

Abstract
The amyloid precursor protein (APP) is a type I transmembrane protein translocated to neuronal terminals, whose function is still unknown. The C-terminus of APP mediates its interaction with cellular adaptor and signaling proteins, some of which signal to the stress-activated protein kinase (SAPK) pathway. Here we show that ASK1, a MAPKKK that activates two SAPKs, c-Jun N-terminal-kinase (JNK) and p38, is present in a complex containing APP, phospho-MKK6, JIP1 and JNK1. In primary neurons deprived of growth factors, as well as in brains of (FAD)APP-transgenic mice, ASK1 was upregulated in neuronal projections, where it interacted with APP. In non-transgenic brains, ASK1 and APP associated mainly in the ER. Our results indicate that recruitment of ASK1 to stress-signaling complexes assembled with APP may be triggered and enhanced by cellular stress. Thus, ASK1 may be the apical MAPKKK in a signaling complex assembled with APP as a response to stress.
Keywords: amyloid precursor protein, apoptosis signaling kinase, stress signaling complex, neurodegeneration, Alzheimer’s disease
INTRODUCTION
APP is a type I transmembrane protein that undergoes extra- and intra-membranous cleavages by α-, β-, and γ-secretases which give rise, in different combinations, to the soluble, large extracellular domain (sAPP), the amyloid beta (Aβ) peptides, the soluble p3 peptide, and a 99-amino acid carboxyterminal fragment as major cleavage products (Nunan, J. and Small, D.H., 2002). Among these species, the Aβ peptides Aβ 1–1–42 and Aβ 1–40 have been most extensively studied, and are believed to be causally involved in the pathogenesis of Alzheimer’s disease (AD) (Naslund, J. et al., 2000; Walsh, D.M. and Selkoe, D.J., 2004; Tanzi, R.E. and Bertram, L., 2005). In spite of the fact that the signaling resulting from the proteolytic processing of APP is being elucidated, the function of APP and those of its proteolytic products remain largely unknown (Selkoe, D.J., 2002).
APP is a synaptic protein (Selkoe, D.J., 2002) that is anterogradely transported to nerve terminals (Koo, E.H. et al., 1990; Sisodia, S.S. et al., 1993; Buxbaum, J.D. et al., 1998). Together with the secretory and endocytic pathways, it has been proposed that a site for production, release and accumulation of the Aβ peptide may be the synapse itself (DeKosky, S.T. and Scheff, S.W., 1990; Lyckman, A.W. et al., 1998; Selkoe, D.J., 2002). Consistent with this idea, recent data indicate that generation of Aβ may be increased by synaptic activity (Kamenetz, F. et al., 2003; Cirrito, J.R. et al., 2005) and in turn inhibit synaptic transmission (Kamenetz, F. et al., 2003). Moreover, a wealth of evidence points to the synapse as one of the major sites for Aβ toxicity (Masliah, E., 1998; Masliah, E. et al., 2001; Selkoe, D.J., 2002). APP shows the signature of a membrane receptor and is possibly a member of the dependence receptor family (Bredesen, D.E. et al., 2005). Its C-terminal, intracellular domain plays a critical role in the intracellular trafficking of the protein and contains signals (YENPTY) that are also required for the delivery of APP to protease-containing compartments and for its retrieval from the plasma membrane by endocytosis (Perez, R.G. et al., 1999; Soriano, S. et al., 1999). This motif has also been implicated in the assembly of signaling complexes with intracellular adaptors and transcriptionally active complexes (Yang, Y. et al., 1998; Cao, X. and Sudhof, T.C., 2001; Matsuda, S. et al., 2001; Sabo, S.L. et al., 2001; Baek, S.H. et al., 2002; Biederer, T. et al., 2002; Ho, C.S. et al., 2002; Scheinfeld, M.H. et al., 2002; Taru, H. et al., 2002; Sabo, S.L. et al., 2003), linking APP to a variety of cellular processes. The C-terminal domain of APP can interact with several adaptor proteins containing phosphotyrosine-binding (PTB) domains such as Fe65 (Fiore, F. et al., 1995; Cao, X. and Sudhof, T.C., 2001), X11 (Biederer, T. et al., 2002; Ho, C.S. et al., 2002), and JNK-interacting protein 1 (Matsuda, S. et al., 2001; Scheinfeld, M.H. et al., 2002; Taru, H. et al., 2002; Inomata, H. et al., 2003), an adaptor-scaffolding protein that has a crucial role in activating the JNK cascade and in axonal vesicle transport (Torroja, L. et al., 1999; Kamal, A. et al., 2001). In addition, it has been shown that APP and the APP family member APLP2 can be phosphorylated by JNK as a response to cellular stress in a process facilitated by Mint2, an adaptor protein that interacts with the C-terminal domain of APP, as well (Taru, H. and Suzuki, T., 2004). Moreover, it was proposed that axonal transport of APP is mediated by direct binding of the C-terminal domain of APP to the light chain subunit of kinesin-I (Kamal, A. et al., 2001), and this interaction may be enhanced by the JIP1-dependent phosphorylation of Thr668 (APP695 numbering) (Inomata, H. et al., 2003; Muresan, Z. and Muresan, V., 2005). Similar to the processing of the Notch receptor (Schweisguth, F., 2004), the cytoplasmic domain of APP released after secretase cleavage forms a multimeric complex with Fe65 and the histone acetyltransferase TIP60, and activates transcription (Cao, X. and Sudhof, T.C., 2001; Baek, S.H. et al., 2002; Biederer, T. et al., 2002).
The mitogen-activated protein (MAP) kinase signaling cascade is evolutionarily conserved, and is typically composed of three protein kinases that include a MAP kinase (MAPK), a MAPK kinase (MAPKK), and a MAPKK kinase (MAPKKK) (Morrison, D.K. and Davis, R.J., 2003). The MAPK cascade plays a fundamental role in the transduction of environmental changes into changes in cell function. In general, it is thought that a MAPKKK activates a MAPKK by phosphorylation, which in turn phosphorylates and activates an effector MAPK. MAPKs then regulate the activity of downstream transcription factor(s) or kinase(s). Thus, activation of a MAPK system will generally result in the activation of a complex cellular response. In mammalian systems, two classes of MAPK (the c-Jun aminoterminal kinases (JNKs) and the p38 MAPKs, referred to collectively as the stress-activated protein kinases or SAPKs) are specifically activated when cells are exposed to a variety of inflammatory cytokines or to a diverse array of cellular insults such as UV radiation, heat and osmotic shock, and growth factor withdrawal (Ichijo, H., 1999). ASK1 is an apical MAPKKK that transduces the signal from ligand-activated tumor necrosis factor receptor (TNFR) to the programmed cell death system (Hoeflich, K.P. et al., 1999) by its interaction with the adaptor protein TRAF2, mediates apoptotic cell death induced by genotoxic stress through the JNK1 and p38 pathways (Chen, Z. et al., 1999), and is potently activated by glucose deprivation, a response that is inhibited by Akt (Song, J.J. and Lee, Y.J., 2005b, a). ASK1 has also been implicated in the regulation of signal transduction by Fas through Daxx, by its sequestration in the cytoplasm (Ko, Y.G. et al., 2001). The interaction of ASK1 with TRAF2 also links ASK1 to the signal transduction activated in the unfolded protein response through IRE1α (Nishitoh, H. et al., 2002). ASK1 has been shown to be essential for ER stress-induced neuronal cell death triggered by expanded polyglutamine repeats (Nishitoh, H. et al., 2002) and has been implicated in Aβ-induced neuronal apoptosis directly through the upregulation of an inhibitor of proteasome activity, E2–25K/Hip-2 (Song, S. et al., 2003). Moreover, it has been suggested by several studies that neuronal apoptosis induced by Aβ depends on JNK1 and p38 (Morishima, Y. et al., 2001; Savage, M.J. et al., 2002; Minogue, A.M. et al., 2003; Hashimoto, Y. et al., 2004), the two effector MAPKs regulated by ASK1 activity. Interestingly, the recent demonstration that Aβ can bind directly to its precursor APP (Lorenzo, A. et al., 2000 Lu, D.C. et al., 2003a), accelerate its multimerization at the cell surface, and induce the interaction of the intracellular domain with caspase-8 (an initiator caspase mediating apoptosis (Lu, D.C. et al., 2003a), suggests that at least some of the toxicity attributed to Aβ may proceed through signaling via its precursor/receptor, APP. In support of this notion, it was demonstrated that Aβ toxicity requires APP in vitro (Lu, D.C. et al., 2003a), suggesting that one mode of action of Aβ may involve binding APP directly in a receptor-ligand fashion (Lorenzo, A. et al., 2000; Lu, D.C. et al., 2003a). Consistent with a potentially crucial role of APP dimerization in the induction of neuronal cell death, epidermal growth factor (EGF) treatment potently induced cell death in cells of neuronal origin expressing fusion constructs of the EGF receptor (EGFR) ectodomain fused to the intracellular domain of APP (APP hybrid) in a manner sensitive to caspase inhibitors and dependent on ASK1 activity (Hashimoto, Y. et al., 2003). In these studies, APP hybrid-induced cell death could also be blocked by a specific JNK inhibitor, and required binding of the intracellular domain of APP to the adaptor JIP1b.
