

SUMMARY
Poxviruses are large DNA viruses that cause human smallpox and molluscum contagiosum in addition to several zoonoses. Here we show that viral mRNA and transcription factors together with cellular RNA binding protein heterodimer G3BP/Caprin-1 (p137), translation initiation factors eIF4E and eIF4G, and ribosomal proteins are concentrated in subdomains of cytoplasmic DNA factories. Coordination of transcription and translation within the factory was demonstrated by localization of a reporter protein and the resolution of individual cyan and yellow factories when cells were co-infected with recombinant vaccinia viruses expressing a core protein fused to cyan or yellow fluorescent protein. Thus, the poxvirus factory is the site of viral RNA and protein synthesis as well as genome replication and virion assembly. In addition to enhancing the efficiency of virus replication, hijacking the translation apparatus likely contributes to the suppression of host protein synthesis facilitating virus take over and global obstruction of host responses.
INTRODUCTION
Poxviruses are large DNA viruses that cause human smallpox and molluscum contagiosum in addition to several zoonoses. They are distinguished from other DNA viruses by replicating exclusively in the cytoplasm (Moss, 2007). Most of what we know about poxvirus replication derives from studies of vaccinia virus (VACV), which was used as the vaccine to eradicate smallpox, a disease caused by a closely related family member. VACV contains nearly 200 genes, which encode proteins that function in disabling host defenses, enabling replication and transcription of the viral genome, and assembling virus particles. The genes can be divided into early, intermediate and late classes based on their time of expression. The multisubunit viral DNA-dependent RNA polymerase and factors needed for the early stage of transcription are packaged within the infectious particle, allowing synthesis of mRNA soon after entry into the cytoplasm, whereas intermediate and late transcription require de novo viral protein synthesis and genome replication. Genome replication and assembly of virus particles occur in cytoplasmic domains often referred to as DNA factories (Cairns, 1960; Dales and Siminovitch, 1961; Harford et al., 1966). Early after infection (< 4 h), many of the DNA factories are bounded by rough endoplasmic reticulum, which becomes dispersed at the start of intermediate or late viral protein synthesis and virus assembly (Tolonen et al., 2001).
Poxviruses employ a variety of specific and global mechanisms to evade host defenses. The former include the synthesis of viral proteins that target mediators of innate immunity such as interferons, tumor necrosis factors, interleukins, complement and chemokines as well as intracellular signal transduction pathways (Seet et al., 2003). On a broader level, poxviruses down regulate the synthesis of many cellular gene products at intermediate and late times. Thus, microarray analyses indicate substantial decreases of most host mRNAs (Brum et al., 2003; Guerra et al., 2003), consistent with earlier data (Rice and Roberts, 1983). Rapid degradation of cellular and viral mRNAs is initiated by a recently discovered poxvirus-encoded decapping enzyme (Parrish et al., 2007; Shors et al., 1999). Augmented translation of viral mRNAs has been suggested from in vitro studies, though a structural explanation of this has not been determined (Bablanian et al., 1991). In the current study, we provide a mechanism for preferential expression of viral genes based on compartmentalization of viral mRNA and transcription factors with cellular translation factors within the virus factory. Furthermore, evidence for viral transcription and translation within the factory was obtained by simultaneously infecting cells with two recombinant poxviruses; individual factories within a cell contained unique protein products of each recombinant demonstrating the origin of a factory from a single viral genome and the linkage of transcription and translation sites. Hijacking elements of the translation apparatus allows the coordination of viral mRNA and protein synthesis at sites of virus assembly and can contribute to the suppression of cellular protein synthesis, facilitating virus take over and global obstruction of host responses.
RESULTS
Localization of RNA in Subdomains of the Viral DNA Factory
Viral DNA factories are commonly visualized by fluorescence microscopy using 4′,6-diamidino-2-phenylindole (DAPI) or Hoechst stain as one or more juxtanuclear structures. Acridine orange, in contrast to these DNA-specific fluorochromes, can be used to visualize RNA as well as DNA (Zelenin, 1999). Acridine orange gives bright green fluorescence bound to DNA in the nucleus and deep red fluorescence bound to RNA in the cytoplasm and nucleoli. When VACV-infected cells were stained with acridine orange, the factories appeared green as expected. However, the DNA staining was not uniform and in some factories there was one large cavity and in others many small ones. Interestingly, these cavities stained red (Figure 1, Row A) suggesting the presence of RNA, since large amounts of single-stranded VACV DNA have not been reported.
Figure 1. Localization of RNA within virus factories.
