.
Induction of Cell Death.
Apoptosis in wing discs was induced by incubation of HS-hid larvae (5) during 30 min at 37°C. Irradiation of larvae with x-rays (either at a dose of 4,000 rads in wild-type larvae or 1,000 rads in T(2, 3)SM6a-TM6b/+ larvae) induces apoptosis by DNA damage and by generation of aneuploids (17, 18). Gene targeted cell death was induced using the GAL4/UAS-ricin toxin A chain gene (RTA) system (14). hsp70-flp; PUFWTRA; UAS-lacZ flies were crossed to different GAL4 lines and F1 larvae were incubated 1 h at 37°C. In flies carrying the PUFWTRA construct the RTA gene is separated from the UAS control element by a fragment of DNA flanked by two FLP recombination target sites in the same orientation. Thus, RTA activation by the GAL4 protein takes place only after a recombination event catalyzed by yeast FLP recombinase. We have also made use of RAcs (13). UAS-lacZ; UAS-RAcs flies were crossed to different GAL4 lines; F1 larvae were grown at 25°C and shifted to 31°C for different periods of time to allow functioning of the toxin in the cells where the GAL4 driver is being expressed.
Visualization of Cell Death.
Three techniques were used to visualize dead cells in the imaginal discs: (i) staining with the vital dye acridine orange that specifically identifies apoptotic cells (17); (ii) the TUNEL technique, which labels fragmented DNA in dying cells (19); and (iii) Hoechst 33258 staining to visualize condensed DNA of dying cells. After induction of apoptosis (by overexpression of hid or x-rays irradiation) or of necrosis (by mechanically breaking the discs) there is a great increase in the number of TUNEL-labeled cells in imaginal discs in relation to control untreated discs, thus demonstrating the labeling of apoptotic and necrotic nuclei by this technique. All the results presented in this report have been obtained using TUNEL in fixed imaginal discs, which allows the most accurate quantitative and topological analysis of cell death. Similar results have been obtained by staining with acridine orange.
Clonal Analysis.
f36a/+; en-GAL4/+; UAS-RAcs/+ larvae and control f36a/+; Bc Gla/+; UAS-RAcs/+ larvae were maintained 24 h at 31°C (from 36–60 h after egg laying) before induction of mitotic recombination by x-ray irradiation as described in ref. 20). Forty to 50 clones were examined in each experiment.
RESULTS
Cell Death Patterns in the Imaginal Wing Disc.
To estimate the possible contribution of cell death to wing and notum morphogenesis we have analyzed the pattern and frequency of dead cells in the epithelium of the imaginal wing disc during the second and third larval instars and pupal development. Apoptotic nuclei, visualized either by their DNA condensation or fragmentation (as revealed by Hoechst staining or TUNEL, respectively) are located in the basal end of the wing disc epithelium (data not shown). Irrespective of their size (100–1,500 cells), early wing imaginal discs show a constant proportion (average 1.4%) of TUNEL-labeled cells (Fig. 1B) (except at the larval molts where there is a great increase in dying cells; Fig. 1A and refs. 8 and 16). During early larval stages dead cells appear throughout the entire disc without any noticeable pattern and either as single cells or in compact clusters of cells (Fig. 1B). The average size of these clusters is 3.4 cells (n = 120 clusters of 2–10 cells). The density of dead cells within the clusters is very similar to that of the rest of epithelial cells, indicating that TUNEL identifies clusters of neighboring dead cells. DNA fragmentation takes place at the earliest stages of apoptosis (19), hence TUNEL identifies clusters of synchronized neighboring cells entering apoptosis. Indeed, it has been shown in the Drosophila embryo that reaper, a gene that triggers apoptosis, is expressed in small clusters of cells that are going to undergo apoptosis 2–3 h later (21).
Figure 1.
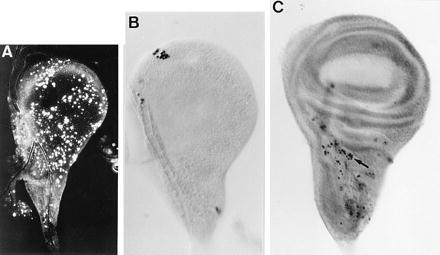
Patterns of apoptotic cells in wild-type wing discs. Apoptotic cells were identified by acridine orange staining (A) and TUNEL method (B and C). (A) High frequency of apoptotic cells in a wing disc from an early third instar larva immediately after the second molt. (B) Imaginal wing disc from early third instar larva. Note the cluster of apoptotic cells. (C) Distribution of apoptotic cells in late third instar wing disc. Arrow points to a cluster of apoptotic cells at the wing/notum border region. All discs are reproduced at the same magnification. In all figures anterior is to the left and ventral is up.