We had previously shown that APP can be immunoprecipitated with MKK6 and with its upstream MAPKKK, ASK1 (Peel, A.L. et al., 2004). In the present work we sought to further explore the association of ASK1 with APP and the assembly of APP-containing stress-signaling complexes, both in vitro and in vivo. We showed that APP and ASK1 are present in protein complexes formed in transfected HEK293 cell, in cultured primary neurons (Figure 1) and in mouse brains (Figures 4 and 6). In cultured cells (Figure 1) and in synaptic vesicles purified from mouse brains (Figure 5), APP-ASK1 complexes contained the adaptor JIP1b, activated MKK6 and activated JNK. The 68-amino acid fragment Asp597–Asp664, encompassing Aβ1–42 and an additional 26 residues carboxyterminal to it (therefore spanning APP’s transmembrane domain and the first 16 amino acids of its intracellular domain) was sufficient for ASK1 binding (Figure 2). The extreme C-terminal sequences required for the interaction of APP with the adaptor JIP1 (embedded in the last 31 amino acids of the cytoplasmic domain), and the regulatory N-terminal domain of ASK1 (Figure 2), however, were not. These observations suggest that the C-terminal domain of ASK1, which contains the Ser/Thr kinase domain, was sufficient for its interaction with the cytoplasmic domain of APP. Consistent with this hypothesis, the kinase activity of ASK1 was required for its interaction with APP-containing complexes (Figure 2). ASK1 was upregulated and formed a complex with APP in vesicle-like structures in neuronal projections, both in primary neuronal cultures deprived of trophic factors (Figure 3) and in brains from mice modeling Alzheimer’s disease (Figure 4). The association of ASK1 and APP in non-transgenic brains, in contrast, was mainly restricted to the neuronal endoplasmic reticulum (Figures 4 and 5). Given the involvement of the ASK1-downstream effectors JNK and p38 in Aβ-induced neurotoxicity, and recent observations indicating that Aβ binds to and multimerizes APP at the cell surface (Lorenzo, A. et al., 2000), our data suggest that ASK1 may be the apical MAPKKK in a stress-activated signaling complex assembled at the C-terminus of APP in response to stress.
Figure 1. ASK1 and APP are immunoprecipitated together from HEK293 cell lysates.
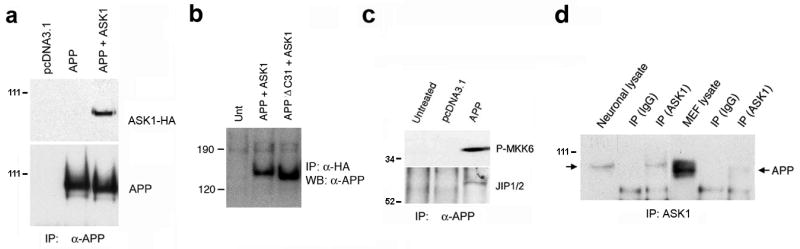
a. Complexes immunoprecipitated with an anti-APP antibody (5A3/1G7) from lysates of HEK293 cells transfected with the indicated constructs were electrophoretically separated, transferred to membranes, and probed with anti-HA antibody. b. Complexes immunoprecipitated with anti-HA antibodies were processed as in (a) and probed with anti-APP 5A3/1G7 antibodies. c. The same complexes were probed with antibodies specific for phosphorylated MKK6 (P-MKK6) and JIP1/2. d. Complexes immunoprecipitated with anti-ASK1 antibodies (DAV) from lysates of primary cortical neuronal cultures or mouse embryonic fibroblasts (MEF), and total lysates, were resolved in gels and probed with anti-APP (5A3/1G7) antibody. Arrows indicate APP-immunoreactive bands.
Figure 4. ASK1 is upregulated and forms a complex with APP in transgenic mouse brains.
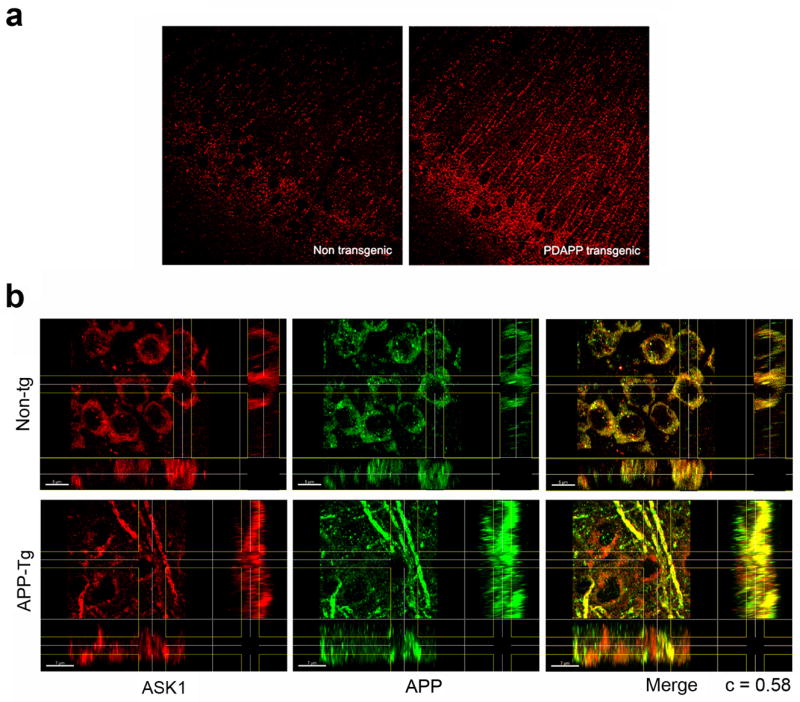
a. Confocal images of hippocampal sections of brains of non-transgenic and APP-transgenic mice stained with rabbit anti-ASK1 (H300) and Texas Red-conjugated anti-rabbit antibodies. Magnification = 600X. b. X, Y and Z projections of three-dimensional reconstructions of confocal z-stacks obtained from hippocampal sections of brains of non-transgenic and APP-transgenic mice stained with anti-ASK1 (H300) and anti-APP (22C11) antibodies.
Figure 6. ASK1 and APP are associated with neuronal ER in mouse brains.
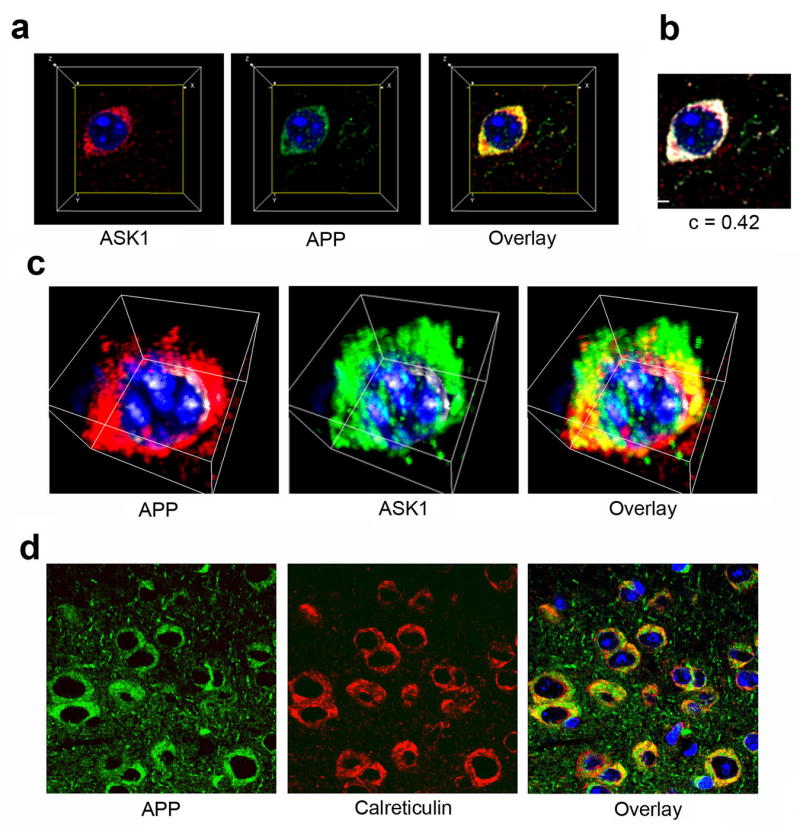
a–d. Three-dimensional reconstruction of confocal z-stacks from hippocampal sections of non-transgenic mouse brains stained with rabbit anti-ASK1 (H300) and mouse anti-APP (22C11) antibodies. Perpendicular projections of each channel (a) and of the colocalization channel (b) are shown. c. Volumetric projection of three-dimensional reconstructions. d. Confocal images of hippocampal sections of non-transgenic mouse brains stained with anti-APP and anti-calreticulin antibodies.
Figure 5. APP is associated with stress-signaling proteins in synaptic vesicles.
