

Abstract
We have previously shown that doxorubicin sensitizes prostate cancer cells to TNF-Related Apoptosis Inducing Ligand (TRAIL). Sensitization correlated with decreased expression of the anti-apoptotic protein cFLIPS. The decrease in cFLIPS could not be explained by transcriptional regulation or increased degradation, leading us to focus on translational mechanisms. In this study, we found that doxorubicin caused strong and sustained phosphorylation of elongation factor 2 (EF-2), which interferes with protein elongation. Phosphorylation of EF-2 appeared to occur in a kinase-independent manner. Treatment with hydrogen peroxide recapitulated the events observed after doxorubicin treatment. In addition, cells treated with hydrogen peroxide expressed less XIAP and survivin which, like cFLIPS, are short half-life proteins with an anti-apoptotic function while expression levels of DR5, caspases-8, -9, -3, and Bax are maintained. The doxorubicin-mediated decrease in cFLIPS and XIAP as well as TRAIL-induced apoptosis was prevented by pretreatment with an iron chelator, indicating that expression of these proteins was affected by free radical generation upon interaction of iron with doxorubicin. In conclusion, our data suggest that free radicals can affect the phosphorylation of EF-2 resulting in a net loss of short half-life proteins such as cFLIPS and XIAP, leaving a cell more vulnerable to apoptotic stimuli.
Keywords: apoptosis, doxorubicin, hydrogen peroxide, translation, c-FLIP (cellular FLICE inhibitory protein), elongation factor 2, cancer
Introduction
Doxorubicin (brand named Adriamycin) has been a staple of chemotherapy for decades. Cancer patients who receive doxorubicin treatment can expect multiple side effects which may include hair loss, anemia, nausea, reduced immunity, and general fatigue [1]. In severe cases, doxorubicin side effects may include cardiac toxicity [2, 3]. The mechanism of doxorubicin action has been linked to DNA damage, topoisomerase inhibition, and iron sequestration with subsequent free radical generation [4–7]. Although the DNA damaging aspects of doxorubicin are the most widely accepted explanation of the drug’s effects, both therapeutic and detrimental, the free radical hypothesis has been revisited recently by several groups. When doxorubicin is administered it binds to cellular iron, generating free radicals including hydrogen peroxide through Fenton chemistry [8]. These free radicals are believed to directly degrade the anti-apoptotic protein cFLIP, rendering the treated cells more vulnerable to apoptosis [9]. This free radical mechanism hypothesis is summarized in Figure 1.
Figure 1. Current model of doxorubicin-mediated sensitization to death ligand-induced apoptosis.

This model is a combination of previously proposed mechanisms (9). Generation of reactive oxygen species (ROS) occurs following interaction of doxorubicin with iron, an effect that can be ameliorated by iron chelators or anti-oxidants. ROS directly negatively impact expression levels of cFLIP, an inhibitor of caspase-8 in the apoptotic pathway. Decreased cFLIP expression makes cells vulnerable to death ligand induced apoptosis.
cFLIP is a caspase-8 homolog and has as many as 11 splice variants [10]. Two variants have been well characterized, called cFLIPL and cFLIPS based on the length of the mRNA and protein product. Previously, we have described a link between doxorubicin treatment and the downregulation of the short isoform of cFLIP [11]. cFLIPS is enzymatically inactive and binds to caspase-8 preventing homo-dimer formation and the autocatalytic cleavage step required for caspase activation [12]. By inhibiting the activation of caspase 8, FLIPS plays an important role in preventing death-ligand induced apoptosis.
We have previously demonstrated that upon doxorubicin treatment, prostate carcinoma cells which are resistant to the death ligand TNF Related Apoptosis Inducing Ligand (TRAIL) become TRAIL-sensitive, which correlated with a decrease in cFLIPS levels [11]. In addition, PC3 cells can be sensitized with simple downregulation of cFLIPS through siRNA, suggesting that the myriad other actions of doxorubicin may be superfluous to this sensitization process [13]. Identifying this key protein change, we wished to determine how doxorubicin was accomplishing the downregulation of cFLIPS.
The regulation of cFLIPS by doxorubicin does not appear to be mediated at the levels of transcription or proteasomal degradation [13]. Therefore, in this study we examined the possibility of translational control of cFLIPS. Protein translation consists of several discrete steps, each mediated by a set of specific translation factors. The initiation step of translation involves loading ribosomes with mRNA and the complexing of these loaded ribosomes into aggregates known as polysomes [14]. The elongation step of translation follows the ribosomal loading and is mediated by Elongation Factor 2 (EF-2), which moves the mRNA forward through the ribosome from codon to codon as the nascent protein grows. EF-2 activity is largely determined by its phosphorylation status; phosphorylation deactivates the enzyme and is typically controlled by a dedicated kinase (EF-2 kinase) [15]. However, EF-2 is also vulnerable to oxidative phosphorylation via free radical activity [16].
Normally, phosphorylation of EF-2 occurs when the cell is resource starved or when the cell is undergoing mitosis [17, 18]. The resulting decrease in protein synthesis is an important mechanism that allows the cell to conserve energy or direct energy to other cellular functions. Recent research has demonstrated that free radical treatments, which would presumably force a phosphorylation state of EF-2, result in a significant G2/M phase arrest [19]. Interestingly, doxorubicin treatment also results in a G2/M phase arrest [20]. This correlation suggests that free radical production by doxorubicin may play a role in causing a G2/M arrest although the effect has long been thought to result from doxorubicin-induced DNA damage [21].
In this study, we demonstrate that free radical generation is sufficient to explain doxorubicin-mediated sensitization to TRAIL. We have expanded the current model of doxorubicin-mediated events to include translational regulation and discuss the possible clinical ramifications of the data.
Materials and methods
Cell culture and reagents
The human prostate cancer cell line PC3 was purchased from ATCC and maintained in RPMI 1640 media supplemented with 10% FBS and antibiotic/antimycotic from Gibco, Grand Island, NY, at 37°C with 5% CO2. Doxorubicin, Desferoxamine (DFO) and hydrogen peroxide were obtained from the MUSC pharmacy. KillerTRAIL was purchased from Axxora, San Diego CA.
35S-methionine incorporation
After overnight plating, PC3 cells were treated with increasing concentrations of doxorubicin or 10 μg/ml cycloheximide (as a positive control) for 4–24 hours as indicated in Figure 2. Cells were incubated in methionine-deficient RPMI1640 containing 10% FBS one hour prior to adding 50 μCi of L-[35S]-Methionine (in vivo cell labeling grade) (Amersham, Piscataway, NJ). After a 2 hours pulse, the medium was removed, cells rinsed with PBS and protein prepared in RIPA buffer (1% NP-40, 0.1% SDS and 0.5% deoxycholate in PBS) containing freshly added protease inhibitors (P8340, Sigma, St. Louis, MO). To separate unreacted amino acid from protein products, 10 μl lysate was spotted on Whatman 3mm paper for TCA precipitation. TCA-precipitated 35S-labeled protein was quantitated by scintillation counting. All assays were performed in duplicate.