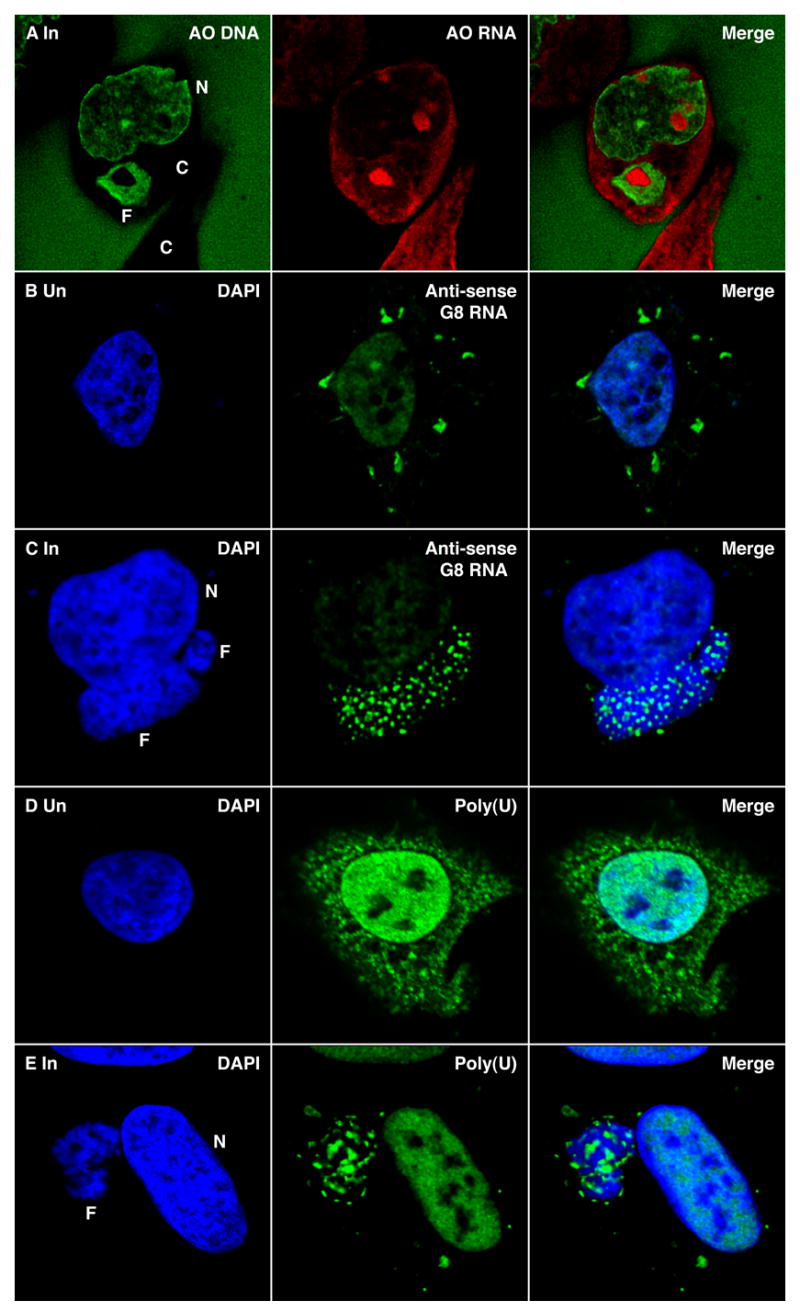
(Row A) HeLa cells were infected with VACV for 6 h. The cells were then washed, fixed, stained with acridine orange (AO) and viewed by confocal microscopy to visualize DNA (green) and RNA (red). One entire cell (center) and the cytoplasm of a second cell (bottom) are seen. The green stain outside the cell is due to AO bound to the glass cover slip. N, nucleus; C, cytoplasm; F, viral factory. (Row B) Uninfected HeLa cells were transfected with anti-sense G8R RNA labeled with digoxigenin UTP. After 6 h, the cells were washed, fixed and stained with sheep anti-digoxigenin-fluorescein, followed by Alexa Fluor 488 donkey anti-sheep (green) and DAPI (blue). (Row C) HeLa cells were infected with VACV and transfected 1 h later with anti-sense G8R RNA labeled with digoxigenin UTP. At 6 h after infection, the cells were analyzed as in Row B. (Row D) Uninfected HeLa cells were transfected with biotinylated poly (U). After 6 h, the cells were stained with streptavidin Alexa Fluor 488, followed by anti-Alexa Fluor 488 rabbit IgG (green) and DAPI (blue). (Row E) HeLa cells were infected with VACV and transfected 1 h later with biotinylated poly (U) and processed as in Row D.
To further investigate the presence of RNA in virus factories, we employed a technique of introducing anti-sense oligonucleotide probes into live cells (Carmo-Fonseca et al., 1991; Politz et al., 1995), which was previously used to locate early VACV transcripts in the cytoplasm of infected cells by confocal microscopy (Mallardo et al., 2002; Mallardo et al., 2001). Digoxigenin-labeled anti-sense intermediate stage G8R RNA was transfected in the present study. In uninfected cells, the probe lightly stained the nucleus and appeared as bright aggregates within the cytoplasm (Figure 1, Row B). In infected cells, the anti-sense probe was concentrated in the factory coincident with multiple areas of low DNA staining (Figure 1, Row C). Serial optical sections showed that cavities containing the probe extended through the entire factory and may be open to the cytoplasm (Figure S1). In factories of some infected cells, the anti-sense viral RNA probe was predominantly in one large cavity, similar to that seen in the example of acridine orange staining in Figure 1, Row A, or at the factory interface with the cytoplasm (not shown). However, little or no probe was detected elsewhere in the cytoplasm. In contrast, when infected cells were transfected with a digoxigenin-labeled anti-sense human glyceraldehydes 3-phosphate dehydrogenase RNA, cytoplasmic staining was seen (not shown). Nevertheless, we could find some cells in which the probe was associated with factories. The latter association could be due to the presence of RNA binding proteins in the factories, as will be shown in a following section.