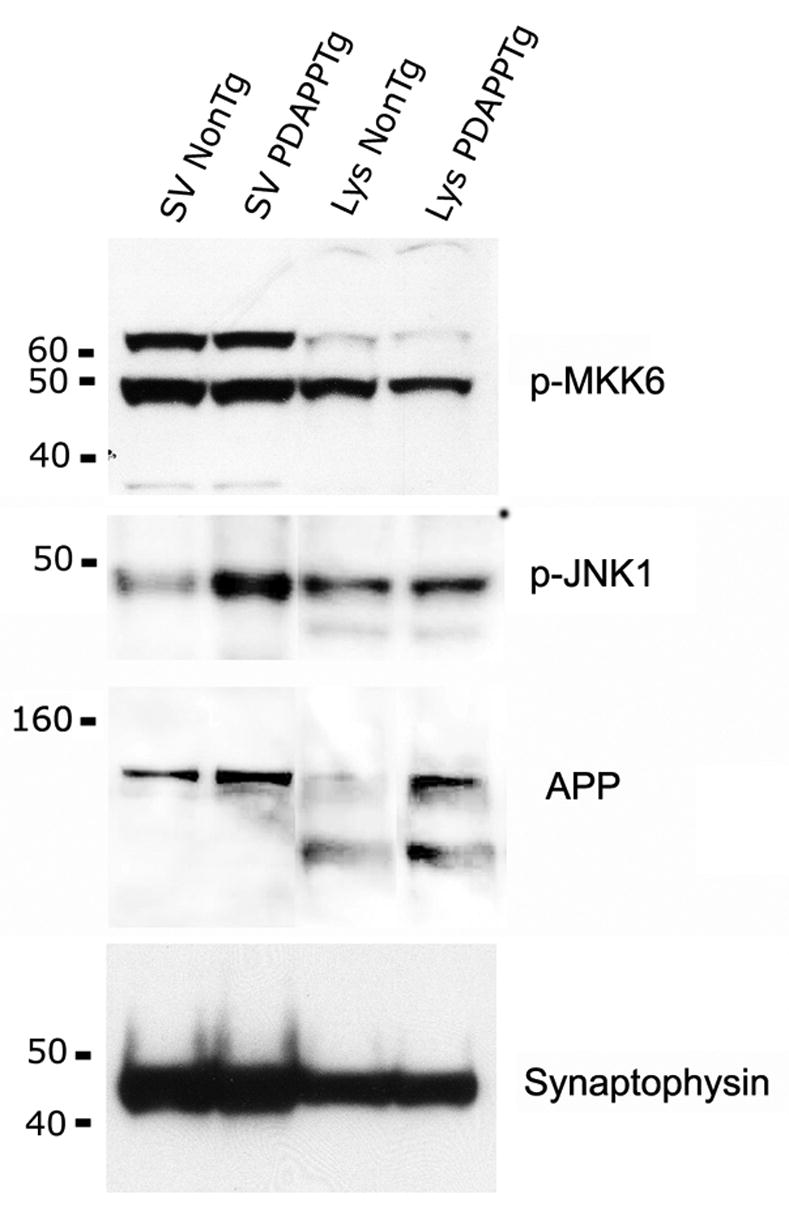
Synaptic vesicles were fractionated from non-transgenic and transgenic brains as described in Methods, resolved in gels, and probed with the indicated antibodies.
Figure 2. Residues 597–664 in APP cytoplasmic domain are sufficient to mediate binding to ASK1.
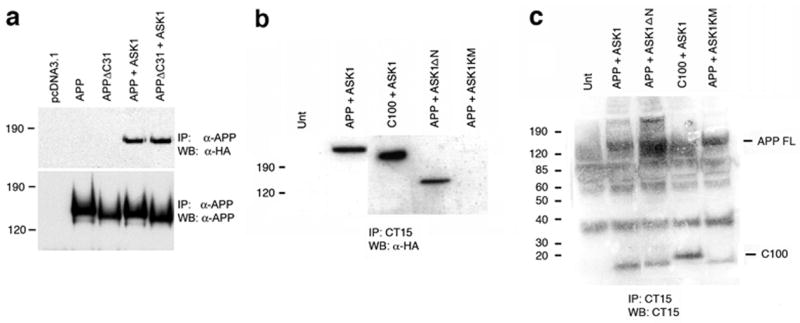
a. Complexes immunoprecipitated with an anti-APP antibody (5A3/1G7) from lysates of HEK293 cells transfected with the indicated constructs were probed with anti-HA or anti-APP antibodies. b. Complexes immunoprecipitated with an antibody that recognizes the last 15 amino acids of the cytoplasmic domain of APP (CT15, (Sisodia, S.S. et al., 1993)) from lysates of HEK293 cells transfected with the indicated constructs were probed with anti-HA 5A3/1G7 antibodies. c. The same complexes were probed with anti-APP (5A3/1G7) antibody.
Figure 3. ASK1 and APP form a complex in cultured primary neurons.
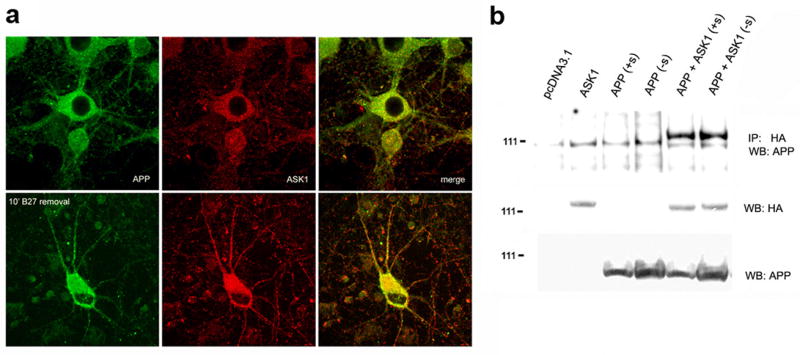
a. Confocal images of primary hippocampal neuronal cultures that were either left untreated or incubated in media without growth factors (B27) for 10′, fixed, and stained with mouse anti-APP (22C11) and rabbit anti-ASK1 (H300) antibodies. Texas Red-conjugated anti-mouse and FITC-conjugated anti-rabbit were used. b. Complexes immunoprecipitated with anti-HA antibody from lysates of HEK293 cells transfected with the indicated constructs and maintained in complete media or in serum-free media for 2 h were resolved in gels and probed with the indicated antibodies.
MATERIALS AND METHODS
Cell culture and transient transfections
Human embryonic kidney (HEK) 293 cells were grown in 100 mm dishes at 37°C in 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) and penicillin/streptomycin (P/S) (Cellgro, Mediatech, Kansas, MO). 293 cells grown in 100 mm dishes were transiently transfected with 3–5 or 6–8 μg of the indicated plasmid constructs respectively using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Lysates were collected 13–15 hours after transfection. In all transfection experiments, the amount of plasmid DNA transfected was equalized by the addition of pcDNA3.1 vector DNA. Mouse embryonic fibroblasts were cultured in a 1:1 mixture of DME and F12K media supplemented with 10% heat-inactivated FBS, 14 mM HEPES, 4.5 g/l glucose, non-essential amino acids and P/S. Hippocampal or cortical neurons derived from 17-day old mouse embryos were plated in Neurobasal media supplemented with 5% horse serum. Three days later, the cultures were treated with 10 mM cytosine arabinoside (AraC) and media was changed to Neurobasal media supplemented with B27 (Gibco, Carlsbad, CA).
DNA constructs and antibodies
Hemagglutinin-tagged wild-type and kinase-mutant human ASK1 (ASK1-HA and ASK1KD-HA) and rabbit anti-ASK1 (DAV) were described previously (Nishitoh, H. et al., 1998; Nishitoh, H. et al., 2002). ASK1KD-HA is the kinase-inactive mutant form of ASK1 (ASK1-K709R). Residues 1–648 of full-length ASK1, encompassing its N-terminal regulatory (inhibitory) domain, have been deleted in ASK1ΔN (ASK1-649-1375), yielding a constitutively-active form of the kinase. Wild-type human APP695, the C-terminal deletion APPΔC31, and the APP-C100 constructs have been described previously (Lu, D.C. et al., 2000). The sequences C-terminal to the Asp664 cleavage site (the carboxy-terminal 31 amino acids of APP, 665–695, corresponding to the APP-C31 peptide (Gervais, F.G. et al., 1999; Lu, D.C. et al., 2000) have been deleted in APPΔC31. APP-C100 consists of the signal peptide sequence of APP fused to the C-terminal 99 amino-acid residues beginning at the Asp residue of Aβ. Anti-HA antibodies (HA.11) were purchased from BabCO (Richmond, CA). Rabbit anti-ASK1 (Ct) (ab16505), mouse anti-APP MAB348 (formerly Roche 1285262) (22C11) were purchased from Chemicon (Temecula, CA) and used for immunohistochemistry. Anti-ASK1 (H-300) and protein A/G-sepharose were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-calreticulin (SPA-600) was purchased from Stressgen (Victoria, BC). Immunoprecipitation of APP was with the 5A3/1G7 monoclonal (recognizing the ectodomain of APP) or with CT15 (Sisodia, S.S. et al., 1993) kindly provided by Dr. Edward Koo, University of California, San Diego. All HRP-conjugated secondary antibodies, FITC-conjugated anti-goat and Texas Red-conjugated anti-rabbit were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA) and AP-conjugated anti-rabbit and AP-conjugated anti-mouse were purchased from Promega (Madison, WI). Anti-phospho-SAPK/JNK(Thr183/Tyr185) was purchased from Cell Signaling (Danvers, MA). Rabbit anti JIP1/2 was purchased from Zymed (So. San Francisco, CA). Alexa Fluor488 donkey anti-mouse, Alexa Fluor488 donkey anti-rabbit IgGs (H+L) were purchased from Molecular Probes (Eugene, OR). Nuclei were visualized using TOTO-3 iodide (642/660) nuclear counterstain (Molecular Probes, Carlsbad, CA).