Figure 2. Doxorubicin inhibits incorporation of S35-methionine in a dose- and time-dependent manner.
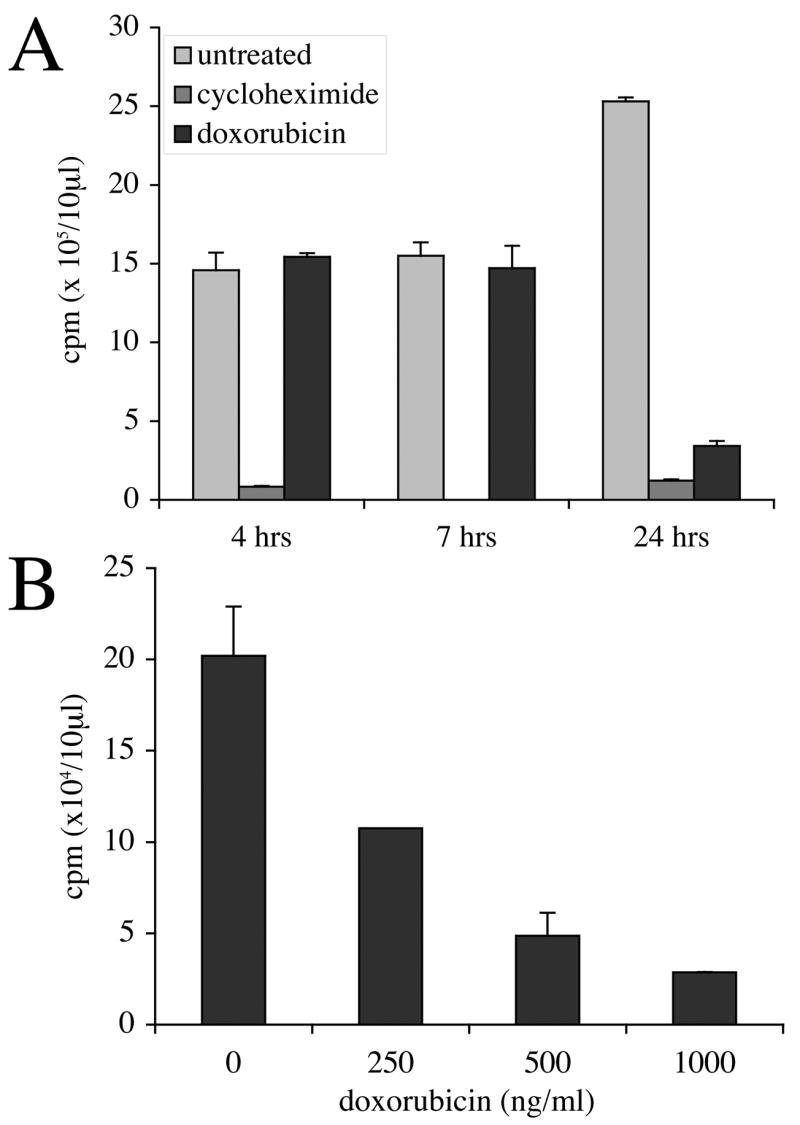
PC3 cells were incubated with 10 μg/ml cycloheximide or 1000 ng/ml doxorubicin (A) or with the indicated concentrations of doxorubicin for 20 hours (B). The incorporation of 35S-methionine into protein was determined as described in the Materials and Methods. Data shown are the mean ± SEM from experiments performed in duplicate.
Polysome profiles
PC3 cells were left untreated or treated with 250 ng/ml doxorubicin for 20 hours and then rinsed three times on ice with ice-cold phosphate-buffered saline to which 100 μg/ml cycloheximide was added to arrest polypeptide chain elongation. Cells were scraped from the plates in 10 ml of phosphate-buffered saline/cycloheximide, pelleted by centrifugation, and resuspended in 1 ml of resuspension buffer (10 mM Tris, pH 7.5, 250 mM KCl, 2 mM MgCl2, 0.5% (v/v) Triton X-100). The resuspended cells were homogenized with 18 strokes of a glass A pestle Dounce homogenizer and transferred to a chilled 1.5-ml microtube. 150 μl of a solution containing 10% (v/v) Tween 80, 5% (w/v) deoxycholate was added, and the homogenate was vortexed and incubated on ice for 15 min. The lysates were then layered on a 15–50% sucrose gradient containing 200mM Tris (pH 7.5), 2.5M KCl, and 100mM MgCl2, and ultracentrifuged at 35000 RPM for 100 minutes at 4°C. Traces were obtained by running the gradients through an ISCO fractionator with upward displacement, set to continuously monitor at 254 nm. Polysome data shown is representative of two independent experiments.
Western Blotting
PC3 cells were plated 5 × 105 per well in 6 well plates for all experiments with the exception of the TRAIL toxicity experiment (Figure 6) in which cells were plated 1.5 × 105. After 24 hours, media was changed and cells were treated as indicated. Following treatment, cells were scraped into media and centrifuged at 1500 rpm for 5 minutes at 4°C. Supernatant was discarded and protein was prepared in RIPA buffer containing freshly added mammalian protease and phosphatase inhibitor cocktails (P8340, P2850, P5726, Sigma, St. Louis, MO). Lysates were then centrifuged for 20 minutes at 13,000 rpm at 4°C and supernatants for western blot analysis stored at −20°C. Protein was separated on 4–12% Bis/Tris NuPage gels in MES buffer, transferred to nitrocellulose for 90 minutes at 30 V and blocked in 5% milk. Sources of antibodies were as follows: Antibodies against cleaved PARP, Bax, XIAP, Survivin, EF-2, phospho-EF-2, and EF-2 kinase were purchased from Cell Signaling Technologies, Danvers, MA. The NF-6 hybridoma supernatant against FLIP was generously provided by Dr. Marcus Peter, University of Chicago. Antibodies against DR5, caspase-8 and caspase-3 were purchased from Axxora, San Diego, CA. Anti-actin was purchased from Sigma. After blocking, membranes were probed with primary antibody overnight at 4°C (1;1000 or 1:2000 in TBS-Tween with 5% milk), except NF-6, which was used at room temperature at 1:5 in TBS-Tween without milk. Following three washes with TBS-Tween, membranes were incubated with the anti-mouse (1:5000) or anti-rabbit (1:50000) HRP-conjugated secondary antibodies (SantaCruz Biotechnologies, Santa Cruz, CA) for 1 hour at room temperature in TBS-Tween with 5% milk. Membranes were washed three times in TBS-Tween followed by chemiluminescent detection of the secondary conjugates with DuraWest Supersignal (Pierce, Rockford, IL). In some cases membranes were stripped with Re-Blot mild solution (Chemicon, Temecula, CA) and reprobed. All Western blot data shown is representative of at least three independent experiments.
Figure 6. DFO pretreatment inhibits sensitization to TRAIL by doxorubicin.