Fluorochrome-labeled oligo(dT) was previously used to target poly(A)-containing RNA in live cells (Politz et al., 1995). For the same purpose, we transfected biotin-labeled poly(U). In uninfected cells, the poly(U) probe was visualized in the cytoplasm and nucleus (Figure 1, Row D). In infected cells, the probe was also found in the nucleus but was largely absent from the cytoplasm except for the factory region (Figure 1E). Moreover, the factory localization of the poly(U) probe was similar to that of the anti-sense viral RNA. In contrast, transfected biotin-labeled poly(C) was not found in factories but was detected in the nucleus (not shown).
Taken together, the localization of acridine orange staining and anti-sense viral RNA and poly(U) probes suggested the presence of mRNA in previously uncharacterized cavities within the viral factories. The apparent absence of polyadenylated mRNA elsewhere in the cytoplasm at 6 h after infection was consistent with degradation of host messages by this time.
Presence of RNA Binding Proteins and Transcription Factors in RNA Subdomains
E3 is a VACV-encoded double-stranded RNA binding protein that inhibits the interferon-induced double-stranded RNA-activated protein kinase (PKR) and the 2′, 5′-oligoisoadenylate synthetase-dependent RNase (RNase L) (Chang et al., 1992; Rivas et al., 1998; Yuwen et al., 1993). E3 is expressed early after infection and at 2 h had a diffuse cytoplasmic distribution with some nuclear staining (Figure 2, Row A). However, by 4 h and later times, corresponding to intermediate and late viral gene expression, E3 was largely relocated within and at the surface of the DNA factories (Figure 2, Row B). The redistribution could reflect the location of the large amount of double-stranded viral RNA produced as a consequence of read through transcription at intermediate and late times (Boone et al., 1979).
Figure 2. Localization of Viral RNA-Binding Protein and Transcription Factors within Virus Factories.
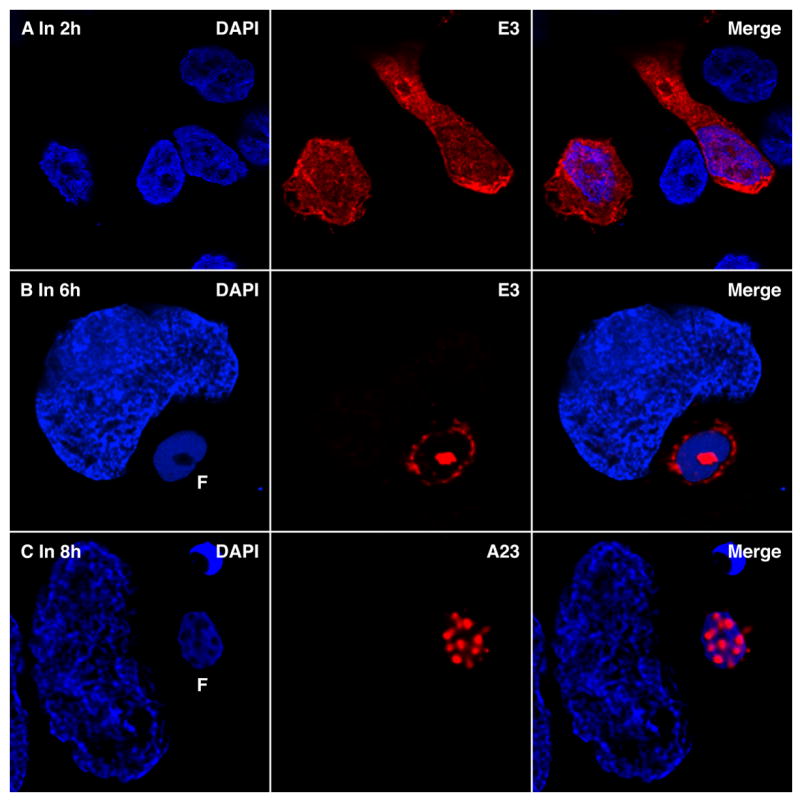
(Row A) HeLa cells were infected with VACV and after 2 h processed and stained with anti-E3 mouse MAb (red), followed by Alexa Fluor 488 goat anti-mouse IgG and DAPI (blue). Two infected and several uninfected cells are present in the field. (Row B) Same as Row A except that cells were processed at 6 h after infection. (Row C) VACV infected HeLa cells were fixed after 8 h, processed and stained with rabbit polyclonal anti-A23 (red), followed by Alexa Fluor 488 goat anti rabbit IgG and DAPI (blue). F, virus factory.
The RNA depots detected in the factories could represent sites of synthesis, accumulation or degradation. In order to investigate these possibilities, we determined the distribution of the A23 protein subunit of the intermediate transcription factor VITF-3 encoded by VACV (Sanz and Moss, 1998). VITF-3 also localized in cavities of the factory as shown in Figure 2, Row C and as serial optical sections in Figure S2.