In late third instar wing discs and pupal wings, the size of the clusters of dead cells is similar to that found in early discs and the percentage of dead cells remains very low (Fig. 1C). However, dead cells appear nonrandomly distributed. In third instar wing discs they are preferentially located at the wing/notum border (Fig. 1C). In early pupal wings (0–12 h APF) dead cells are located at the wing hinge and, at later pupal stages (12–20 h APF), they can be also found in the pupal wing blade without any noticeable pattern with respect to the veins or interveins (data not shown). In 20–24 h APF wings, dead cells are distributed along the periphery of the wing blade and at the wing margin (data not shown). There is no detectable cell death after 24 h APF. In contrast to the increase of cell death at larval molts, the percentage of dead cells does not change at pupariation and pupation (data not shown).
Turnover of Apoptotic Cells.
Cells undergoing apoptosis are usually engulfed by circulating phagocytes very rapidly (in <1 h; see ref. 1). In the wing disc epithelium the increased frequency of apoptotic cells found after the larval molts (larvae were selected individually when in ecdysis) lasts for at least 4–6 h thereafter, suggesting that apoptotic cells remain that long in the epithelium before being engulfed. Clearance time can be more precisely determined by inducing cell death and following dead cell disappearance. Targeted cell death was induced by the GAL4/UAS-RAcs system (13). Thus, in wing discs from 734-GAL4; UAS-lacZ; UAS-RAcs larvae, lacZ and RAcs genes are expressed in the same cells and according to the pattern of expression of GAL4 (Fig. 2A). When such larvae are grown at 25°C, the toxin is inactive and their discs do not show more cell death in the region expressing RAcs than wild-type discs. After a shift to 31°C during 8 h, there is an abrupt increase in the frequency of dead cells in the domain of expression of GAL4 (Fig. 2A). When these larvae are placed at 17°C to inactivate the toxin and cell death patterns are analyzed subsequently, after 1.5 h of chase we find a high frequency of dying cells in the lacZ expressing regions but 4 h later this frequency decreases to the low normal levels. The same experiment carried out with other weak GAL4 lines (to avoid possible nonautonomous effects, see below) gives similar results.
Figure 2.
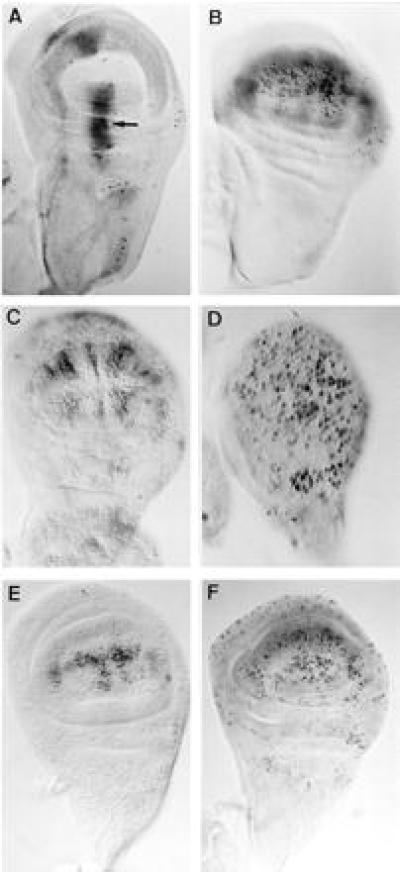
(A and B) Targeted induction of cell death in GAL4; UAS-lacZ; UAS-RAcs wing discs. Larvae were grown at 31°C during 8 h and labeled by 4-bromo-3-chloro-2-indolyl β-d-galactoside staining (light gray staining) and TUNEL (black dots). (A) 734-GAL4; UAS-lacZ; UAS-RAcs. (B) Sd-GAL4; UAS-lacZ; UAS-RAcs. Arrow in A points to a cluster of dead cells. (C–F) Response of imaginal disc cells to induction of apoptosis. (C) Imaginal wing disc from a late third instar HS-hid larva stained with TUNEL 45 min after a pulse of 30 min at 37°C. (D–F) T(2, 3) SM6a-TM6b/+ early third instar (D) and late third instar (E, F) wing discs irradiated at a dose of 1,000 rads and stained by TUNEL 8 h (D and E) or 24 h (F) thereafter. (D is twice magnified.)