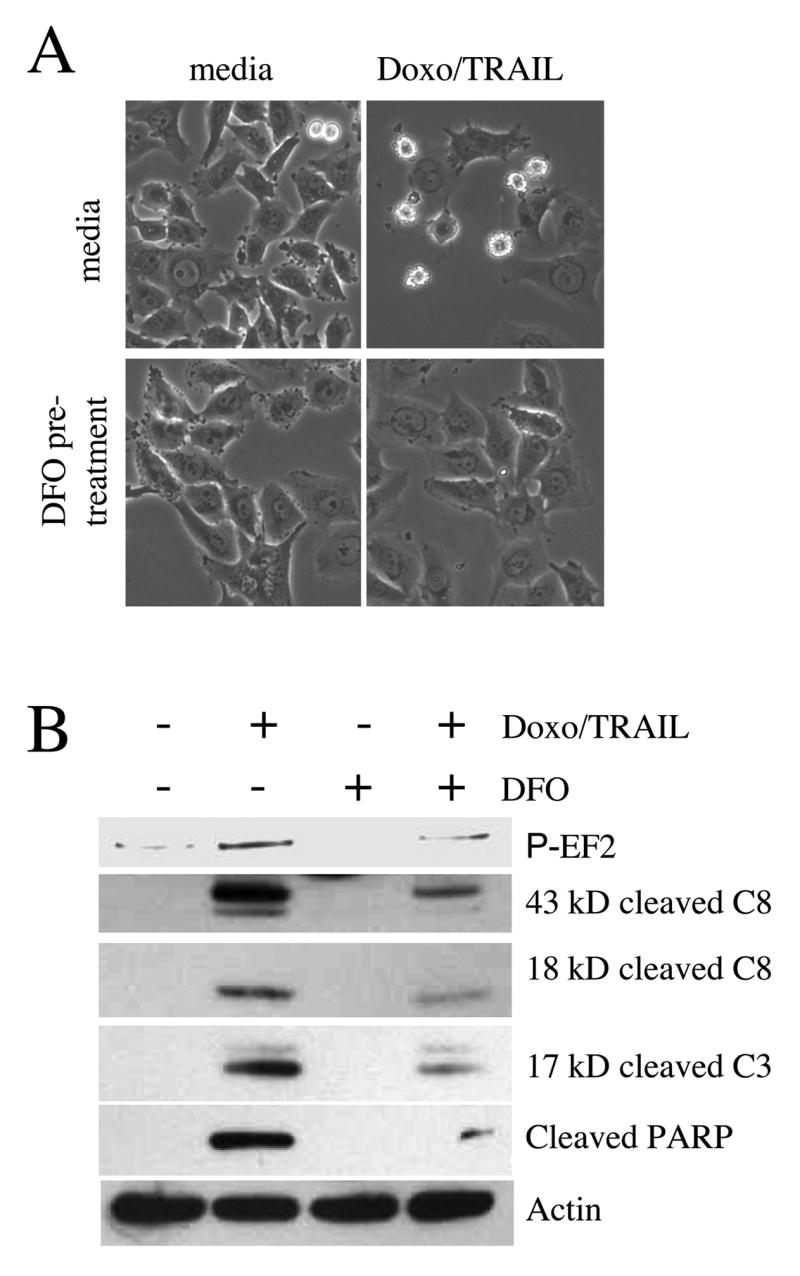
Cells were either treated with 40 μM DFO for three hours or left untreated before the addition of 250 ng/ml doxorubicin for 16 hours were then exposed to two hours of 50 ng/ml TRAIL. (A) Morphological analysis of cells treated as indicated at 200X magnification. (B) Western blot analysis of phospho-EF2, caspase-8 (C8), caspase-3 (C3) and PARP in lysates made from pictured cells. This experiment was repeated with similar results.
Caspase assay
PC3 cells were plated in triplicate at 1 × 104/well in a 96 well plate. Cells were left untreated, treated with 100 ng/ml TRAIL, treated with 25 μM hydrogen peroxide, or a combination of both. Caspase activity was determined at 4 hours using the Apo-ONE homogeneous Caspase-3/7 assay (Promega, Madison WI) according to manufacturer’s instructions. The fluorescence readings were taken on a FluostarOptima plate reader with 485nm excitation and 530nm emission wavelengths (BMG technologies, Durham, NC). Background fluorescence was determined by incubating media with substrate alone and subtracting these values from wells containing cells. Percent increase was determined by dividing the fluorescence intensity from TRAIL treated samples by their respective control either media or hydrogen peroxide alone.
Flow Cytometry
PC3 cells were plated at 3 ×105/well in 6-well plates. Cells were serum starved for 60 hours and then treated with 250 ng/ml doxorubicin or 25 μM hydrogen peroxide in normal growth media. After two hours of treatment, normal growth media replaced the treatment media. The cells were harvested after 24 hours for cell cycle analysis by incubating the cells with Cell Stripper (Mediatech, Inc, Herndon, VA) at 37°C for 5–10 minutes. Cells were then transferred to a 15 ml conical tube containing their spent media and any non-adherent cells and centrifuged at 4°C and 1500 rpm for 5 minutes. Supernatant was discarded and the cells resuspended in 100 μL PBS. 2 mL ice-cold 70% ethanol was added drop-wise to each sample while vortexing and the samples were incubated at 4°C for one hour. Cells were then repelleted and washed with PBS twice. The pellets were resuspended in 100 μL of PBS followed by addition of 100 μL of RNAase (1 mg/mL) and 200 μL of propidium iodide (0.1 mg/ml). Samples were incubated in the dark for 30 minutes before quantification on a FACalibur (Beckton Dickinson, Bedford, MA) in the MUSC flow cytometry core facility. A minimum of 10,000 events was scored for each sample. Data were analyzed with the CellQuest software.
Results
Doxorubicin treatment halts protein synthesis
Our previous study indicated that a doxorubicin dose as low as 250 ng/ml could effectively downregulate cFLIPS but that the effect is not mediated by transcriptional control or by ubiquitination [13]. Therefore, we next investigated translational control. To assess the effect of doxorubicin on protein synthesis, we initially performed S35-methionine incorporation assays. As shown in Figure 2A, unlike our positive control cycloheximide, which nearly completely inhibited incorporation of the radiolabel by four hours, a high dose of doxorubicin (1000 ng/ml) did not globally affect incorporation of 35S-methionine at four or seven hour time points. However, within 24 hours doxorubicin suppressed 35S-methionine incorporation to a similar extent as cycloheximide. Based on morphological analysis and our earlier data demonstrating that exposure of PC3 cells to doxorubicin does not induce apoptosis nor significantly affect viability within 24 hours, reduced incorporation of 35S-methionine was attributed to inhibition of protein synthesis rather than cell death [22, 23]. The lack of doxorubicin-mediated inhibition of 35S-methionine incorporation at the earlier time points suggests that protein synthesis may be affected indirectly as a consequence of other cellular events. A dose response curve demonstrates that 250 ng/ml doxorubicin reduces incorporation of 35S-methione by about half within 20 hours of treatment (Figure 2B). These experiments confirm that treatment of PC3 cells with doxorubicin results in global downregulation of protein synthesis in a dose and time-dependent manner. We hypothesized that cFLIPS, a protein with a reported half-life of about 40 minutes [24], would disappear quickly, giving the impression that it had been preferentially downregulated, when in fact it was merely an early indication of a systemic change. Therefore, we changed the focus of our investigation from cFLIPS-specific translational control to global translational control.