Two cellular proteins, G3BP and Caprin-1 (p137), recently shown to be a heterodimer (Katsafanas and Moss, 2004; Solomon et al., 2007), are required for in vitro transcription of VACV intermediate genes (Katsafanas and Moss, 2004). These proteins were distributed within the cytoplasm of uninfected cells (Figure 3, Row A) but were relocated to the factory subdomains in infected cells (Figure 3, Row B; Figure S3 movie). Furthermore, G3BP co-localized with poly(A)-containing RNA within the factory (Figure 3, Row C). The viral RNA polymerase was also located in the virus factory but was distributed throughout (data not shown) like virion structural proteins, probably because most of the enzyme is destined for packaging into assembling virus particles. The presence of transcription factors at the same location as the mRNA, suggested that the cavities are sites of RNA synthesis and accumulation.
Figure 3. Localization of G3BP and Caprin 1 (p137) in Virus Factories.
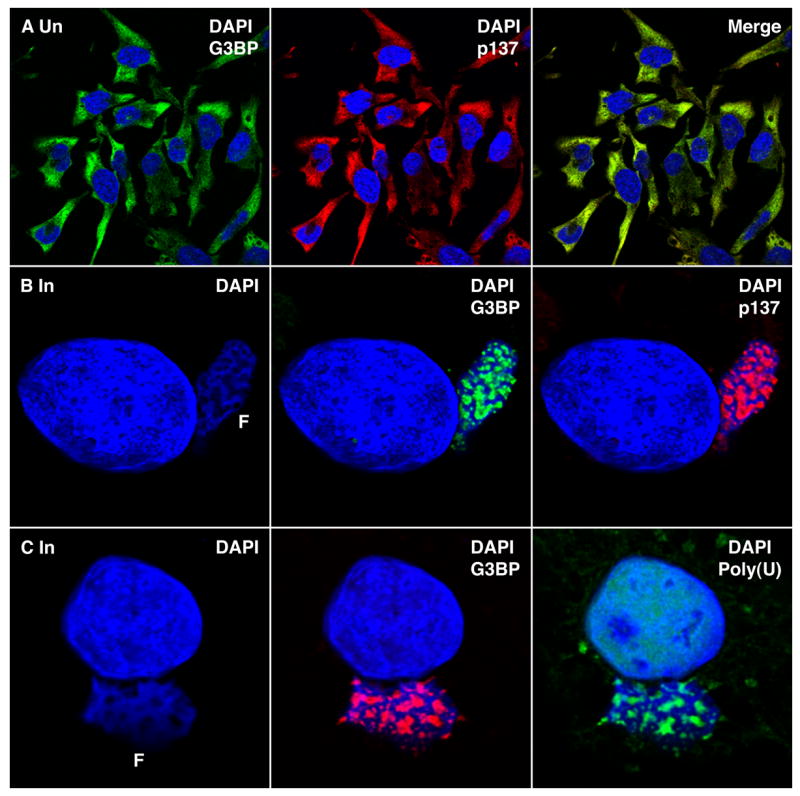
(Row A) Uninfected HeLa cells were fixed and sequentially stained with mouse MAb anti-G3BP (green), rabbit polyclonal anti-p137 (red), Alexa Fluor 488 goat anti-mouse IgG, and Alexa Fluor 594 goat anti-rabbit IgG and DAPI (blue) and viewed by confocal microscopy. (Row B) HeLa cells were infected with VACV and after 8 h were processed as in Row A. (Row C) HeLa cells were infected with VACV and transfected 1 h later with biotinylated poly (U). After 6 h the cells were fixed and stained sequentially with streptavidin Alexa Fluor 488 (green), mouse MAb anti-G3BP (red), anti-Alexa Fluor 488 rabbit IgG fraction, Alexa Fluor 594 goat anti-mouse IgG and DAPI (blue).
The co-localization of cellular G3BP and Caprin-1 (p137) with viral mRNA was consistent with previous evidence for a role in VACV transcription. Nevertheless, it raised the important question of whether cellular proteins might relocate to these subdomains non-specifically. We therefore determined the effect of VACV infection on several different proteins including NF-kB and CRKL. The major form of NF-kB resides in the cytoplasm in an inactive state and is activated and transported to the nucleus in response to a variety of stimuli (Baeuerle and Baltimore, 1988). CRKL is an adapter protein that can translocate to different regions of the cell (ten Hoeve et al., 1993). Following VACV infection, NF-kB retained a cytoplasmic distribution and CRKL was distributed throughout the cell (Figure S4). Thus, the presence of certain viral and cellular proteins within the cavities seems specific.
Localization of Translation Initiation Factors within the Factories
The presence of viral mRNA exclusively within the factory suggested that it is translated nearby. Indeed, we found that cellular translation initiation factors eIF4G (Figure 4, Row A) and eIF4E (Figure 4, Row B) were re-located from the cytoplasm to the factories where they co-localized with G3BP in the cavities. Serial optical sections demonstrating the factory distribution of eIF4E are shown in Figure S5. Although antibody to the large ribosomal subunit P proteins stained the factories exclusively in the cavities, there was similar intensity staining of the cytoplasm outside of the factories (data not shown). These data indicated that a smaller proportion of the ribosomes relocated to factories compared to the translation factors.
Figure 4. Localization of Translation Initiation Factors within Virus Factories.