These turnover results were confirmed when targeted cell death was induced by the “flip-out” technique in several hs-FLP; PUFWTRA/GAL4; UAS-lacZ lines (see ref. 14 and Materials and Methods). The results described below were obtained with the Sd-GAL4 driver (Fig. 2B) and similar results are obtained employing other GAL4 lines. In that experiment hs-FLP/Sd-GAL4; PUFWTRA; UAS-lacZ larvae were incubated 1 h at 37°C and 0–1.5 h after such treatment recombinant dead cells (30–40 cells per wing pouch) are observed within the lacZ expressing regions (data not shown). However, 4 h later there are no more dying cells than in control discs. Thus, the observed 1.4% of dead cells in a given instant represents the sum of the actual numbers of dying cells plus those that have died at least in the preceding 2–4 h. Evidently, 2–4 h may be an overestimation of the turnover time if massive induced cell death somehow saturates the clearance capacity of the phagocytes.
Dying Cells Do Not Migrate.
The topographical distribution of apoptotic cells in late third instar wing discs and pupal wings may be due either to localized cell death or to their passive transport to specific regions of the disc. To sort out this alternative, we have induced targeted cell death in GAL4; UAS-lacZ; UAS-RAcs larvae by incubation at 31°C for 24 h and then monitored both cell death and lacZ expression. Since the turnover of the β-galactosidase is longer than the 2–4 h it takes dead cells to disappear and expression of lacZ occurs within cells showing apoptotic morphology (even after engulfment by macrophage-like cells (ref. 21 and data not shown), we can analyze the putative migration of dead cells by monitoring the pattern of cells containing β-galactosidase. We have compared the patterned distribution of lacZ expressing cells in wing discs of several GAL4; UAS-lacZ; UAS-RAcs lines pulsed at 31°C with the corresponding GAL4; UAS-lacZ lines in the absence of induced cell death. The results obtained are similar in both experiments indicating that dead cells do not migrate. Moreover, dead cells are distributed throughout the expression domain of the GAL4 driver. Thus, dead cells are engulfed in place.
Sensitivity of Imaginal Disc Cells to Induced Apoptosis.
It has been shown that overexpression of hid induces apoptosis in the Drosophila embryo (5). After generalized induction of hid by incubation of second instar HS-hid larvae during 30 min at 37°C there is a great increase in the number of cells identified by TUNEL in relation to control wild-type heat shocked larvae (data not shown). In late third instar HS-hid larvae, heat treatment is followed by the appearance of apoptotic cells in the intervein territories (Fig. 2C). Since in HS-hid 0–8 h pupal wings all the cells (both G1 arrested wing margin cells and G2 arrested wing blade cells, see ref. 20) undergo apoptosis after the treatment (data not shown), the patterned sensitivity of third instar disc cells to hid does not seem to be related to differences in cell cycle stage and may be attributable to patterned expression of apoptosis regulators. However, it will be shown below that nonautonomously induced apoptosis occurs mainly in G1 cells.
Apoptosis can also be induced by short irradiation of larvae with x-rays (17). A high dose irradiation (4,000 rads) in late third instar wild-type wing discs is followed 8 h later by an abrupt increase in the number of apoptotic cells (data not shown) preferentially localized at the wing margin (interestingly, part of the hid nonsensitive cells). Wild-type larvae irradiated with a lower dose (1,000 rads) show no increase in cell death frequency. However, this dose of irradiation, when applied to discs of cells heterozygous for multiple inversions [T(2, 3) SM6a-TM6b/+], causes cell death by aneuploidy following chromosome segregation in cell division (18). When irradiation is applied to T(2, 3) SM6a-TM6b/+ early imaginal discs, and cell mortality is visualized 8 h later, to allow for at least one round of cell division (16), cell death appears throughout the disc (Fig. 2D) but when applied to late third instar wing discs, apoptotic cells appear preferentially located at the wing margin (Fig. 2E). Thus, in both early and late wing discs there is a correlation between the pattern of cells sensitive to cell death induced by irradiation and that of mitotically active cells (as visualized by the pattern of expression of the stg gene; see ref. 16). There is a similar correlation in eye-antenna discs where cell death appears preceding the morphogenetic furrow (data not shown).