Translation initiation is not inhibited by doxorubicin
To examine initiation of translation, PC3 cells treated with 250 ng/ml doxorubicin or left untreated for 20 hours were harvested and spun through a sucrose gradient for polysome profile analysis. A loss of polysome formation was expected if doxorubicin negatively affected initiation of translation. However, as shown in Figure 3A, traces obtained from polysome analysis of untreated and doxorubicin treated samples were similar, indicating that doxorubicin treatment does not result in loss of polysome formation. Therefore, we chose to focus next on translation elongation.
Figure 3. Doxorubicin-induced inhibition of protein synthesis occurs at the elongation step of translation.
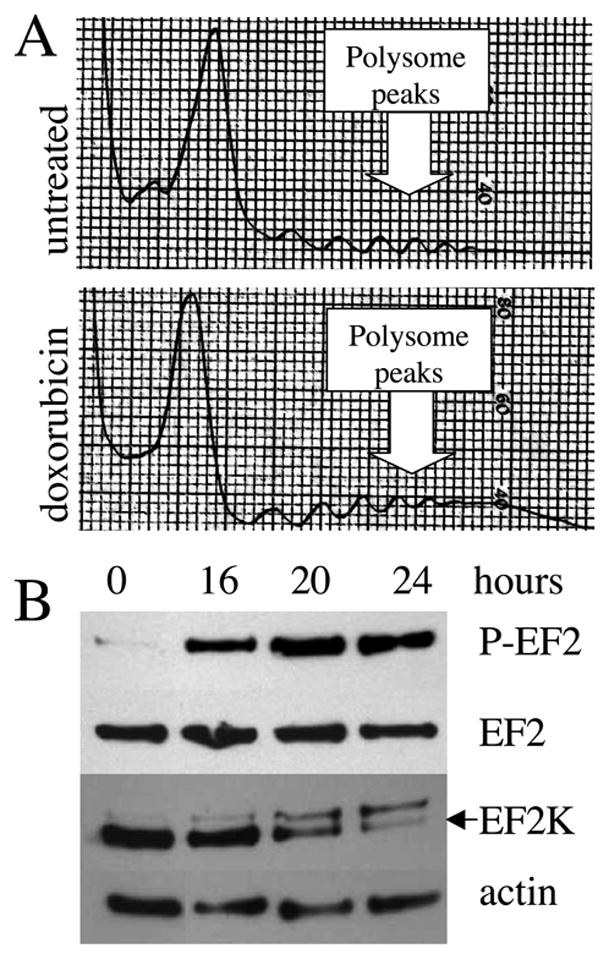
(A) Polysome analysis of untreated cells and cells treated with 250 ng/ml doxorubicin for 20 hours. (B) Western blot analysis of untreated cells and cells treated with 250 ng/ml doxorubicin using antibodies against phosphorylated EF-2 (P-EF-2), total EF-2 (EF-2) and EF-2 kinase (EF-2K). Note: The EF-2-P membrane was stripped and reprobed for EF-2K. The upper band on the EF-2K membrane is residual EF-2-P signal. Actin serves as a loading control.
Translation elongation is inhibited by doxorubicin via phosphorylation of EF-2
In order to investigate the possible effect of doxorubicin on elongation we probed doxorubicin-treated lysates for phosphorylation of EF-2. As shown in Figure 3B, EF-2 was strongly phosphorylated 16 hours after initiation of doxorubicin treatment and remained phosphorylated until 24 hours. We also investigated the effect of doxorubicin on EF-2 kinase, the enzyme responsible for phosphorylation of EF-2. As shown in Figure 3B, the levels of EF-2 kinase are in fact downregulated at the same time that phosphorylation of EF-2 is strongest. EF-2 kinase has a half-life of six hours and expression levels may therefore be quickly affected by inhibition of translation [25]. These data suggested that a kinase-independent mechanism might explain the continued and increased EF-2 phosphorylation induced by doxorubicin.
Free radicals can phosphorylate EF-2 and reduce cFLIPS expression
Oxidative phosphorylation does not require the action of a kinase and free radicals have been shown to phosphorylate EF-2 at relatively high doses [26]. We show here that the free radical source hydrogen peroxide can both phosphorylate EF-2 and downregulate cFLIPS in PC3 cells (Figure 4A). The hydrogen peroxide dose curve indicates that at doses as low as 25 μM, the protein profile of doxorubicin treatment is recapitulated. This profile, as we have shown in previous work [13] and in Figure 3B, is characterized by a reduction in cFLIPS, preservation of cFLIPL and strong phosphorylation of EF-2. We therefore hypothesized that hydrogen peroxide could, like doxorubicin, sensitize PC3 cells to TRAIL. As shown in Figure 4B, microscopic analysis of PC3 cells reveals that combined treatment with hydrogen peroxide and TRAIL results in apoptotic morphology and loss of cells. The apoptotic nature of cell death was confirmed in a caspase-3/7 activity assay. The TRAIL-induced increase in caspase-3/7 activity was nearly three times higher in cells that were co-treated with hydrogen peroxide (Fig. 4C).
Figure 4. Treatment with free radical source hydrogen peroxide mimics the effects of doxorubicin.
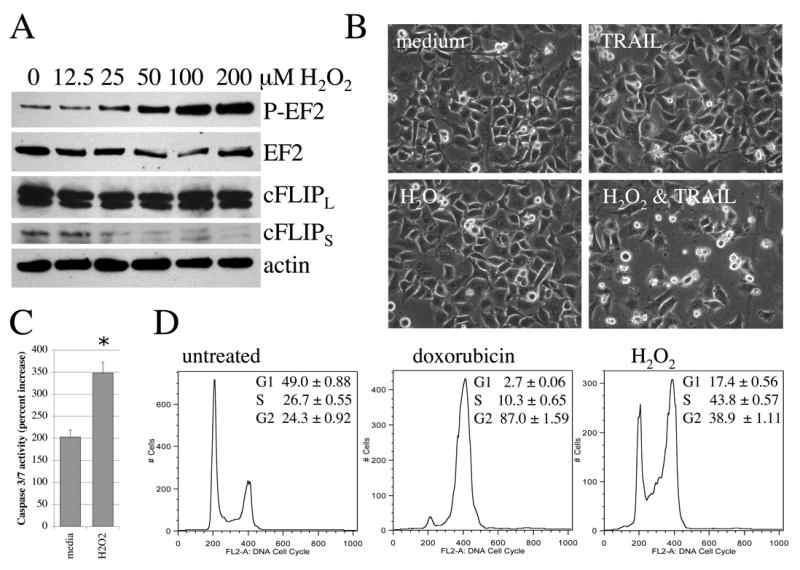
(A) Cells were treated with increasing concentrations of hydrogen peroxide for 4 hours followed by western blot analysis of phosphorylated and total EF-2 and FLIP isoforms. Actin served as a loading control. (B, C) Cells were treated with 25 μM hydrogen peroxide (H2O2), 50 ng/ml TRAIL or both and effects assessed at 4 hours after treatment initiation. (B) Morphological analysis of untreated and treated PC3 cells. Magnification = 200x. (C) Caspase-3/7 activity is shown as an increase in TRAIL-treated samples over their respective controls. *p<0.0001. (D) Following serum starvation for 60 hours, cells were exposed to 25 μM hydrogen peroxide or 250 ng/ml doxorubicin in growth medium for two hours. Cells were incubated an additional 22 hours in growth medium in the absence of treatments and then analyzed by flow cytometry analysis for cell cycle distribution as described in the Materials and Methods. Representative cell cycle distributions are shown. Inserts indicate the mean ± SD of triplicate determinations.