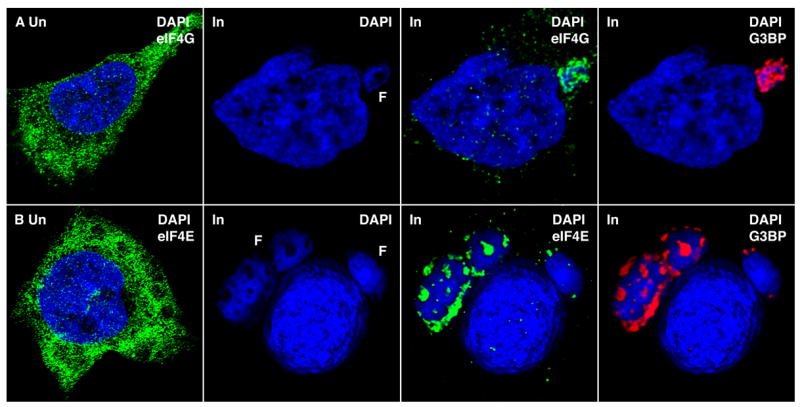
(Row A) Uninfected (Un) HeLa cells were fixed and stained with goat polyclonal anti-eIF4G (green), Alexa Fluor 488 donkey anti-goat IgG and DAPI (blue). VACV infected (In) HeLa cells were processed after 8 h and stained sequentially with goat polyclonal anti-eIF4G (green), mouse MAb to G3BP (red), Alexa Fluor 488 donkey anti-goat IgG, Alexa Fluor 594 goat anti-mouse IgG and DAPI (blue) (Row B) Uninfected (Un) HeLa cells were stained with mouse MAb to eIF4E (green), Alexa Fluor 488 goat anti-mouse IgG and DAPI (blue). VACV infected (In) HeLa cells were stained sequentially with mouse MAb to eIF4E (green), rabbit polyclonal anti-G3BP (red), Alexa Fluor 488 goat anti-mouse IgG, and Alexa Fluor 594 goat anti-rabbit IgG.
Viral Proteins are Synthesized within the Factory
The intracellular redistribution of translation factors led us to consider that viral protein synthesis might occur within the factory rather than in the surrounding cytoplasm. To investigate this hypothesis, we determined the location of newly synthesized proteins encoded by the viral genome. A recombinant VACV with the Escherichia coli lacZ gene regulated by a VACV intermediate promoter was chosen, as it seemed unlikely that β-galactosidase would be targeted to the virus factory if it were synthesized outside of it. Remarkably, at 6 h after infection, β-galactosidase was predominantly in the factory (Figure 5, Rows A, B). Later, as the factories became less discrete, the protein was more diffusely distributed in the cytoplasm (not shown).
Figure 5. Protein Synthesis within Virus factories.
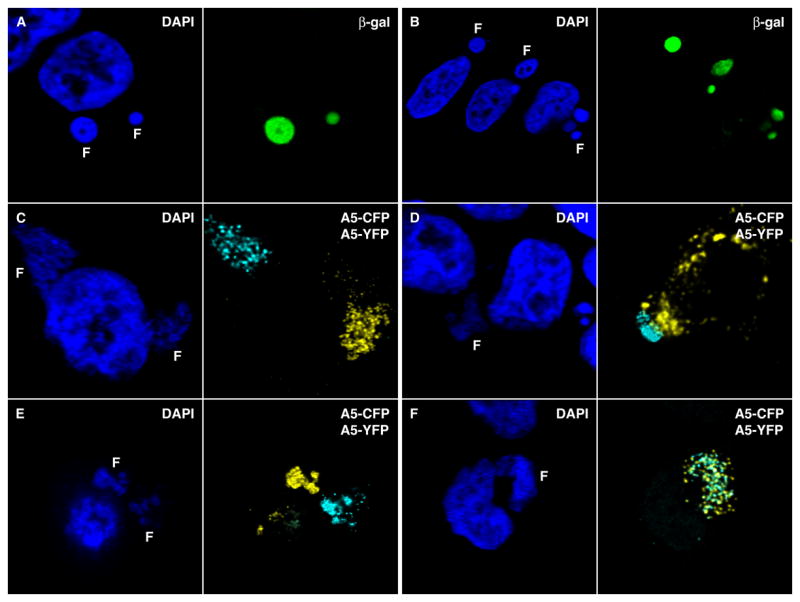
(Rows A, B) HeLa cells were infected with 1 PFU per cell of VACV expressing β–Galactosidase driven by the G8R intermediate promoter. After 6 h the cells were fixed and processed for confocal microscopy. Cells were stained with rabbit polyclonal anti β–Galactosidase (green), followed by Alexa Fluor 488 goat anti-rabbit IgG and DAPI. In row B, the nuclei and adjacent factories are in three separate cells (Rows C–F) Hela cells were infected with 0.5 PFU of vA5L-Cyan and 0.5 PFU of vA5L-Yellow. After 8 to 12 h, the cells were fixed and florescence was monitored at the appropriate channels using confocal microscopy. In row D, the field contains one infected cell with factories and parts of several uninfected cells.