Secondary Effects of Induced Apoptosis.
Interestingly, the frequency of apoptotic cells induced by x-ray treatment (1,000 rads) in T(2, 3) SM6a-TM6b/+ larvae (Fig. 2 D and E) is much higher than the known frequency of mitotic recombination events induced by such doses of irradiation (100–200 recombinant cells for the whole disc at the stage shown in Fig. 2E; see ref. 18). Twenty-four hours after irradiation, apoptotic cells appear throughout the whole disc, still in frequencies higher than expected for aneuploid segregation (Fig. 2F). The above results suggest a nonautonomous induction of cell death in cells other than those where genetic lethality has been induced.
We have confirmed this spreading effect of nonautonomous cell death after targeted induction of cell death in GAL4; UAS-lacZ; UAS-RAcs discs. Expression of active RAcs driven by the en-GAL4 line during 8–24 h at 31°C causes the appearance of apoptotic cells in the posterior (P) compartment (Fig. 3A). However, after a chase period of 8–24 h at 17°C, causing inactivation of the toxin, ectopic cell death is now observed in the anterior (A) compartment even though it has largely decreased in the P compartment (Fig. 3B). Different discs show apoptotic cells at different distances to the compartment border, suggesting a temporal evolution in the spreading effect. The low frequency of dead cells in the region where cell death had been induced excludes the interpretation that ectopic cell death results from apoptotic diffusable signals arising from the induced dead cells. Ectopic cell death was also observed when cell death is induced in other regions of the disc (i.e., wing hinge in line MS209, A/P compartment boundary in line 734 and dorsal compartment in ap-GAL4, see Fig. 3F).
Figure 3.
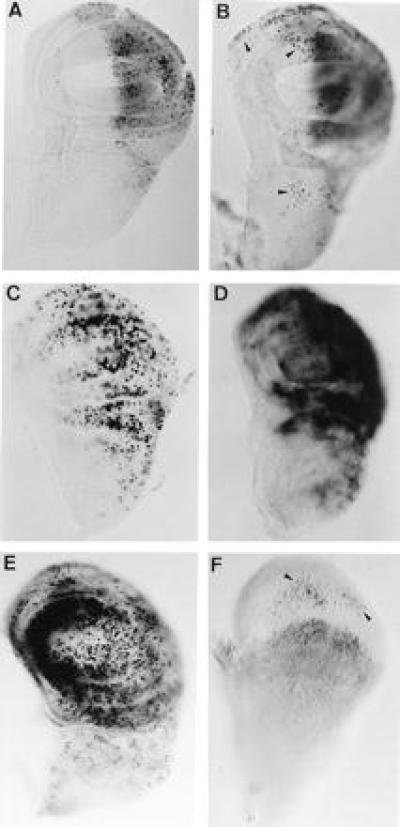
Autonomous and nonautonomous induction of cell death and subsequent cell proliferation recovery. Imaginal wing discs from en-GAL4/UAS-lacZ; UAS-RAcs (A–E) and ap-GAL4; UAS-lacZ; UAS-RAcs (F) larvae. Larvae grown at 31°C during 24 h were stained with TUNEL and 4-bromo-3-chloro-2-indolyl β-d-galactoside immediately after a switch to 17°C (A) or after 24 h at 17°C (B and F). Note in B and F the presence of apoptotic cells (some of them are indicated by arrowheads) in the region not expressing lacZ (gray staining). (C and E) BrdUrd incorporation and (D) in situ hybridization with a stg probe in larvae grown at 31°C during 24 h and stained immediately after the switch (C and D) or after 8 h at 17°C (E).
Cell Proliferation Patterns After Induced Cell Death.