Immunoprecipitation and immunoblotting
Cells were collected, washed once in phosphate-buffered saline (PBS) and disrupted in lysis buffer (1 mM EDTA, 20 mM TrisHCL pH 7.5, 12 mM β-glycerophosphate, 1 mM sodium orthovanadate, 150 mM NaCl, 5 mM EGTA, 10 mM NaF, 1% Triton X-100, 0.5% sodium deoxycholate, 3 mM DTT with the addition of Mini protease inhibitors (Roche)). In the cases of immunoprecipitation experiments involving primary mouse embryonic fibroblast or cultured neuronal cells, 1% Triton X-100 was replaced by 0.2% NP-40, 0.5% sodium deoxycholate and 0.1% Triton X-100. Lysates were spun at 16,000 × g, supernatants were precleared by incubation with Protein A/G-agarose for 30′ at 4°C and proteins were immunoprecipitated by incubation in appropriate antibodies followed by protein A/G PLUS-agarose. Pellets were washed 3 times with lysis buffer and then twice with kinase buffer (25 mM HEPES pH 7.6, 20 mM MgCl, 2 mM DTT, 1mM sodium orthovanadate and 20 mM β-glycerophosphate). Complexes were disrupted in Laemmli buffer (Invitrogen) and resolved in 7%, 3–8% Tris-acetate gels or in 10% NuPage gels (Invitrogen). The separated polypeptides were transferred to PVDF membranes (Schleicher and Schuell, Keene, NH), blocked in 5% non-fat milk and immunoblotted with the indicated antibodies. Immunoreactive proteins were visualized by enhanced chemiluminescence (Amersham Pharmacia, Piscataway, NJ) and detected using Kodak Biomax MR film (Kodak Eastman, New Haven, CT).
Synaptic vesicle preparation
All tissues, buffers, and tubes were kept cold (5°C) or on ice during the preparation. Whole brains from 6–8 adult transgenic and non-transgenic PDAPP mice (Hsia, A.Y. et al., 1999; Mucke, L. et al., 2000) were diced and then homogenized 25 strokes in a 20 ml Potter Elvehjem (PE) homogenizer in “Hg” buffer (0.32M sucrose, 150mM NaCl, 5mM Hepes, pH 7.4,; 1mM EDTA) containing 2 tablets of Complete Protease Inhibitor (Roche) at a ratio of 10 ml of buffer per 3 brains. Homogenates were centrifuged at 1000 × g for 10 min and supernatants only were centrifuged over a 2M sucrose, 5mM Hepes, pH 7.4 “cushion” in 12 ml Ultraclear tubes (Beckman) at 17.3K × g for 10 min in an SW41 Ti Beckman rotor. The interface was resuspended in Hg buffer, and the 17.3K × g centrifugation repeated. The interface was resuspended in “GB” 5mM Hepes, 150mM NaCl, pH 7.4 to a volume of 15 ml and 2.5 ml of this was layered on top of (6) stepped gradients comprised of 2 ml 2M sucrose, 2.5 ml 1.2M sucrose, 2.5 ml 1.0M sucrose, and 2.5 ml 1.85M sucrose, all in GB. The gradients were centrifuged at 82.5K × g for 2 hours with slow acceleration and brake.
Synaptosomes were enriched in interfaces between 1.2M and 1.0M sucrose as well as 1.0M and 0.85M sucrose. These two fractions were combined and resuspended in GB to 23 mls, split to 4 polyallomer (Beckman) 12 ml tubes and centrifuged at 100K × g for 30 min. To rupture synaptosomes by osmotic shock, thereby releasing synaptic vesicles (SV), pellets were taken one at a time, resuspended in 25 ml ice-cold Milli-Q (Millipore) filtered H2O in a 25 ml PE homogenizer using 30 strokes; to restore buffered conditions, samples were transferred to a beaker on ice to which 2.5 ml 50mM Hepes, 1.5M NaCl, pH 7.4 was added. The ruptured synaptosomes were left to sit on ice for 30 min. To remove synaptosome membranes and mitochondria, these samples were centrifuged 11 ml at a time over a 2M sucrose cushion at 25K × g for 20 min. Free SV are in the supernatant.
To concentrate the SV, the supernatants were centrifuged over a 2M sucrose cushion at 150K × g for 2 h. The enriched SV pellet was diluted in GB to 15 mls and 2 ml were layered on top of a gradient comprised of 2ml 2M, 2ml 1.2M,2ml 0.8M, 2ml 0.6M, and 2ml 0.4M sucrose and centrifuged at 64.7K × g for 4 h, with slow acceleration and brake. All interfaces were collected and resuspended in GB and centrifuged at 38K × g for 1 h. The interface between 0.8 and 0.6M sucrose contained the most SV, with some at the 0.6M and 0.4M interface. Purity of the preparation was tested by western blotting using a variety of SV markers, including synaptophysin and synaptobrevin, which showed a dramatic enrichment of SV and the absence of Cytochrome C, a marker for mitochondria.
Immunohistochemistry
Primary neurons were grown on 8-well polylysine-treated chamber slides and treated as indicated. Cells were then fixed by immersion in −20°C methanol for 20 min and blocked in 10% normal donkey serum (Jackson ImmunoResearch Laboratories) and 1% bovine serum albumin (BSA) in 1 X PBS for 1 hour at room temperature. Cells were then incubated in Goat anti-ASK1 (N-19) and rabbit anti-IGF-IR (C-20) in 1% BSA for 1 hour at room temperature, washed three times with PBS and incubated with FITC conjugated, donkey anti-goat-and Texas Red conjugated donkey anti-rabbit antibodies for 1 hour at room temperature. Cells were then washed extensively (8–10 times, 15 min) with PBS at room temperature and mounted in TOTO-3 iodide (642/660). Single confocal images or z-stacks of images were acquired using a Nikon Eclipse-800 microscope and collected using Compix Simple PCI software (Compix Inc., Sewickley, PA). Where indicated, deconvolution was done using a maximum likelihood estimation algorithm with Huygens software and then processed by the Imaris imaging interphase (Bitplane AG, Zurich, Switzerland). All single confocal images and stacks of images were then processed in a SGI Octane R12 computer running Bitplane’s Advanced Imaging Software suite. Analysis of colocalization was done by obtaining Pearson correlation coefficients of colocalized material in the regions of interest considered (c) using the Coloc algorithm of Imaris (Bitplane AG) (Costes, S.V. et al., 2004), which was used as a measure of the degree of colocalization of the signals in each channel.
RESULTS
APP and ASK1 are found in a complex in vitro
A protein complex that includes the adaptor JIP1b (Matsuda, S. et al., 2001; Scheinfeld, M.H. et al., 2002; Inomata, H. et al., 2003) as well as JNK1 (Matsuda, S. et al., 2001) is assembled at the extreme C-terminus of APP in a manner dependent on its YENPTY motif (Matsuda, S. et al., 2001). Since ASK1 is a MAP kinase kinase kinase (MAPKKK) whose substrates, MKK6 and MKK7, phosphorylate the JNK1 and p38 effector stress-activated kinases (Ichijo, H., 1999), we hypothesized that ASK1 might be associated with a signaling complex assembled at the C-terminus of APP. To test this hypothesis, we transiently expressed APP and influenza virus hemagglutinin epitope (HA)-tagged ASK1 (ASK1-HA) in HEK293 cells and performed immunoprecipitation experiments. As we reported previously (Peel, A.L. et al., 2004), ASK1-HA immunoreactive material was present in complexes immunoprecipitated with APP-specific antibodies (Figure 1a). The reciprocal was also true (Figure 1b). Moreover, the complexes immunoprecipitated by anti-APP antibodies also contained the phosphorylated (active) form of MKK6, and a protein band that was reactive for an antibody that recognizes the two JIP family members, JIP1 and JIP2 (Figure 1c). The size of the band recognized by the antibody was consistent with the expected size for human JIP1 (~80 KDa). Thus, consistent with our previous observations (Peel, A.L. et al., 2004), ASK1 was present in protein complexes immunoprecipitated with anti-APP antibodies from lysates of HEK293 cells overexpressing ASK1 and APP. In addition, as reported previously (Matsuda, S. et al., 2001; Scheinfeld, M.H. et al., 2002; Inomata, H. et al., 2003; Peel, A.L. et al., 2004), the complex precipitated with APP antibodies contained the active form of MKK6 and the adaptor JIP1. To determine whether endogenous APP and ASK1 associate, we performed reciprocal immunoprecipitation studies using lysates from mouse primary mouse neurons and fibroblasts. Complexes immunoprecipitated with an anti-ASK1 antibody from primary neurons and fibroblasts contained APP-immunoreactive material (Figure 1d). Notably, most of the APP immunoreactivity detected in total lysates from primary mouse neurons was found in association with ASK1. In contrast, only a small fraction of the total APP present in mouse primary fibroblasts was found in ASK1-containing complexes (Figure 1 d). These results indicate that endogenous ASK1 and APP are present in protein complexes in mouse neurons and fibroblasts, and suggest that this complex may also contain activated MKK6 and the adaptor JIP1.