Free radical treatment results in accumulation of cells in G2/M phase of the cell cycle arrest
Doxorubicin treatment results in numerous cellular responses including G2/M arrest and free radical generation [27, 28]. We wished to perform a direct comparison of the effect of 25 μM hydrogen peroxide or 250 ng/ml doxorubicin on cell cycle distribution in order to examine the possible contribution of free radicals to G2/M arrest seen upon doxorubicin treatment. As shown in Figure 4D, after 24 hours of treatment, cells treated with doxorubicin are almost fully arrested in G2/M. Cells treated with hydrogen peroxide have a 60% increase in G2/M and are trending strongly away from G1 phase (Figure 4D).
Hydrogen peroxide treatment creates a pro-apoptotic phenotype
We have shown that treatment with hydrogen peroxide results in phosphorylation of EF-2, decreased levels of cFLIPS and sensitization to TRAIL. Therefore, we wished to determine the effect of hydrogen peroxide treatment on other proteins in the apoptotic pathway. Because several anti-apoptotic proteins have extremely short half-lives, we predicted their expression to decrease upon hydrogen peroxide-induced EF-2 phosphorylation. In contrast, we predicted that pro-apoptotic proteins would be spared due to their typically longer half-lives [29–32]. The TRAIL-induced apoptotic pathway involves several steps including ligand-receptor binding, activation of the initiator caspase-8, Bax oligomerization at the mitochondria, and subsequent apoptosome formation involving activation of caspases-9 and 3. Antagonists of this pathway include cFLIP (caspase-8 inhibition), Bcl-2 (inhibition of Bax oligomerization), XIAP (inhibition of caspases-9 and –3) and survivin (inhibition of caspase-3).
As expected, following treatment with hydrogen peroxide, the protein machinery for apoptosis, including the TRAIL receptor DR5, caspases-8, -9, and -3, and Bax, was preserved (Fig 5A). The anti-apoptotic counterparts of these proteins, however, did not maintain their basal levels upon EF-2 phosphorylation in response to hydrogen peroxide. In addition to cFLIPS, XIAP (half-life of 120 minutes) and survivin (half-life of 30 minutes) disappeared from cells upon treatment [33, 34] (Figure 5a). Bcl-2 and Mcl-1, which antagonize apoptotic proteins at the mitochondrial level, were below detection limits in these cells (data not shown). Thus, caspases in hydrogen peroxide treated PC3 cells are essentially unopposed.
Figure 5. Effects of free radical generation and iron chelation on proteins of the apoptotic pathway.
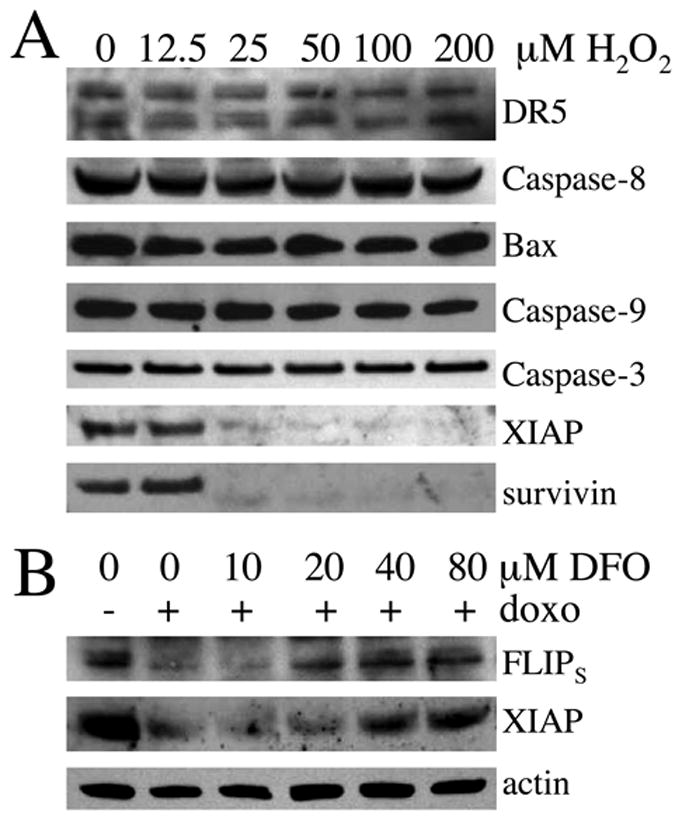
(A) Cells were treated for 4 hours with increasing concentrations of hydrogen peroxide followed by western blotting of the indicated proteins. (B) Cells were pre-treated for 2 hours with increasing concentrations of the iron chelator DFO and then exposed to 250 ng/ml doxorubicin for 24 hours. Actin serves as a loading control.
Pretreatment with the iron chelator DFO antagonizes doxorubicin-induced loss of cFLIPS and XIAP
We next directly tested the idea that doxorubicin downregulates short half-life proteins through free radicals formation following association with intracellular iron (as shown in Figure 1). We pre-treated PC3 cells with increasing doses of the iron chelator desferoxamine (DFO) for two hours before treating with 250 ng/ml doxorubicin for 24 hours. Figure 5B shows the dose-dependent rescue of the anti-apoptotic proteins cFLIPS and XIAP from doxorubicin-induced downregulation.
Pretreatment with DFO protects against doxorubicin-induced TRAIL sensitization
We further pursued the observation that DFO pretreatment protects against the doxorubicin-mediated downregulation of FLIPs and XIAP by testing the TRAIL vulnerability of PC3 cells treated with both DFO and doxorubicin. We have shown previously that doxorubicin pretreatment sensitizes cells to TRAIL (11) and predicted that pretreatment with DFO would have a protective effect due to preservation of anti-apoptotic proteins. To test this hypothesis, cells were pretreated with DFO for three hours, cultured in the presence of doxorubicin for an additional 16 hours and then exposed to TRAIL for 2 hours. As shown in Fig. 6A, DFO pretreatment strongly inhibited the apoptotic response to doxorubicin and TRAIL. Analysis of protein lysates revealed that after only two hours of TRAIL treatment, the doxorubicin treated cells already showed strong cleavage of caspase-8, caspase-3, and PARP. DFO exerted a protective effect on cleavage of these proteins. Importantly, we also confirmed that phosphorylation of EF2 was reduced when an iron chelator precludes doxorubicin interaction with endogenous iron, supporting the hypothesis that protein translation machinery plays an important role in determining the response of a cell to TRAIL by modulating the expression of anti-apoptotic proteins.