After induction of cell death in the en pattern during 8–24 h, there is a substantial increase in the rate of DNA synthesis (Fig. 3C) and of stg expression (Fig. 3D) in the P compartment cells when compared with the wild-type patterns at this developmental stage shown in ref. 16. (Note in Fig. 3C the presence of BrdUrd-labeled cells in the zone of nonproliferating cells; see ref. 16). The frequency of mitotic figures in the P compartment is also increased (data not shown). Surprisingly, the A compartment at that stage shows a substantial reduction in BrdUrd and stg labeling (Fig. 3 C and D) and mitotic figures are absent (data not shown). Similar differences in proliferation patterns are found in both ricin-treated and nontreated territories when RAcs expression is driven by other GAL4 lines (data not shown). After a switch to the permissive temperature, the nontreated territory recovers 8 h later an apparently wild-type pattern of BrdUrd incorporation and 24 h later that of stg expression (Fig. 3E and data not shown). Thus, induction of cell death in the ricin expressing territory causes proliferation arrest and nonautonomous cell death in G1 in the complementary territories.
Induction of cell death in the P compartment during 24 h (and even 48 h) of larval development does not affect the final size of the wings (data not shown) suggesting a restoring proliferative activity in both compartments to compensate for the loss of dead cells. Indeed, mitotic recombination clones initiated immediately after cell death induction (see Materials and Methods) show a size increase in both territories. The average sizes of the clones in the A and P compartments, initiated in 60 h after egg laying larvae after 24 h of induction of cell death in the P compartment are 190 and 270 cells, respectively. These figures are significantly larger than control ones for the same age (125 and 165 cells in the A and P compartments, respectively). Interestingly, the frequency of clones in the P compartment initiated after the treatment (0.12 clones per wing) is larger than in control wings (0.04), whereas in the A compartment it remains at the normal value (0.05 and 0.06, respectively). This confirms the high frequency of cycling cells in the P compartment observed by BrdUrd and stg labeling (Fig. 3 C and D).
While wing discs transitorily exposed to induced cell death recover their normal size, the continuous expression of active RAcs in one region of the wing disc results in wings smaller in number of cells (up to 40% of reduction) than normal ones, although with normal cell size, wing shape, proportions, and vein patterning. Similar wing size reductions are observed when cell death is chronically induced in other territories of the wing (dorsal compartment or wing hinge). The compensating growth between directly and indirectly affected territories to give rise to normally patterned wings will here be called “accommodation.”
Cell Death in Apogenetic Mosaic Mutant Discs.
Genetic mosaics consist in the juxtaposition of normal and mutant cells in the same primordium. Morphogenetic mosaics, in particular those corresponding to homeotic mutants [bithorax (bx, ref. 22); Cbx (12); en (23), and ap (24)] as well as clonal overexpression of signaling genes such as decapentaplegic (dpp) (25), show extra growth of the mutant and normal cells to accommodate for the confrontation of independently determined positional values (ref. 12 and see Discussion). Similarly, in apogenetic mosaics, caused by incomplete expressivity of homeotic mutations (bx, Cbx, and en), in which gene expression spatially correlates with the partial transformation, both auto and allotypic territories lie side by side integrated in a presumed invariant prepattern (12, 22, 26). Accommodation in both types of mosaics occurs by cell proliferation, as a result of confrontation of territories with different proliferation/specification dynamics (12). We inquired here whether cells in such apogenetic mosaics accommodate also through cell death. In CbxM1/+ wing discs or bx34e haltere discs, haltere and wing incompletely transformed territories are juxtaposed in the same disc and compartment during development (12, 22). Fig. 4A and B show generalized apoptosis in both haltere and wing territories (the frequency of apoptotic cells in other discs of CbxM1/+ and bx34e larvae is similar to that of wild-type discs). Similarly, in en1 mutant discs, which show incomplete transformation of the P compartment, cell death appears in both the A and P compartments (data not shown). In the case of dppd5 mutant discs, whose insufficiency occurs at the time of the observation only along the A/P boundary, cell death appears again throughout all the wing discs (Fig. 4C).
Figure 4.
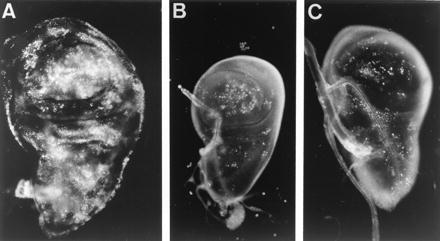
Apoptosis, visualized by acridine orange staining, in late third instar CbxM1/+ (A) and dppd5 (C) wing discs, and bx34e haltere disc (B).