Although the above experiment strongly suggested the coordination of viral transcription and translation within the factory, we devised a more decisive experiment. Previous studies indicated that the number of replication sites in a cell is related to the number of virus particles used for infection (Cairns, 1960; Mallardo et al., 2002). Those data suggested that a virus factory might arise from a single virus particle. We devised a way of demonstrating this and testing our hypothesis regarding the linkage of transcription and translation. Two recombinant VACVs were constructed: one expressed the A5 core protein (Carter et al., 2003) fused to yellow fluorescent protein (YFP) and the other A5 fused to cyan fluorescent protein (CFP). The two fluorescent proteins differ by only a few amino acids, so that the recombinant viruses were nearly identical though easily distinguishable by confocal fluorescent microscopy. Since A5 is a core protein (Maa and Esteban, 1987), it must be present within the virus factory for assembly to occur. This localization could result from synthesis within the factory or synthesis outside the factory followed by targeting to assembly sites. We considered that if a cell was infected with one of each recombinant virus particle and generated two factories, one should exhibit yellow fluorescence and the other cyan provided that each factory was derived from a single genome and translation occurred within the factory. On the other hand, if mRNAs were exported from the factory prior to translation, the factories should contain a mixture of YFP and CFP. We infected cells with a 1:1 mixture of the two viruses and looked for individual cells that exhibited both yellow and cyan fluorescence. Among those cells were many examples with individual yellow and cyan factories (Fig. 5, Rows C–E). In addition, there were factories that exhibited both cyan and yellow fluorescence (Fig. 5F) suggesting an origin from two or more particles. Even in the latter, however, there were distinct yellow and blue granules that presumably represent individual or aggregated virions containing multiple copies of either A5-YFP or A5-CFP.
DISCUSSION
Prokaryotes coordinate gene expression by coupling transcription and translation (French et al., 2007; Miller et al., 1970). In eukaryotes, however, the nuclear membrane separates replication and transcription from translation, a functional division that is maintained following infection by most DNA viruses. Poxviruses are exceptional, however, because they replicate within the cytoplasm in foci called DNA factories that have been likened to mini-nuclei (Schramm and Locker, 2005). Here we showed that the latter term is not appropriate at late times, when transcription and translation occur within the poxvirus DNA factory. In addition to enhancing VACV replication efficiency, linkage of these two processes may give poxviruses a competitive advantage, contributing to the take over of cell functions.
The transcription of VACV early genes occurs within cytoplasmic core particles and the mRNAs are transported via microtubules to distant sites (Mallardo et al., 2001). Although DNA replication and virion assembly were known to occur within poxvirus DNA factories, the location of intermediate and late viral mRNA had not previously been determined. We found that RNA was localized within cavities or tunnels, denoted by diminished DNA staining, and on the surface of the factory. Optical sections and accessibility of a transfected anti-sense probe suggested that the cavities connect with the cytoplasm and in that way increase the factory/cytoplasm interface area. The viral E3 double-stranded RNA binding protein, which prevents activation of PKR and RNase L innate immune defenses (Chang et al., 1992; Rivas et al., 1998), was also present in the cavities. The additional presence of the virus-encoded intermediate transcription factor suggested that the cavities were sites of RNA synthesis and not merely storage depots. Finding the G3BP/Caprin-1 (p137) heterodimer, which we previously showed to complement intermediate transcription in vitro (Katsafanas and Moss, 2004), concentrated in the cavities provided the first in vivo evidence for a role of this host protein in viral transcription. In contrast, cellular NF-kB and CRKL, cytoplasmic regulatory proteins that have not been implicated in VACV transcription were not concentrated in the factory. In addition, our localization of viral and cellular RNA binding proteins and transcription factors in RNA subdomains of the factory contrasts with the partial leakage of some nuclear proteins (e.g. SP1, TBP and YY1) that diffusely localize to VACV replications sites evidently due to their DNA-binding domains (Oh and Broyles, 2005). It will be interesting to determine the intracellular localization of hnRNP A2 and RBM3, RNA binding proteins that have been reported to stimulate late transcription in vitro (Wright et al., 2001).
The recruitment of mRNA to the ribosome is controlled by the activity of the translation factor eIF4E, the component of eIF4F that binds to the 5′-cap structure of mRNA (Kapp and Lorsch, 2004). Translation initiation is dependent upon the interaction of eIF4E with the scaffold protein eIF4G and other factors. Therefore, the finding that eIF4E and eIF4G were concentrated in the cavities, suggested that poxvirus transcription and translation are linked, though probably not directly coupled as in bacteria. The presence of ribosomes in the cavities was suggested by staining with antibody to the P proteins of the large ribosomal subunit. However, ribosomes were still distributed throughout the cytoplasm outside of factory areas. In most situations, translation initiation factors such as eIF4E, rather than ribosomes, are limiting for protein synthesis (Hershey, 1991). The RNA-containing subdomains within poxvirus factories share some features with cytoplasmic RNA granules present in the cytoplasm of uninfected cells (Anderson and Kedersha, 2006). Thus, both cellular stress granules and the factory subdomains contain mRNA, eIF4E, eIF4G and G3BP. However, the stress granules are thought to regulate cellular mRNA translation whereas viral RNA appears to be synthesized and translated in association with the factory subdomains.