Discussion
We show that doxorubicin treatment of PC3 carcinoma cells halts protein translation and that this effect does not occur at initiation but elongation of translation and is mediated by phosphorylation of EF-2. The effect of doxorubicin on EF-2 phosphorylation, loss of cFLIPS and enhanced sensitivity to TRAIL was recapitulated by treatment with hydrogen peroxide. Cell cycle distribution in hydrogen peroxide treated cells also trended towards G2/M arrest. Free radical generation did not only result in reduced levels of cFLIPS but also in the loss of XIAP and survivin, which like cFLIPS have short half-lives and function as antagonists in the apoptotic pathway. Pro-apoptotic proteins including DR5, caspases-8, -9, and –3 as well as Bax were not affected by hydrogen peroxide treatment. Overall these data suggest that free radical generation, phosphorylation of EF-2 and subsequent inhibition of protein elongation results in a preferential loss of short half-life proteins with anti-apoptotic function leaving a cell vulnerable to apoptotic stimuli. Doxorubicin treatment accomplishes this sensitization by generating free radicals upon association with iron, since decreased expression of cFLIPS and XIAP was abrogated by pre-treatment with an iron chelator. Our data suggest that the phosphorylation status of EF-2 is exquisitely sensitive to oxidative stress and inactivation of EF-2 by phosphorylation quickly results in a lowering of defenses against apoptosis. This built-in sensitivity to abnormal free radical abundance may be an important self-diagnostic capability of cells that naturally results in a vulnerable physiological state. Our proposed model of how doxorubicin affects cancer cells is more comprehensive than has been elucidated previously (Figure 6). The inclusion of the EF-2 arm of this pathway opens a new area of inquiry into the role of free radicals in controlling cancer growth.
A rigorous conversation about free radicals, antioxidants, and cancer therapies is well underway and timely. Currently, highly concentrated doxorubicin therapy is sometimes coupled with iron chelation therapy. The iron chelation prevents free radical formation as demonstrated in a variety of studies and confirmed here [3, 9]. The purpose of the dual therapy is to protect cardiac myocytes which are particularly sensitive to free radical damage, perhaps due to a reduced intrinsic ability to process free radicals as compared to other normal cells such as cardiac fibroblasts [35]. The danger inherent in this approach is that the iron chelators may have the potential to be cancer-protective as well as cardiac-protective. Interestingly, our DFO dose curve and TRAIL sensitization studies (Figures 5B and 6) show that a relatively high concentration of DFO is required to protect PC3 cancer cells (at least 40 μM) while cardiac cells are protected by doses as low as 10 μM [36]. These results suggest that there may be a therapeutic window for DFO, which would be important to quantify in order to give patients the maximally effective treatment. Dexrazoxane (DRZ), another iron chelating agent, has been evaluated for cardiac-protective effects when used in combination with ABVE (doxorubicin, bleomycin, vincristine and etoposide) therapy in children with Hodgkin’s disease [37]. The authors found that secondary acute myeloid leukemia and myelodysplastic syndrome occurred more frequently among children in the DRZ arm of the study, which approached but did not reach statistical significance. Two patients in the DRZ group also developed secondary solid tumors earlier than expected. The authors concluded that “the incidence of and the time of onset of the specific leukemias and solid tumors noted after DRZ with doxorubicin and etoposide heightens our concern about using DRZ to reduce long-term cardiopulmonary toxicity in the context of ABVE-based therapy”. Our data suggest a mechanism for this observation that should be considered when designing further studies on amelioration of side effects associated with doxorubicin.
Debate about the role of free radicals in cancer development is ongoing. Our proposed model of doxorubicin’s toxic effect on cancer cells is counter-intuitive to the long-standing belief that free radicals typically cause cancer rather than facilitate eradication of cancer. However, a recent study in Cancer Cell revealed that the ROS-forming compound beta-phenylethyl isothiocyanate, found in vegetables such as broccoli and cabbage, actually destroys cancer in a mouse model through a ROS-mediated mechanism [38]. Another recent mouse model of cancer treatment, which unexpectedly hinged on ROS production is the use of manganese superoxide dismutase (MnSOD) in combination with TRAIL. The MnSOD produces hydrogen peroxide, which was reported to activate caspase-8 and downregulate Bcl-2 [39]. Our data suggest that anti-apoptotic proteins in addition to Bcl-2 may also have been down-regulated by this approach and that caspase-8 activation may have been the result of reduced c-FLIP expression. Furthermore, a group which focused specifically on TRAIL activation of caspase-8 in prostate cancer cells recently published their finding that caspase activation could be blocked by the antioxidants Vitamin C or catalase [40]. Our data suggest the antioxidants may be preventing EF-2 phosphorylation, thereby protecting the cell by preserving anti-apoptotic proteins such as cFLIPS, XIAP, and survivin.
In addition to illuminating a therapeutic mechanism of doxorubicin, our data suggest that EF-2 phosphorylation plays a role in the side effects caused by the drug. For instance, anemia and nausea may be attributed in part to cell cycle arrest in cells, which normally proliferate (blood cells and gut epithelial cells, respectively). Several studies have shown that iron chelators and antioxidants may help to ameliorate such doxorubicin-induced side effects [41–43]. Early work on EF-2 suggested that EF-2 phosphorylation may impact cell cycling by downregulating protein synthesis and thereby clearing the cell of short-half life proteins such as cyclins [17]. The authors of this study state that, “Temporary inhibition of translation may trigger the transition of a cell from one physiologic state into another because of the disappearance of short-lived repressors.” We propose that this mechanism is common between cell cycle modulation and TRAIL sensitization. In this paper we connect phosphorylation of EF-2 by doxorubicin to TRAIL sensitization via changes in the profile of apoptosis-related proteins and also conclude that the phosphorylation of EF-2 by free radicals may contribute to the cell cycle arrest seen upon doxorubicin treatment.
Our model also clarifies some of our previous findings. For instance, we have recently shown that downregulation of cFLIPS was sufficient to sensitize PC3 cells to TRAIL [13]. Because these cells have such low levels of Bcl-2 and Mcl-1, cFLIPS may be the critical determinant of its apoptotic phenotype. In other cell systems, the global downregulation of anti-apoptotic proteins may be required to sensitize cells to TRAIL. We also previously demonstrated that cFLIPS was decreased sufficiently after four hours of doxorubicin treatment to allow TRAIL-induced apoptosis to occur [11]. However, levels of cFLIPS rebounded by 24 hours, suggesting the effect of doxorubicin on inhibition of protein elongation is transient and reversible. Since translational control is an excellent modifier of the protein profile of a cell, we see EF-2 as an interesting new target for cancer researchers.
Figure 7. Proposed model for doxorubicin mechanism of action.