DISCUSSION
Apoptosis in the Wing Primordium.
The pattern of appearance of apoptotic cells during normal development reveals certain regularities. Apoptotic cells occur as single cells or in clusters of neighboring cells with an average size of 3.4 cells, interestingly similar to that of clusters of cells synchronized in the same cell cycle stage (16, 20). Clusters of apoptotic cells are also found in haltere and leg discs (unpublished results). Apoptotic cells remain in the epithelium for 2–4 h before being engulfed in place by hemolymph cells. Apoptosis in wild-type discs is a rather rare phenomenon during both larval and pupal development (1.4% of the anlage cells). At early larval stages apoptotic cells appear throughout the primordium but are topologically localized in characteristic regions at late stages (late larva and pupa). There is no cell death after 24 h APF at the end of the proliferative period of the wing primordium (20).
There is a great increase in the number of apoptotic cells at both larval molts but not at pupariation and pupation. Since molting is associated with changes in hormone titers (ecdysone and/or juvenile hormone) (27) the absence of increased apoptosis at pupariation and pupation could be related to the low titer in juvenile hormone at these stages.
Local and Long-Range Cell Interactions in Apoptosis.
The occurrence of clusters of apoptotic cells in imaginal discs during normal development, in discs with cells dying from ricin expression and in discs with aneuploid cells, suggests local cell-cell interactions leading to cell death. Cell death over long distances occurs in territories adjacent to those where cell death has been induced by ricin synthesis promoted by different GAL4 lines. In addition, cell death appears throughout the disc anlage in dpp mutant discs beyond the region where dpp is normally expressed (see also ref. 28). Cell death also occurs throughout the discs in apogenetic mosaics caused by incomplete homeotic transformations (en, Cbx, and bx). These long range nonautonomous effects in cell death patterns reveal long range accommodation effects through local cell-cell interactions (see below).
Cell mortality caused by a ricin synthesis pulse, driven by several GAL4 lines, affects the GAL4 expressing territory and the adjacent complementary ones in different ways (Fig. 3 and 5). The directly affected territory begins active proliferation immediately after induced cell death. This correlates with significant increases in mitotic recombination frequencies (revealing an increased proportion of cells at the G2 stage; see ref. 29) in this territory. The complementary territory substantially decreases cell proliferation, shows cell death later and resumes S cell cycle stage and G2 stage subsequently. Thus, the proliferation arrest and apoptosis apparently occurs in cells in G1.
While direct cell death caused by toxin activation accounts for itself, cell death in adjacent RAcs nonexpressing territories and changes in proliferation rate in both territories beg an interpretation. Nonautonomous cell death can hardly be explained by the liberation of apoptotic signals by initial dead cells, because mortality in the secondarily affected territory appears when the directly affected one does not express the active toxin and shows little cell death. Thus, indirect delayed cell death could be caused by signal deprivation as it is known to occur in other developmental systems (2). Cell death due to signal deprivation can be explained in the Entelechia model (16, 20). According to this model, cells proliferate, survive, or die not in response to diffusible morphogens, but according to their own positional values following computation with those of neighboring cells (possibly via ligands and activated receptors in the cell surface). They continue to proliferate as long as neighboring cells show differences in positional values, intercalating between a maximal value, at restriction borders, and a minimal value. When differences in positional values between neighboring cells are below a certain threshold, cells stop dividing; when minimal by signal deprivation cells die. We propose that in the present experiments insufficient number of cells along borders between territories results in a lowering in maximal positional values along this border (Fig. 5). This would diminish the differences in positional values between adjacent cells down the slope in the indirectly affected territory (Fig. 5b). Low differences lead to lower proliferation, and eventually, by signal deprivation, to cell death (Fig. 5c). Conversely, in the territory where cell death has been directly induced, reduction in the number of cells leads to higher than normal differences in positional values between adjacent cells, causing a higher than normal proportion of dividing cells (Fig. 5b). Thus, perturbation caused by cell death is corrected in both territories through both cell proliferation and apoptosis, leading later to proportioned primordia following a normal Entelechia process (Fig. 5d), as confirmed by the larger than control size of the clones in both territories. Whereas discs, treated for a period of up to 48 h in growing larvae, regenerate to normally sized and proportioned adult wings, chronically treated larvae (from late larval stages to adult) give rise to smaller but correctly proportioned wings.