Since the interaction of the adaptor JIP1 with the cytoplasmic domain of APP (residues 649–695) is dependent on the GYENPTY motif embedded in the last 31 amino acids of APP a toxic peptide that is cleaved off the APP precursor by caspases (Gervais, F.G. et al., 1999; Lu, D.C. et al., 2000; Galvan, V. et al., 2002) we sought to determine whether the interaction of ASK1 with APP would require these sequences in APP by overexpressing a truncated form of APP in which the last 31 amino acids of its cytoplasmic domain had been deleted (APPΔC31, (Lu, D.C. et al., 2000)) together with full-length ASK1 in HEK293 cells. Deletion of the C-terminal 31 amino acids of APP, which include the GYENPTY motif, did not abolish ASK1 binding (Figure 2a). We then sought to determine whether sequences of the APP cytoplasmic domain N-terminal to Asp664 (the last amino acid in the APPΔC31 construct) would be involved in ASK1 binding. To this aim, we overexpressed the cytoplasmic domain of APP from a construct in which the APP signal sequence was placed directly N-terminal to the first residue of the Aβ1–42 sequence, which lies 28 residues N-terminal to the first residue of the transmembrane domain, thus encompassing the complete transmembrane and cytoplasmic domains of APP (APP-C100, (Lu, D.C. et al., 2000)) in HEK293 cells. The presence of the native APP signal sequence and its complete transmembrane domain in this construct insures that the truncation is correctly sorted during synthesis and is presented in the ER- and plasma membrane-associated domains. As shown in Figure 2 b and c, complexes immunoprecipitated from APP-C100-overexpressing HEK293 cells by CT15, an antibody that recognizes the last 15 amino acids of APP (Sisodia, S.S. et al., 1993) contained ASK1. Thus, taken together, these observations indicate that residues 597–664 in APP cytoplasmic domain are sufficient to mediate binding to ASK1. A C-terminal truncation of C100 (deleting the motif required for JIP1 binding to APP) could not be used in immunoprecipitation experiments due to the paucity of antibodies recognizing the C-terminal domain of APP at sequences N-terminal to its last amino acids. The only antibody recognizing sequences C-terminal to Asp664 available to us, α-I (Selkoe, D.J. et al., 1988) was not suitable for use in immunoprecipitation experiments in our hands (data not shown).
ASK1 contains a C-terminal serine/threonine protein kinase domain (residues 680 to 938) and a regulatory (inhibitory) N-terminal domain (Nishitoh, H. et al., 1998). To determine whether the N-terminal regulatory domain of ASK1 is required for its interaction with the cytoplasmic domain of APP we performed immunoprecipitation experiments in lysates of HEK293 using constructs in which the N-terminal domain of ASK1 (residues 1–648) had been deleted (ASK1ΔN, (Nishitoh, H. et al., 1998)). The C-terminal domain of ASK1 is a constitutively active form of the kinase (Nishitoh, H. et al., 1998). As shown in Figure 2b and c, deletion of the N-terminal domain of APP did not affect its ability to associate with APP. The N-terminal domain of ASK1 is therefore not required for its interaction with APP, suggesting that the catalytic C-terminal domain of ASK1 is sufficient to mediate the interaction of ASK1 the cytoplasmic domain of APP.
We next determined whether the kinase activity of ASK1 was required for its interaction with APP. The interaction of ASK1 kinase-dead mutants (ASK1KM, (Nishitoh, H. et al., 1998) with APP was examined in lysates of HEK293 cells transiently overexpressing these proteins. HA-specific immunoreactive material was significantly decreased in complexes immunoprecipitated with anti-APP antibodies (Figure 2b and c), suggesting that the kinase activity of ASK1 is required for its association with APP-containing protein complexes.
APP and ASK1 interact in intact neurons in vitro
Immunoprecipitation experiments involve the disruption of cellular compartments. Thus, protein-protein interactions detected using immunoprecipitation can in some cases be artifactual, since cellular compartmentalization may preclude such interactions in vivo. Therefore, to determine whether ASK1 and APP associate in intact cells, we performed immunohistochemical studies of primary neuronal cultures using anti-APP and anti-ASK1 antibodies. While only a small fraction of the total APP and ASK1 immunoreactivity colocalized in vesicle-like structures in primary neurons maintained in complete media, brief (10-minute) removal of trophic factors (B27 supplement as described in Methods) from the culture media induced an apparent increase in the abundance of both APP and ASK1 in neuronal bodies, and the marked relocalization of both proteins to neuronal projections (Figure 3a). These results suggest that the association of ASK1 in a complex containing APP may be enhanced by cellular stress (as in this case by trophic factor removal) and that these APP-ASK1 complexes are transported to neuronal projections under conditions of cellular stress.
To determine whether removal of serum from the culture media would also affect the association of APP and ASK1 in HEK293 cells transiently over-expressing the proteins, we immunoprecipitated HA-ASK1 from lysates of 293 cells that over-expressed HA-ASK1 and APP and had been cultured in the presence or absence of serum for 2 hours. Since we aimed to examine the interaction between extremely abundant overexpressed proteins 13 h after transfection, we allowed for 2 hours of stimulation of 293 cells by serum deprivation to maximize the number of protein complexes recovered. In contrast to our observations in intact primary neuronal cultures, we found no difference in the amount of APP-immunoreactive material present in complexes immunoprecipitated with HA-ASK1 from lysates of HEK293 cells in the presence or absence of serum (Figure 3b). These observations suggest that the increased association of APP and ASK1 observed in trophic factor-deprived primary cultures might require the presence of intact cellular compartments, and that these compartments may be membrane-associated. Alternatively, it is possible that transiently over-expressed APP and ASK1 are not subject to the same regulatory processes as their endogenous counterparts, conceivably due to their abundance. Thus, stress induced by growth factor removal can induce the formation of complexes containing endogenous APP and ASK1 in primary neuronal cultures, but not in HEK293 cells transiently over-expressing both proteins.
ASK1 is upregulated in neuronal projections in brains of APP transgenic mice
Since the formation of APP and ASK1-containing complexes was strongly regulated by trophic factor removal-induced stress in primary neuronal cultures, we sought to determine whether the location or the levels of endogenous ASK1 would be affected in brains of APP transgenic mice that constitute a well-defined animal model of AD(Hsia, A.Y. et al., 1999; Mucke, L. et al., 2000). Comparison of ASK1-specific immunoreactivity in hippocampal sections from brains of non-transgenic and transgenic PDAPP mice revealed a pronounced upregulation of endogenous ASK1 in neuronal bodies and projections in the granular layer of CA1 in brains of APP transgenic mice (Figure 4a). Thus, consistent with our observations in primary neuronal cultures, endogenous ASK1 was upregulated in neuronal projections in brains of APP transgenic mice.
APP and ASK1 colocalize in brains of APP transgenic mice
Since ASK1 was upregulated in brains from APP transgenic mice, we asked whether ASK1 would be found in association with APP both in APP-transgenic and in non-transgenic mouse brains. Three-dimensional reconstruction of confocal z-stacks collected from CA1 of non-transgenic and APP-transgenic brains revealed that APP- and ASK1-specific immunoreactivity colocalized in punctate structures both in neuronal bodies and in neuronal projections (Figure 4b). In non-transgenic animals the interaction was mostly restricted to punctations in neuronal bodies, while in APP transgenic mouse brains APP and ASK1 were also prominently present and colocalized in neuronal projections and in fibers (Figure 4b). The Pearson correlation coefficient of colocalized material in the region of interest (ROI) considered (c, (Costes, S.V. et al., 2004)) was obtained using Imaris (Bitplane, Switzerland) and used as a measure of the degree of colocalization of the signals in each channel. Quantitation of c for the colocalization of APP and ASK1 immunoreactive signals revealed that the two signals colocalized to a high degree (c= 0.58). These observations indicate that APP and ASK1 were associated in complexes both in non-transgenic and in APP-transgenic mouse brains. Moreover, the ASK1-immunoreactive material increased in neuronal projections in brains of APP transgenic mice was associated with APP.