Free radicals are generated upon doxorubicin binding iron resulting in phosphorylation of EF-2 and subsequent inhibition of translation. Short-half life proteins quickly disappear from the cell, leaving it vulnerable to death ligand-induced apoptosis.
Acknowledgments
The authors would like to gratefully acknowledge Drs. David Kurtz and Semyon Rubinchik for their thoughtful comments on the draft manuscript. Thanks also to Rick Peppler for performing the flow cytometry. Dr. Marcus Peter kindly provided the NF-6 antibody. This work was supported by National Institutes of Health (NIH RO1 CA102218).
Abbreviations
- TRAIL
TNF-Related Apoptosis Inducing Ligand
- EF-2
Elongation Factor 2
- cFLIP
cellular FLICE-Like Inhibitor Protein
- XIAP
X-linked Inhibitor of Apoptosis Protein
- DFO
Desferoxamine
- DRZ
Dexrazoxane
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 2.Panjrath GS, Jain D. Monitoring chemotherapy-induced cardiotoxicity: Role of cardiac nuclear imaging. J Nucl Cardiol. 2006;13:415–426. doi: 10.1016/j.nuclcard.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Kalyanaraman B, Joseph J, Kalivendi S, Wang S, Konorev E, Kotamraju S. Doxorubicin-induced apoptosis: Implications in cardiotoxicity. Mol Cell Biochem. 2002;234–235:119–124. [PubMed] [Google Scholar]
- 4.Swift LP, Rephaeli A, Nudelman A, Phillips DR, Cutts SM. Doxorubicin-DNA adducts induce a non-topoisomerase ii-mediated form of cell death. Cancer Res. 2006;66:4863–4871. doi: 10.1158/0008-5472.CAN-05-3410. [DOI] [PubMed] [Google Scholar]
- 5.Seiter K. Toxicity of the topoisomerase II inhibitors. Expert Opin Drug Saf. 2005;4:219–234. doi: 10.1517/14740338.4.2.219. [DOI] [PubMed] [Google Scholar]
- 6.Barnabe N, Zastre JA, Venkataram S, Hasinoff BB. Deferiprone protects against doxorubicin-induced myocyte cytotoxicity. Free Radic Biol Med. 2002;33:66–275. doi: 10.1016/s0891-5849(02)00873-0. [DOI] [PubMed] [Google Scholar]
- 7.Le NT, Richardson DR. Potent iron chelators increase the mrna levels of the universal cyclin-dependent kinase inhibitor p21(cip1/waf1), but paradoxically inhibit its translation: A potential mechanism of cell cycle dysregulation. Carcinogenesis. 2003;24:1045–1058. doi: 10.1093/carcin/bgg042. [DOI] [PubMed] [Google Scholar]
- 8.Wagner BA, Evig CB, Reszka KJ, Buettner GR, Burns CP. Doxorubicin increases intracellular hydrogen peroxide in PC3 prostate cancer cells. Arch Biochem Biophys. 2005;440:181–190. doi: 10.1016/j.abb.2005.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nitobe J, Yamaguchi S, Okuyama M, Nozaki N, Sata M, Miyamoto T, Takeishi Y, Kubota I, Tomoike H. Reactive oxygen species regulate flice inhibitory protein (FLIP) and susceptibility to Fas-mediated apoptosis in cardiac myocytes. Cardiovasc Res. 2003;57:119–128. doi: 10.1016/s0008-6363(02)00646-6. [DOI] [PubMed] [Google Scholar]
- 10.Djerbi M, Darreh-Shori T, Zhivotovsky B, Grandien A. Characterization of the human Flice-inhibitory protein locus and comparison of the anti-apoptotic activity of four different flip isoforms. Scand J Immunol. 2001;54:180–189. doi: 10.1046/j.1365-3083.2001.00941.x. [DOI] [PubMed] [Google Scholar]
- 11.Kelly MM, Hoel BD, Voelkel-Johnson C. Doxorubicin pretreatment sensitizes prostate cancer cell lines to trail induced apoptosis which correlates with the loss of c-FLIP expression. Cancer Biol Ther. 2002;1:520–527. doi: 10.4161/cbt.1.5.169. [DOI] [PubMed] [Google Scholar]
- 12.Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S. Cellular Flice-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem. 2001;276:20633–20640. doi: 10.1074/jbc.M101780200. [DOI] [PubMed] [Google Scholar]
- 13.White SJ, Lu P, Keller GM, Voelkel-Johnson C. Targeting the short form of cFLIP by RNA interference is sufficient to enhance TRAIL sensitivity in PC3 prostate carcinoma cells. Cancer Biol Ther. 2006;5:1618–1623. doi: 10.4161/cbt.5.12.3352. [DOI] [PubMed] [Google Scholar]
- 14.Dever TE. Gene-specific regulation by general translation factors. Cell. 2002;108:545–556. doi: 10.1016/s0092-8674(02)00642-6. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Wang X, Proud CG. Activation of mRNA translation in rat cardiac myocytes by insulin involves multiple rapamycin-sensitive steps. Am J Physiol Heart Circ Physiol. 2000;278:H1056–1068. doi: 10.1152/ajpheart.2000.278.4.H1056. [DOI] [PubMed] [Google Scholar]
- 16.Ayala A, Parrado J, Bougria M, Machado A. Effect of oxidative stress, produced by cumene hydroperoxide, on the various steps of protein synthesis. Modifications of elongation factor-2. J Biol Chem. 1996;271:23105–23110. doi: 10.1074/jbc.271.38.23105. [DOI] [PubMed] [Google Scholar]
- 17.Celis JE, Madsen P, Ryazanov AG. Increased phosphorylation of elongation factor 2 during mitosis in transformed human amnion cells correlates with a decreased rate of protein synthesis. Proc Natl Acad Sci U S A. 1990;87:4231–4235. doi: 10.1073/pnas.87.11.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M. Activation of amp-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol. 2002;12:1419–1423. doi: 10.1016/s0960-9822(02)01077-1. [DOI] [PubMed] [Google Scholar]
- 19.Bataller M, Portugal J. Apoptosis and cell recovery in response to oxidative stress in p53-deficient prostate carcinoma cells. Arch Biochem Biophys. 2005;437:151–158. doi: 10.1016/j.abb.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 20.Siu WY, Yam CH, Poon RY. G1 versus G2 cell cycle arrest after adriamycin-induced damage in mouse swiss3t3 cells. FEBS Lett. 1999;461:299–305. doi: 10.1016/s0014-5793(99)01481-7. [DOI] [PubMed] [Google Scholar]
- 21.Islaih M, Halstead BW, Kadura IA, Li B, Reid-Hubbard JL, Flick L, Altizer JL, Thom Deahl J, Monteith DK, Newton RK, Watson DE. Relationships between genomic, cell cycle, and mutagenic responses of TK6 cells exposed to DNA damaging chemicals. Mutat Res. 2005;578:100–116. doi: 10.1016/j.mrfmmm.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 22.Norris JS, Hyer ML, Voelkel-Johnson C, Lowe SL, Rubinchik S, Dong JY. The use of Fas ligand, TRAIL and Bax in gene therapy of prostate cancer. Curr Gene Ther. 2001;1:123–136. doi: 10.2174/1566523013348977. [DOI] [PubMed] [Google Scholar]