Figure 5.
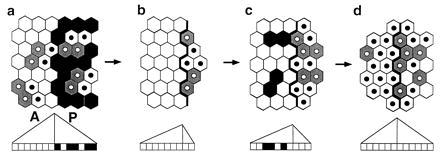
Apoptosis and cell proliferation patterns during accommodation. Ricin induced apoptosis (black cells in a) in the P compartment of the wing disc is followed (b–d) by increased cell proliferation in this territory and nonautonomous proliferation arrest in G1 (white cells) and cell death (black cells) in the A compartment. Gray cells and black nuclei correspond to stg expressing and S-phase cells, respectively. Below each figure, a representation of positional values in a gradient with relative maxima at the compartment borders is shown. Notice the change of slope of positional values during the accommodation process in both compartments.
Apogenetic mosaics of alleles with incomplete transformations such as Cbx (12), bx (22), or en (26) viable mutations have extensive cell death in the mutant discs. This cell death could be caused (i) by differences in cell adhesivity/cell recognition (as in genetic mosaics of the same homeotic genes (30, 31) or in re-aggregates of cells from different segments or compartments (32); or (ii) from confrontation of cells with abrupt positional value differences leading to accommodation of disparate territories to generate (through cell proliferation and apoptosis) a continuous landscape of positional values. Global cell death in dpp mutant discs can be also explained by accommodation rather than by insufficient morphogens released by A/P border cells.
The observed nonautonomously induced cell death may affect our interpretation of cell lethality patterns in many types of mutant discs, because lethality may not reflect exclusively local autonomous cell death caused by the mutation but also accommodation effects in the rest of the mutant disc.
Acknowledgments
We are most grateful to S. Cohen, J. L. Gómez-Skarmeta, J. Modolell, and M. Raff for constructive suggestions on the manuscript; to A. Brand, M. Calleja; C. J. O’Kane, and H. Steller for providing fly stocks; and A. López, P. Martín, and R. Hernández for technical assistance. M.M. is a postdoctoral fellow of the Fundación Rich. This work was supported by Grants PB92-0036 (to A.G.-B.) and PB93-0181 (to J. Modolell) from The Dirección General de Investigación Científica y Técnica and an institutional grant from The Fundación Ramón Areces to the Centro de Biología Molecular Severo Ochoa.
ABBREVIATIONS
- BrdUrd
5-bromo-2′-deoxyuridine
- RAcs
cold-sensitive form of ricin A
- TUNEL
terminal deoxynucleotidyltransferase-mediated UTP end labeling
- APF
after puparium formation, RTA, ricin toxin A chain
- bx
bithorax
- Cbx
Contrabithorax
- dpp
decapentaplegic
- en
engrailed
- stg
string
- ap
apterous
- Sd
Scalloped
- HS
hsp70
- hid
head involution defective
- A
anterior
- P
posterior
- UAS
upstream activation sequence
References
- 1.Jacobson M D, Weil M, Raff M C. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 2.Raff M C. Nature (London) 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- 3.Wolff T, Ready F. Development (Cambridge, UK) 1991;113:825–839. doi: 10.1242/dev.113.3.825. [DOI] [PubMed] [Google Scholar]
- 4.White K, Grether M E, Abrams J M, Young L, Farrell K, Steller H. Science. 1994;264:677–683. doi: 10.1126/science.8171319. [DOI] [PubMed] [Google Scholar]
- 5.Grether M E, Abrams J M, Agapite J, White K, Steller H. Genes Dev. 1995;9:1694–1708. doi: 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- 6.Chen P, Nordstrom W, Gish B, Abrams J M. Genes Dev. 1996;10:1773–1782. doi: 10.1101/gad.10.14.1773. [DOI] [PubMed] [Google Scholar]