APP, JNK1 and MKK6 are associated with synaptic vesicles
APP is a transmembrane protein that is transported anterogradely from its site of synthesis in the neuronal soma to synaptic sites (Koo, E.H. et al., 1990; Sisodia, S.S. et al., 1993; Buxbaum, J.D. et al., 1998). Since some of the punctate structures stained by anti-APP and anti-ASK1 antibodies in mouse brains were reminiscent of synaptic boutons in size and shape, we purified synaptic vesicles from non-transgenic and APP transgenic mouse brains and probed the preparations with synaptophysin, JNK1, MKK6 and APP-specific antibodies. Synaptic vesicle fractions from both non-transgenic and APP-transgenic mice contained high amounts of synaptophysin, a canonical synaptic protein (Figure 5). Consistent with the punctate distribution of APP and ASK1 immunoreactivity in mouse brain sections and with previously published data (Sapirstein, V.S. et al., 1994; Marquez-Sterling, N.R. et al., 1997), we found that APP was enriched in preparations of synaptic vesicles from both non-transgenic and APP-transgenic brains (Figure 5). Also, phosphorylated MKK6 was abundant in synaptic vesicles of both non-transgenic and APP-transgenic mice, while phosphorylated JNK1 was prominent in synaptic vesicle preparations from APP-transgenic, but not from non-transgenic, mouse brains (Figure 5). The evidence for the presence of ASK1-specific immunoreactivity in synaptic preparations from mouse brains, however, was ambiguous, so whether ASK1 is associated with synaptic vesicle preparations could not be determined.
APP and ASK1 interact in neuronal ER in mouse brains
APP, like other transmembrane proteins, is folded in the endoplasmic reticulum (ER) compartment. In addition, APP undergoes extensive glycosylation in the Golgi (Georgopoulou, N. et al., 2001). ASK1 has been implicated in the transduction of stress signals emanating from the ER in conditions of cellular stress as part of the unfolded protein response (Nishitoh, H. et al., 2002). Since most of the APP- and ASK1-immunoreactive signals in non-transgenic mouse brains were found colocalized in a compartment juxtaposed to the nucleus in bodies of neurons, we next sought to determine whether APP and ASK1 associate in the neuronal ER in mouse brains. Examination of high-magnification confocal z-stacks collected from mouse brain sections stained with anti-ASK1 and anti-APP antibodies revealed that a large fraction of total ASK1 and APP immunoreactivity colocalize in a compartment juxtaposed to the nucleus in cells of non-transgenic mouse brains (Figure 6a–d). Quantitation of c (Costes, S.V. et al., 2004) for the colocalization of APP and ASK1 immunoreactive signals in this compartment revealed that the two signals colocalized to a high degree (c=0.42). To determine whether APP and ASK1 are present in the ER, we stained contiguous brain sections with an APP-specific antibody together with an antibody specific for calreticulin, an ER-resident protein. APP-specific immunoreactivity in cellular bodies was present in the same compartment stained by a calreticulin-specific antibody, confirming that APP is localized to the ER in neuronal bodies in non-transgenic mouse brains (Figure 6e). The presence of ASK1 immunoreactivity in the calreticulin-containing compartment could not be directly verified since both primary antibodies had been raised in the same host. Antibodies to other ER-specific proteins such as PDI or BiP that were available to us did not stain mouse brain sections. However, the observations that (i) APP and ASK1 colocalize in a compartment juxtaposed to the cell nucleus in mouse brain sections and (ii) APP and calreticulin colocalize in the same compartment in adjacent brain sections provide indirect evidence that ASK1 and APP interact in a calreticulin-containing compartment, likely ER, of neurons in mouse brains.
DISCUSSION
In a previous report, we described the interaction of APP and MKK6, and of APP and ASK1, in transiently transfected cells (Peel, A.L. et al., 2004). In the present study, we have extended our previous observations and verified that the interaction of endogenous APP and ASK1 occurs in primary neuronal cultures (Figure 3) and in mouse brains (Figure 4 and 6). ASK1 is a MAPKKK that is involved in the transduction of the response to cellular stress through the activation of the MAPKKs MKK6, 7, 4 and 3 to the effector SAPKs JNK and p38. ASK1 is strongly stimulated by oxidative stress (Matsukawa, J. et al., 2004), and is required for the sustained activation of the UPR induced by protein aggregation (Nishitoh, H. et al., 2002) and by glucose starvation (Song, J.J. and Lee, Y.J., 2005a). ASK1 has an important role in transducing apoptosis signals and may play a crucial role in neuronal cell death in neurodegeneration (Sekine, Y. et al., 2006) but also in the regulation of immune responses (Hayakawa, T. et al., 2006). Moreover, it was shown that dimerization of a fusion of the EGFR and the cytoplasmic domain of APP by EGF strongly induced cell death in a continuous neuronal cell line, and that this response was dependent on kinase-active ASK1 and could be inhibited by specific JNK inhibitors (Hashimoto, Y. et al., 2003). Interestingly, in these studies the C-terminus of APP was shown to serve as a scaffold for the assembly of ternary complexes containing the intracellular domain of APP, JIP1b and ASK1, but in contrast to our findings, the interaction between ASK1 and APP depended on JIP1b. The observation that brief removal of trophic factors from the culture media in primary neuronal cultures (Figure 3) or the expression of a human APP transgene in vivo (Figure 4) resulted in the upregulation of ASK1 at neuronal projections where it was found in a complex with APP (Figures 3 and 4), together with the observation that the APP-ASK1 complex also contained the adaptor protein JIP1 and activated MKK6 in cultured cells (Figure 1), and activated JNK1 in mouse synaptic vesicles (Figure 5) suggests that ASK1 may be an apical MAPKKK in a signal transduction cascade activated by stress due to trophic factor deprivation or to the overexpression of a FAD-mutant human APP transgene in mouse brains. In non-stressed conditions, such as in primary neurons maintained in defined media (Figure 3) or in non-transgenic mouse brains (Figure 4b), the interaction of APP and ASK1 was mainly restricted to a perinuclear compartment, likely ER, of neuronal cells.
To define the region of APP that mediates its interaction with ASK1-containing complexes, we used a construct in which the last 31 amino acids of APP are deleted (APPΔC31) and a construct in which APP’s signal sequence is placed directly N-terminal to the transmembrane domain of APP (amino acids 625–648) and is followed by its cytoplasmic domain, to ensure the correct sorting and presentation of the truncated protein in association with the ER and plasma membranes. Transient overexpression experiments using these truncated forms of APP showed that the region in the C-terminal domain of APP that was required for interaction with ASK1-containing complexes encompassed the fragment 597–664 (Figures 1 and 2), of which amino acids 625–648 constitute the transmembrane domain. Taken together, our results indicate that the first 16 residues (649–664) of the cytoplasmic domain of APP are sufficient to mediate the formation of ASK1-containing complexes. Interestingly, the motif required for interaction of the JIP1 adaptor with the cytoplasmic domain of APP (GYENPTY), which is contained in the last 31 amino acids of APP, was not required for the interaction of APP with ASK1 (Figure 1), arguing that JIP1 binding may not be required for the assembly of ASK1 in a complex with the cytoplasmic domain of APP. Since it has been shown that JIP1b mediates the assembly of a ternary complex comprising the intracellular domain of APP, JIP1b and ASK1 (Hashimoto, Y. et al., 2003), our results suggest that additional protein-protein contact(s) may occur between the sequences immediately N-terminal to the JIP1b binding site on the APP cytoplasmic domain and ASK1.
The present studies expand the current knowledge about the proteins that mediate signaling from APP, and are consistent with a proposed function of APP at synaptic sites (Koo, E.H. et al., 1990; Sisodia, S.S. et al., 1993; Buxbaum, J.D. et al., 1998). Moreover, our results provide in vivo evidence that lends further support to the hypothesis that APP has a role in activating intracellular signaling cascades, possibly through ligand binding (McLoughlin, D.M. and Miller, C.C., 1996; Kimberly, W.T. et al., 2001; Matsuda, S. et al., 2001 Scheinfeld, M.H. et al., 2002; Minogue, A.M. et al., 2003 Sabo, S.L. et al., 2003). Although the ligands for APP have not yet been fully described, it was recently demonstrated that oligomers of APP’s toxic proteolytic product, Aβ, interacts with and oligomerizes APP, leading to complex formation and cleavage at Asp664 (Lu, D.C. et al., 2003a; Lu, D.C. et al., 2003b). Moreover, it was shown that APP mediates a significant component of Aβ toxicity (Lu, D.C. et al., 2003a; Lu, D.C. et al., 2003b; Shaked, G.M. et al., 2006) through its interaction with Aβ. Consistent with a putative role of the APP/Aβ interaction in synaptic function, it was shown that Abeta production is strongly upregulated by synaptic activity and that accumulated Aβ in turn negatively modulates synaptic function (Kamenetz, F. et al., 2003; Cirrito, J.R. et al., 2005). This hypothesis is strongly supported by the studies of Hashimoto et al. that demonstrated that enforced dimerization of the cytoplasmic domain of APP strongly induces ASK1- and JNK-dependent death in cells of neuronal origin (Hashimoto, Y. et al., 2003). Taken together, these observations suggest that APP may play an important role at the synapse, possibly transducing an Aβ-induced toxic signal through the association of its C-terminus with effectors of the SAPK cascade. It is likely, however, that signaling from APP in vivo may also have trophic effects, since a recovery in synaptic number and function and in AD-like deficits was observed in transgenic mice in which the C-terminal sequences required for assembly of signaling complexes were stabilized by a point mutation (Galvan, V. et al., 2006). That APP has a role in cell adhesion (Small, D.H. et al., 1992; Small, D.H. et al., 1994) and migration (Sabo, S.L. et al., 2001, 2003) and may regulate neuronal survival (Araki, W. et al., 1991; Mattson, M.P. et al., 1993; Furukawa, K. et al., 1996; Mattson, M.P., 1997) had been suggested previously. Trophic signals emanating from APP may arise from complexes assembled either with the monomeric form of the molecule, or with oligomers binding to other as-yet unknown ligands. The present study provides additional information regarding the mechanism(s) by which signaling cascades emanating from the C-terminus of APP may have an important role in the regulation of neuronal function.