Although the presence of translation initiation factors and ribosomal proteins provided circumstantial evidence that protein synthesis occurs in the factory, more compelling data was needed. First, we infected cells with a recombinant VACV expressing the β-galactosidase reporter gene and found that the protein was located in the factory despite the improbability of specific targeting signals. Evidently, the matrix of the factory prevented the rapid diffusion of β-galactosidase from its site of synthesis into the surrounding cytoplasm. Even more compelling evidence that viral proteins are synthesized in the factory was obtained by infecting cells simultaneously with two recombinant viruses, one expressing a VACV core protein fused to CFP and the other to YFP. The finding of individual yellow and cyan factories in the same cell could be explained if a factory formed from a single virus particle and both transcription and translation occurred within the factory boundary. This result would be difficult to explain if viral mRNAs were translated in the surrounding cytoplasm where they would be likely to mix. Not surprisingly, we also found factories containing both CFP and YFP. In some cases yellow and cyan sectoring indicated that the two factories had fused while in others there was mixing of yellow and cyan suggesting a factory had formed from two particles. The latter could arise from aggregated particles, cores that traffic along microtubules to a similar juxtanuclear destination (Carter et al., 2003), or the merging of replication sites.
The synthesis of viral RNA and proteins within the factory in which genome replication and virion assembly occur provides obvious production efficiency. However, there may be an additional benefit. Poxviruses encode a variety of proteins that limit specific host responses at early times of infection. At later stages of infection, there is a global inhibition of host gene expression. How the latter occurs has been subject to much speculation over the years (Moss, 2007). One mechanism is mRNA destabilization, a process that is initiated by a decapping enzyme that is encoded by all poxviruses (Parrish et al., 2007). The present study provides evidence for a second mechanism enabled by the sequestering of crucial translation initiation factors within cytoplasmic DNA factories, where viral transcription and translation occur.
EXPERIMENTAL PROCEDURES
Preparation of Cells for Confocal Microscopy
Uninfected or infected (1 PFU/cell) HeLa cells on 12 mm round glass coverslips were washed (3x for 5 min) in Dulbecco’s phosphate buffered saline, fixed in 4% paraformaldhyde for 20 min, washed again, permeabilized with 0.1% Triton X-100 in phosphate buffered saline (PBS) for 15 min, washed again, and incubated at room temperature for 1 h in PBS with 1% bovine serum albumin. Primary antibodies were diluted 1:100 in PBS with 1% bovine serum albumin and incubated at room temperature for 6 h or overnight at 4°C. After washing, secondary antibodies were applied at a 1:100 dilution as above. When cells were dual labeled the procedure was repeated. Nuclei and factories were stained with DAPI at a concentration of 5 μg/ml in H2O at room temperature for 15 min and washed. Coverslips were mounted on 75 × 25 mm slides using Mowiol as the mounting medium.
Acridine Orange Staining
Cells prepared as above were stained with 6 μg/ml of acridine orange (Molecular Probes, Eugene, OR), washed and mounted on glass slides. The metachromatic dye was excited at 488 nm and detector slits were set between 510 – 549 nm and 626 – 656 nm to visualize double and single stranded nucleic acids, respectively.
Confocal Microscopy and Image Processing
Images were collected on a Leica SP2 AOBS confocal microscope (Leica Microsystems, Bannockburn, IL) using a 63x oil immersion objective NA 1.32 or 1.4 zoom X. Fluorochromes were excited using 357 or 405 nm for DAPI, 488 nm for Alexa 488, 458 for CFP, 514 nm for YFP, and 594 nm excitation for Alexa 594. Detector slits were configured to minimize crosstalk between the channels. Images were collected sequentially in a z series, deconvolved using Huygens 2.10 (Scientific Volume Imaging, Hilversum, The Netherlands) and processed using Imaris 5.5.1 (Bitplane AG, Zurich, Switzerland) and Adobe Photoshop CS (Adobe Systems, San Jose, CA).
Digoxigenin-Labeled Antisense RNA and Biotinylated Poly(U)
The G8R gene was amplified by PCR using an N-terminal primer: 5′-GCGCGTCGACATGAGCATCCGTATAAAAATCGATAAACTGCGCC-3′ and a C-terminal primer 5′-AATTGCATGCTAATACGACTCACTATAGGGTTAATCTAAAAACGCCATAAAG ATGTTGATCTTAAAGG-3′, which included the sequence of the bacteriophage T7 RNA polymerase promoter. The 785 nucleotide product was purified with a QIAquick PCR purification kit (Qiagen, Fremont, CA) and 1 μg was mixed with 2 μl of 10x DIG labeling mix (10 mM ATP, 10 mM CTP, 10 mM GTP, 6.5 mM UTP, 3.5 mM digoxigenin-11-UTP, pH 7.5), 2 μl of 10x transcription buffer (0.4 M Tris-HCl, pH 8.0, 60 mM MgCl2, 0.1 M dithiotreitol, 20 mM spermidine), 2 μl of T7 RNA polymerase in a total volume of 20 μl (reagents from Roche Applied Science, Indianapolis, IN). This mixture was incubated for 2 h at 37°C and the reaction stopped with 2 μl of 0.2 M EDTA (pH 8.0). The transcript was shown to be pure and of the correct size by electrophoresis of 1 μl on a 6% Tris borate EDTA urea gel and staining with ethidium bromide. Nucleic acid concentration was determined by absorbance and 350 ng mixed with lipofectamine (Invitrogen, Carlsband, CA) was used to transfect uninfected or infected HeLa cells that had been plated on 12 mm round cover slips.