- 7.Zhou L, Hashimi H, Schwartz L M, Nambu J R. Curr Biol. 1995;5:784–790. doi: 10.1016/s0960-9822(95)00155-2. [DOI] [PubMed] [Google Scholar]
- 8.Williams J A, Paddock S W, Carroll S B. Development (Cambridge, UK) 1993;117:571–584. doi: 10.1242/dev.117.2.571. [DOI] [PubMed] [Google Scholar]
- 9.Ng M, Diaz-Benjumea F J, Cohen S M. Development (Cambridge, UK) 1995;121:589–599. doi: 10.1242/dev.121.2.589. [DOI] [PubMed] [Google Scholar]
- 10.Brand A H, Perrimon N. Development (Cambridge, UK) 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 11.Calleja M, Moreno E, Pelaz S, Morata G. Science. 1996;274:252–255. doi: 10.1126/science.274.5285.252. [DOI] [PubMed] [Google Scholar]
- 12.González-Gaitán M A, Micol J L, García-Bellido A. Genetics. 1990;126:139–155. doi: 10.1093/genetics/126.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moffat K G, Gould J H, Smith H K, O’Kane C J. Development (Cambridge, UK) 1992;114:681–687. doi: 10.1242/dev.114.3.681. [DOI] [PubMed] [Google Scholar]
- 14.Smith H K, Roberts I J H, Allen M J, Connolly J B, Moffat K G, O’Kane C J. Dev Genes Evol. 1996;206:14–24. doi: 10.1007/s004270050026. [DOI] [PubMed] [Google Scholar]
- 15.Lindsley D, Zimm G G. The Genome of Drosophila melanogaster. San Diego: Academic; 1992. [Google Scholar]
- 16.Milán M, Campuzano S, García-Bellido A. Proc Natl Acad Sci USA. 1996;93:640–645. doi: 10.1073/pnas.93.2.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abrams J M, White K, Fessler L I, Steller H. Development (Cambridge, UK) 1993;117:29–43. doi: 10.1242/dev.117.1.29. [DOI] [PubMed] [Google Scholar]
- 18.Merriam J R, García-Bellido A. Mol Gen Genet. 1972;115:302–313. doi: 10.1007/BF00333169. [DOI] [PubMed] [Google Scholar]
- 19.Wyllie A H. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 20.Milán M, Campuzano S, García-Bellido A. Proc Natl Acad Sci USA. 1996;93:11687–11692. doi: 10.1073/pnas.93.21.11687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nordstrom W, Chen P, Steller H, Abrams J M. Dev Biol. 1996;180:213–226. doi: 10.1006/dbio.1996.0296. [DOI] [PubMed] [Google Scholar]
- 22.Cabrera C V, Botas J, García-Bellido A. Nature (London) 1985;318:569–571. [Google Scholar]
- 23.Tabata T, Schwartz C, Gustavson E, Ali Z, Kornberg T B. Development (Cambridge, UK) 1995;121:3359–3369. doi: 10.1242/dev.121.10.3359. [DOI] [PubMed] [Google Scholar]
- 24.Diaz-Benjumea F J, Cohen S M. Cell. 1993;75:741–752. doi: 10.1016/0092-8674(93)90494-b. [DOI] [PubMed] [Google Scholar]
- 25.Zecca M, Basler K, Struhl G. Development (Cambridge, UK) 1995;121:2265–2278. doi: 10.1242/dev.121.8.2265. [DOI] [PubMed] [Google Scholar]
- 26.García-Bellido A, Santamaría P. Genetics. 1972;72:87–101. doi: 10.1093/genetics/72.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wigglesworth V B. The Physiology of Insect Metamorphosis. Cambridge, U.K.: Cambridge Univ. Press; 1954. [Google Scholar]
- 28.Bryant P J. Dev Biol. 1988;128:386–395. doi: 10.1016/0012-1606(88)90300-4. [DOI] [PubMed] [Google Scholar]
- 29.González-Gaitán M A, Capdevila M P, García-Bellido A. Mech Dev. 1994;46:183–200. doi: 10.1016/0925-4773(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 30.García-Bellido A. Dev Biol. 1966;14:278–306. doi: 10.1016/0012-1606(66)90017-0. [DOI] [PubMed] [Google Scholar]
- 31.García-Bellido A. Wilhelm Roux’s Arch Entwicklungsmech Org. 1967;158:212–217. doi: 10.1007/BF00573398. [DOI] [PubMed] [Google Scholar]
- 32.García-Bellido A. Genetics. 1968;60:181. (abstr.). [Google Scholar]