Acknowledgments
We thank Drs. Alexei Kurakin, Christopher Link, and Anders Olsen for helpful discussions and insights; Dr. Lennart Mucke for the PDAPP transgenic mice and Dr. Edward Koo for valuable reagents. We also thank Molly Susag for revising and editing the manuscript and Marina Ataie, Diba Ataie and Wei Huang for excellent technical assistance. This work was supported in part by National Institutes of Health Grants (AG05131, AG12282, NS33376 and NS45093) and the Joseph Drown Foundation to DB, and the Alzheimer’s Association NIRG-04-1054 and John D. French Alzheimer’s Foundation to VG. VG specially thanks Mrs. Eloise Goodhew Barnett for her support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Araki W, Kitaguchi N, Tokushima Y, Ishii K, Aratake H, Shimohama S, Nakamura S, Kimura J. Trophic effect of beta-amyloid precursor protein on cerebral cortical neurons in culture. Biochem Biophys Res Commun. 1991;181:265–271. doi: 10.1016/s0006-291x(05)81412-3. [DOI] [PubMed] [Google Scholar]
- Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG. Exchange of n-cor corepressor and tip60 coactivator complexes links gene expression by nf-kappab and beta-amyloid precursor protein. Cell. 2002;110:55–67. doi: 10.1016/s0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- Biederer T, Cao X, Sudhof TC, Liu X. Regulation of app-dependent transcription complexes by mint/x11s: Differential functions of mint isoforms. J Neurosci. 2002;22:7340–7351. doi: 10.1523/JNEUROSCI.22-17-07340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredesen DE, Mehlen P, Rabizadeh S. Receptors that mediate cellular dependence. Cell Death Differ. 2005;12:1031–1043. doi: 10.1038/sj.cdd.4401680. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Thinakaran G, Koliatsos V, O’Callahan J, Slunt HH, Price DL, Sisodia SS. Alzheimer amyloid protein precursor in the rat hippocampus: Transport and processing through the perforant path. J Neurosci. 1998;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] active complex of app with fe65 and histone acetyltransferase tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Chen Z, Seimiya H, Naito M, Mashima T, Kizaki A, Dan S, Imaizumi M, Ichijo H, Miyazono K, Tsuruo T. Ask1 mediates apoptotic cell death induced by genotoxic stress. Oncogene. 1999;18:173–180. doi: 10.1038/sj.onc.1202276. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in alzheimer’s disease: Correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- Fiore F, Zambrano N, Minopoli G, Donini V, Duilio A, Russo T. The regions of the fe65 protein homologous to the phosphotyrosine interaction/phosphotyrosine binding domain of shc bind the intracellular domain of the alzheimer’s amyloid precursor protein. J Biol Chem. 1995;270:30853–30856. doi: 10.1074/jbc.270.52.30853. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Barger SW, Blalock EM, Mattson MP. Activation of k+ channels and suppression of neuronal activity by secreted beta-amyloid-precursor protein. Nature. 1996;379:74–78. doi: 10.1038/379074a0. [DOI] [PubMed] [Google Scholar]
- Galvan V, Chen S, Lu D, Logvinova A, Goldsmith P, Koo EH, Bredesen DE. Caspase cleavage of members of the amyloid precursor family of proteins. J Neurochem. 2002;82:283–294. doi: 10.1046/j.1471-4159.2002.00970.x. [DOI] [PubMed] [Google Scholar]
- Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, Carlson E, Sagi SA, Chevallier N, Jin K, Greenberg DA, Bredesen DE. Reversal of alzheimer’s-like pathology and behavior in human app transgenic mice by mutation of asp664. Proc Natl Acad Sci U S A. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgopoulou N, McLaughlin M, McFarlane I, Breen KC. The role of post-translational modification in beta-amyloid precursor protein processing. Biochem Soc Symp. 2001:23–36. doi: 10.1042/bss0670023. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of alzheimer’s amyloid-beta precursor protein and amyloidogenic a beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Kaneko Y, Tsukamoto E, Frankowski H, Kouyama K, Kita Y, Niikura T, Aiso S, Bredesen DE, Matsuoka M, Nishimoto I. Molecular characterization of neurohybrid cell death induced by alzheimer’s amyloid-beta peptides via p75ntr/plaidd. J Neurochem. 2004;90:549–558. doi: 10.1111/j.1471-4159.2004.02513.x. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Chiba T, Tsukamoto E, Kadowaki H, Nishitoh H, Yamagishi Y, Ishizaka M, Yamada M, Nawa M, Terashita K, Aiso S, Ichijo H, Nishimoto I. The cytoplasmic domain of alzheimer’s amyloid-beta protein precursor causes sustained apoptosis signal-regulating kinase 1/c-jun nh2-terminal kinase-mediated neurotoxic signal via dimerization. J Pharmacol Exp Ther. 2003;306:889–902. doi: 10.1124/jpet.103.051383. [DOI] [PubMed] [Google Scholar]
- Hayakawa T, Matsuzawa A, Noguchi T, Takeda K, Ichijo H. The ask1-map kinase pathways in immune and stress responses. Microbes Infect. 2006;8:1098–1107. doi: 10.1016/j.micinf.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Ho CS, Marinescu V, Steinhilb ML, Gaut JR, Turner RS, Stuenkel EL. Synergistic effects of munc18a and x11 proteins on amyloid precursor protein metabolism. J Biol Chem. 2002;277:27021–27028. doi: 10.1074/jbc.M201823200. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Yeh WC, Yao Z, Mak TW, Woodgett JR. Mediation of tnf receptor-associated factor effector functions by apoptosis signal-regulating kinase-1 (ask1) Oncogene. 1999;18:5814–5820. doi: 10.1038/sj.onc.1202975. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in alzheimer’s disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichijo H. From receptors to stress-activated map kinases. Oncogene. 1999;18:6087–6093. doi: 10.1038/sj.onc.1203129. [DOI] [PubMed] [Google Scholar]
- Inomata H, Nakamura Y, Hayakawa A, Takata H, Suzuki T, Miyazawa K, Kitamura N. A scaffold protein jip-1b enhances amyloid precursor protein phosphorylation by jnk and its association with kinesin light chain 1. J Biol Chem. 2003;278:22946–22955. doi: 10.1074/jbc.M212160200. [DOI] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires app. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. App processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kimberly WT, Zheng JB, Guenette SY, Selkoe DJ. The intracellular domain of the beta-amyloid precursor protein is stabilized by fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem. 2001;276:40288–40292. doi: 10.1074/jbc.C100447200. [DOI] [PubMed] [Google Scholar]
- Ko YG, Kang YS, Park H, Seol W, Kim J, Kim T, Park HS, Choi EJ, Kim S. Apoptosis signal-regulating kinase 1 controls the proapoptotic function of death-associated protein (daxx) in the cytoplasm. J Biol Chem. 2001;276:39103–39106. doi: 10.1074/jbc.M105928200. [DOI] [PubMed] [Google Scholar]
- Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo A, Yuan M, Zhang Z, Paganetti PA, Sturchler-Pierrat C, Staufenbiel M, Mautino J, Vigo FS, Sommer B, Yankner BA. Amyloid beta interacts with the amyloid precursor protein: A potential toxic mechanism in alzheimer’s disease. Nat Neurosci. 2000;3:460–464. doi: 10.1038/74833. [DOI] [PubMed] [Google Scholar]
- Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- Lu DC, Shaked GM, Masliah E, Bredesen DE, Koo EH. Amyloid beta protein toxicity mediated by the formation of amyloid-beta protein precursor complexes. Ann Neurol. 2003a;54:781–789. doi: 10.1002/ana.10761. [DOI] [PubMed] [Google Scholar]
- Lu DC, Soriano S, Bredesen DE, Koo EH. Caspase cleavage of the amyloid precursor protein modulates amyloid beta-protein toxicity. J Neurochem. 2003b;87:733–741. doi: 10.1046/j.1471-4159.2003.02059.x. [DOI] [PubMed] [Google Scholar]
- Lyckman AW, Confaloni AM, Thinakaran G, Sisodia SS, Moya KL. Post-translational processing and turnover kinetics of presynaptically targeted amyloid precursor superfamily proteins in the central nervous system. J Biol Chem. 1998;273:11100–11106. doi: 10.1074/jbc.273.18.11100. [DOI] [PubMed] [Google Scholar]