- 23.Voelkel-Johnson C. An antibody against DR4 (TRAIL-R1) in combination with doxorubicin selectively kills malignant but not normal prostate cells. Cancer Biol Ther. 2003;2:283–290. doi: 10.4161/cbt.2.3.398. [DOI] [PubMed] [Google Scholar]
- 24.Poukkula M, Kaunisto A, Hietakangas V, Denessiouk K, Katajamaki T, Johnson MS, Sistonen L, Eriksson JE. Rapid turnover of c-flipshort is determined by its unique C-terminal tail. J Biol Chem. 2005;280:27345–27355. doi: 10.1074/jbc.M504019200. [DOI] [PubMed] [Google Scholar]
- 25.Arora S, Yang JM, Hait WN. Identification of the ubiquitin-proteasome pathway in the regulation of the stability of eukaryotic elongation factor-2 kinase. Cancer Res. 2005;65:3806–3810. doi: 10.1158/0008-5472.CAN-04-4036. [DOI] [PubMed] [Google Scholar]
- 26.Patel J, McLeod LE, Vries RG, Flynn A, Wang X, Proud CG. Cellular stresses profoundly inhibit protein synthesis and modulate the states of phosphorylation of multiple translation factors. Eur J Biochem. 2002;269:3076–3085. doi: 10.1046/j.1432-1033.2002.02992.x. [DOI] [PubMed] [Google Scholar]
- 27.Lee V, Randhawa AK, Singal PK. Adriamycin-induced myocardial dysfunction in vitro is mediated by free radicals. Am J Physiol. 1991;261:H989–995. doi: 10.1152/ajpheart.1991.261.4.H989. [DOI] [PubMed] [Google Scholar]
- 28.Fornari FA, Jr, Jarvis WD, Grant S, Orr MS, Randolph JK, White FK, Mumaw VR, Lovings ET, Freeman RH, Gewirtz DA. Induction of differentiation and growth arrest associated with nascent (nonoligosomal) DNA fragmentation and reduced c-myc expression in MCF-7 human breast tumor cells after continuous exposure to a sublethal concentration of doxorubicin. Cell Growth Differ. 1994;5:723–733. [PubMed] [Google Scholar]
- 29.Tan M, Gallegos JR, Gu Q, Huang Y, Li J, Jin Y, Lu H, Sun Y. Sag/Roc-SCFbeta-TRCP E3 ubiquitin ligase promotes pro-caspase-3 degradation as a mechanism of apoptosis protection. Neoplasia. 2006;8:1042–1054. doi: 10.1593/neo.06568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao X, Deng X, May WS. Cleavage of Bax to p18 Bax accelerates stress-induced apoptosis, and a cathepsin-like protease may rapidly degrade p18 Bax. Blood. 2003;102:2605–2614. doi: 10.1182/blood-2003-01-0211. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Zhu H, Teraishi F, Davis JJ, Guo W, Fan Z, Fang B. Accelerated degradation of caspase-8 protein correlates with trail resistance in a DLD1 human colon cancer cell line. Neoplasia. 2005;7:594–602. doi: 10.1593/neo.04688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nesterov A, Ivashchenko Y, Kraft AS. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) triggers apoptosis in normal prostate epithelial cells. Oncogene. 2002;21:1135–1140. doi: 10.1038/sj.onc.1205151. [DOI] [PubMed] [Google Scholar]
- 33.Dohi T, Okada K, Xia F, Wilford CE, Samuel T, Welsh K, Marusawa H, Zou H, Armstrong R, Matsuzawa S, Salvesen GS, Reed JC, Altieri DC. An IAP-IAP complex inhibits apoptosis. J Biol Chem. 2004;279:34087–34090. doi: 10.1074/jbc.C400236200. [DOI] [PubMed] [Google Scholar]
- 34.Zhao J, Tenev T, Martins LM, Downward J, Lemoine NR. The ubiquitin-proteasome pathway regulates survivin degradation in a cell cycle-dependent manner. J Cell Sci. 2000;113(Pt 23):4363–4371. doi: 10.1242/jcs.113.23.4363. [DOI] [PubMed] [Google Scholar]
- 35.Purdom S, Chen QM. Epidermal growth factor receptor-dependent and -independent pathways in hydrogen peroxide-induced mitogen-activated protein kinase activation in cardiomyocytes and heart fibroblasts. J Pharmacol Exp Ther. 2005;312:1179–1186. doi: 10.1124/jpet.104.077057. [DOI] [PubMed] [Google Scholar]
- 36.Kotamraju S, Chitambar CR, Kalivendi SV, Joseph J, Kalyanaraman B. Transferrin receptor-dependent iron uptake is responsible for doxorubicin-mediated apoptosis in endothelial cells: Role of oxidant-induced iron signaling in apoptosis. J Biol Chem. 2002;277:17179–17187. doi: 10.1074/jbc.M111604200. [DOI] [PubMed] [Google Scholar]
- 37.Tebbi CK, London WB, Friedman D, Villaluna D, De Alarcon PA, Constine LS, Mendenhall NP, Sposto R, Chauvenet A, Schwartz CL. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric hodgkin's disease. J Clin Oncol. 2007;25:493–500. doi: 10.1200/JCO.2005.02.3879. [DOI] [PubMed] [Google Scholar]
- 38.Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Gu J, Zhao L, He L, Qian W, Wang J, Wang Y, Qian Q, Qian C, Wu J, Liu XY. Complete elimination of colorectal tumor xenograft by combined manganese superoxide dismutase with tumor necrosis factor-related apoptosis-inducing ligand gene virotherapy. Cancer Res. 2006;66:4291–4298. doi: 10.1158/0008-5472.CAN-05-1834. [DOI] [PubMed] [Google Scholar]
- 40.Perez-Cruz I, Carcamo JM, Golde DW. Caspase-8 dependent trail-induced apoptosis in cancer cell lines is inhibited by vitamin c and catalase. Apoptosis. 2007;12:225–234. doi: 10.1007/s10495-006-0475-0. [DOI] [PubMed] [Google Scholar]
- 41.Naganuma A, Satoh M, Imura N. Specific reduction of toxic side effects of adriamycin by induction of metallothionein in mice. Jpn J Cancer Res. 1988;79:406–411. doi: 10.1111/j.1349-7006.1988.tb01605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Satoh M, Naganuma A, Imura N. Modulation of adriamycin toxicity by tissue-specific induction of metallothionein synthesis in mice. Life Sci. 2000;67:627–634. doi: 10.1016/s0024-3205(00)00667-6. [DOI] [PubMed] [Google Scholar]
- 43.Malarkodi KP, Sivaprasad R, Varalakshmi P. Effect of lipoic acid on the oxidoreductive status of red blood cells in rats subject to oxidative stress by chronic administration of adriamycin. Hum Exp Toxicol. 2004;23:129–135. doi: 10.1191/0960327104ht428oa. [DOI] [PubMed] [Google Scholar]