- Marquez-Sterling NR, Lo AC, Sisodia SS, Koo EH. Trafficking of cell-surface beta-amyloid precursor protein: Evidence that a sorting intermediate participates in synaptic vesicle recycling. J Neurosci. 1997;17:140–151. doi: 10.1523/JNEUROSCI.17-01-00140.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E. Mechanisms of synaptic pathology in alzheimer’s disease. J Neural Transm Suppl. 1998;53:147–158. doi: 10.1007/978-3-7091-6467-9_13. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of alzheimer’s disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Yasukawa T, Homma Y, Ito Y, Niikura T, Hiraki T, Hirai S, Ohno S, Kita Y, Kawasumi M, Kouyama K, Yamamoto T, Kyriakis JM, Nishimoto I. C-jun n-terminal kinase (jnk)-interacting protein-1b/islet-brain-1 scaffolds alzheimer’s amyloid precursor protein with jnk. J Neurosci. 2001;21:6597–6607. doi: 10.1523/JNEUROSCI.21-17-06597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa J, Matsuzawa A, Takeda K, Ichijo H. The ask1-map kinase cascades in mammalian stress response. J Biochem (Tokyo) 2004;136:261–265. doi: 10.1093/jb/mvh134. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- McLoughlin DM, Miller CC. The intracellular cytoplasmic domain of the alzheimer’s disease amyloid precursor protein interacts with phosphotyrosine-binding domain proteins in the yeast two-hybrid system. FEBS Lett. 1996;397:197–200. doi: 10.1016/s0014-5793(96)01128-3. [DOI] [PubMed] [Google Scholar]
- Minogue AM, Schmid AW, Fogarty MP, Moore AC, Campbell VA, Herron CE, Lynch MA. Activation of the c-jun n-terminal kinase signaling cascade mediates the effect of amyloid-beta on long term potentiation and cell death in hippocampus: A role for interleukin-1beta? J Biol Chem. 2003;278:27971–27980. doi: 10.1074/jbc.M302530200. [DOI] [PubMed] [Google Scholar]
- Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-jun n-terminal kinase pathway and the induction of fas ligand. J Neurosci. 2001;21:7551–7560. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison DK, Davis RJ. Regulation of map kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muresan Z, Muresan V. Coordinated transport of phosphorylated amyloid-beta precursor protein and c-jun nh2-terminal kinase-interacting protein-1. J Cell Biol. 2005;171:615–625. doi: 10.1083/jcb.200502043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. Jama. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. Ask1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H. Ask1 is essential for jnk/sapk activation by traf2. Mol Cell. 1998;2:389–395. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- Nunan J, Small DH. Proteolytic processing of the amyloid-beta protein precursor of alzheimer’s disease. Essays Biochem. 2002;38:37–49. doi: 10.1042/bse0380037. [DOI] [PubMed] [Google Scholar]
- Peel AL, Sorscher N, Kim JY, Galvan V, Chen S, Bredesen DE. Tau phosphorylation in alzheimer’s disease: Potential involvement of an app-map kinase complex. Neuromolecular Med. 2004;5:205–218. doi: 10.1385/NMM:5:3:205. [DOI] [PubMed] [Google Scholar]
- Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W, Selkoe DJ, Chen X, Stokin GB, Koo EH. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including abeta42. J Biol Chem. 1999;274:18851–18856. doi: 10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The alzheimer amyloid precursor protein (app) and fe65, an app-binding protein, regulate cell movement. J Cell Biol. 2001;153:1403–1414. doi: 10.1083/jcb.153.7.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The amyloid precursor protein and its regulatory protein, fe65, in growth cones and synapses in vitro and in vivo. J Neurosci. 2003;23:5407–5415. doi: 10.1523/JNEUROSCI.23-13-05407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapirstein VS, Durrie R, Berg MJ, Marks N. Amyloid precursor protein is enriched in axolemma and periaxolemmal-myelin and associated clathrin-coated vesicles. J Neurosci Res. 1994;37:348–358. doi: 10.1002/jnr.490370307. [DOI] [PubMed] [Google Scholar]
- Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW. Activation of c-jun n-terminal kinase and p38 in an alzheimer’s disease model is associated with amyloid deposition. J Neurosci. 2002;22:3376–3385. doi: 10.1523/JNEUROSCI.22-09-03376.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinfeld MH, Roncarati R, Vito P, Lopez PA, Abdallah M, D’Adamio L. Jun nh2-terminal kinase (jnk) interacting protein 1 (jip1) binds the cytoplasmic domain of the alzheimer’s beta-amyloid precursor protein (app) J Biol Chem. 2002;277:3767–3775. doi: 10.1074/jbc.M108357200. [DOI] [PubMed] [Google Scholar]
- Schweisguth F. Notch signaling activity. Curr Biol. 2004;14:R129–138. [PubMed] [Google Scholar]
- Sekine Y, Takeda K, Ichijo H. The ask1-map kinase signaling in er stress and neurodegenerative diseases. Curr Mol Med. 2006;6:87–97. doi: 10.2174/156652406775574541. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Podlisny MB, Joachim CL, Vickers EA, Lee G, Fritz LC, Oltersdorf T. Beta-amyloid precursor protein of alzheimer disease occurs as 110- to 135-kilodalton membrane-associated proteins in neural and nonneural tissues. Proc Natl Acad Sci U S A. 1988;85:7341–7345. doi: 10.1073/pnas.85.19.7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked GM, Kummer MP, Lu DC, Galvan V, Bredesen DE, Koo EH. Abeta induces cell death by direct interaction with its cognate extracellular domain on app (app 597–624) Faseb J. 2006;20:1254–1256. doi: 10.1096/fj.05-5032fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodia SS, Koo EH, Hoffman PN, Perry G, Price DL. Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J Neurosci. 1993;13:3136–3142. doi: 10.1523/JNEUROSCI.13-07-03136.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small DH, Nurcombe V, Moir R, Michaelson S, Monard D, Beyreuther K, Masters CL. Association and release of the amyloid protein precursor of alzheimer’s disease from chick brain extracellular matrix. J Neurosci. 1992;12:4143–4150. doi: 10.1523/JNEUROSCI.12-11-04143.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small DH, Nurcombe V, Reed G, Clarris H, Moir R, Beyreuther K, Masters CL. A heparin-binding domain in the amyloid protein precursor of alzheimer’s disease is involved in the regulation of neurite outgrowth. J Neurosci. 1994;14:2117–2127. doi: 10.1523/JNEUROSCI.14-04-02117.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JJ, Lee YJ. Cross-talk between jip3 and jip1 during glucose deprivation: Sek1-jnk2 and akt1 act as mediators. J Biol Chem. 2005a;280:26845–26855. doi: 10.1074/jbc.M502318200. [DOI] [PubMed] [Google Scholar]
- Song JJ, Lee YJ. Dissociation of akt1 from its negative regulator jip1 is mediated through the ask1-mek-jnk signal transduction pathway during metabolic oxidative stress: A negative feedback loop. J Cell Biol. 2005b;170:61–72. doi: 10.1083/jcb.200502070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Kim SY, Hong YM, Jo DG, Lee JY, Shim SM, Chung CW, Seo SJ, Yoo YJ, Koh JY, Lee MC, Yates AJ, Ichijo H, Jung YK. Essential role of e2-25k/hip-2 in mediating amyloid-beta neurotoxicity. Mol Cell. 2003;12:553–563. doi: 10.1016/j.molcel.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Soriano S, Chyung AS, Chen X, Stokin GB, Lee VM, Koo EH. Expression of beta-amyloid precursor protein-cd3gamma chimeras to demonstrate the selective generation of amyloid beta(1–40) and amyloid beta(1–42) peptides within secretory and endocytic compartments. J Biol Chem. 1999;274:32295–32300. doi: 10.1074/jbc.274.45.32295. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Taru H, Iijima K, Hase M, Kirino Y, Yagi Y, Suzuki T. Interaction of alzheimer’s beta -amyloid precursor family proteins with scaffold proteins of the jnk signaling cascade. J Biol Chem. 2002;277:20070–20078. doi: 10.1074/jbc.M108372200. [DOI] [PubMed] [Google Scholar]
- Taru H, Suzuki T. Facilitation of stress-induced phosphorylation of beta-amyloid precursor protein family members by x11-like/mint2 protein. J Biol Chem. 2004;279:21628–21636. doi: 10.1074/jbc.M312007200. [DOI] [PubMed] [Google Scholar]
- Torroja L, Chu H, Kotovsky I, White K. Neuronal overexpression of appl, the drosophila homologue of the amyloid precursor protein (app), disrupts axonal transport. Curr Biol. 1999;9:489–492. doi: 10.1016/s0960-9822(99)80215-2. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in alzheimer’s disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Yang Y, Turner RS, Gaut JR. The chaperone bip/grp78 binds to amyloid precursor protein and decreases abeta40 and abeta42 secretion. J Biol Chem. 1998;273:25552–25555. doi: 10.1074/jbc.273.40.25552. [DOI] [PubMed] [Google Scholar]