For localization of human glyceraldehyde 3- phosphate dehydrogenase, in-vitro transcriptions were carried out as above using the linearized pTRI-GAPDH-plasmid, which includes the sequence of the bacteriophage T7 RNA polymerase promoter (Ambion, Austin, TX). Absorbance was determined and 350 ng of the 387 nucleotide product was used to transfect uninfected or infected HeLa cells that had been plated on 12 mm round coverslips.
To localize polyadenylated mRNAs, a 5′ biotinylated 40mer poly(U) was chemically synthesized (Dharmacon, Lafayette, CO) and 1 μg was used for transfection as described for the anti-sense G8R RNA.
Construction of VACV Recombinants
Open reading frames encoding CFP and YFP were amplified using plasmid templates from Clonetech (Mountain View, CA). Fusion of the open reading frames to the N-terminal codon of A5L was achieved by overlap extension PCR using VACV WR genomic DNA as the template. 1 μg of each PCR product was transfected into HeLa cells that had been infected with VACV vRPO22C10H (Katsafanas and Moss, 1999). The cell lysate of each separate transfection/infection was plated onto BS-C-1 cells and plaques were picked by their ability to fluoresce. Virus in cyan and yellow plaques were each clonally purified 4-times on BS-C-1 cells. The recombinant viruses were amplified in HeLa cells to achieve high titer virus stocks.
Sources of Antibodies and Conjugated Streptavidin
Antibodies and their sources were: anti-G3BP mouse MAb (BD Pharmingen, San Diego, CA); rabbit polyclonal anti-G3BP and rabbit polyclonal anti Caprin-1 (p137) (Katsafanas and Moss, 2004); Mouse MAb anti-E3 (Yuwen et al., 1993); Rabbit polyclonal anti-A23 (unpublished); rabbit polyclonal anti-β galactosidase (Abcam, Inc, Cambridge, MA); goat polyclonal anti-eIF4G, rabbit polyclonal anti-NF-kB and CRKL, mouse MAb anti-eIF4E, (Santa Cruz Biotechnology, Santa Cruz, CA); sheep anti-digoxigenin-fluorescein, (Roche Applied Sciences); Alexa Fluor 488 and Alexa Fluor 594 conjugated to anti-IgG of appropriate species (Molecular Probes). Alexa 488 conjugated to streptavidin and anti-Alexa 488 rabbit IgG was from Molecular Probes.
Supplementary Material
{kind=link}
Acknowledgments
We thank Owen Schwartz and Juraj Kabat for training with confocal microscopy and imaging software and Ted Pierson for comments on the manuscript. The study was supported by the Division of Intramural Research, NIAID, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson P, Kedersha N. RNA granules. J Cell Biol. 2006;172:803–838. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bablanian R, Goswami SK, Esteban M, Banerjee AK, Merrick WC. Mechanism of selective translation of vaccinia virus mRNAs: differential role of poly(A) and initiation factors in the translation of viral and cellular mRNAs. J Virol. 1991;65:4449–4460. doi: 10.1128/jvi.65.8.4449-4460.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle PA, Baltimore D. Activation of DNA-binding activity in an apparently cytoplasmic precursor of the NF-kappa B transcription factor. Cell. 1988;53:211–217. doi: 10.1016/0092-8674(88)90382-0. [DOI] [PubMed] [Google Scholar]
- Boone RF, Parr RP, Moss B. Intermolecular duplexes formed from polyadenylated vaccinia virus RNA. J Virol. 1979;30:365–374. doi: 10.1128/jvi.30.1.365-374.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum LM, Lopez MC, Varela JC, Baker HV, Moyer RW. Microarray analysis of A549 cells infected with rabbitpox virus (RPV): a comparison of wild-type RPV and RPV deleted for the host range gene, SPI-1. Virology. 2003;315:322–334. doi: 10.1016/s0042-6822(03)00532-4. [DOI] [PubMed] [Google Scholar]
- Cairns J. The initiation of vaccinia infection. Virology. 1960;11:603–623. doi: 10.1016/0042-6822(60)90103-3. [DOI] [PubMed] [Google Scholar]
- Carmo-Fonseca M, Pepperkok R, Sproat BS, Ansorge W, Swanson MS, Lamond AI. In vivo detection of snRNP-rich organelles in the nuclei of mammalian cells. EMBO J. 1991;10:1863–1873. doi: 10.1002/j.1460-2075.1991.tb07712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter GC, Rodger G, Murphy BJ, Law M, Krauss O, Hollinshead M, Smith GL. Vaccinia virus cores are transported on microtubules. J Gen Virol. 2003;84:2443–2458. doi: 10.1099/vir.0.19271-0. [DOI] [PubMed] [Google Scholar]
- Chang HW, Watson JC, Jacobs BL. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci USA. 1992;89:4825–4829. doi: 10.1073/pnas.89.11.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dales S, Siminovitch L. The development of vaccinia virus in Earle’s L strain cells as examined by electron microscopy. J Biophys Biochem Cytol. 1961;10:475–503. doi: 10.1083/jcb.10.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French SL, Santangelo TJ, Beyer AL, Reeve JN. Transcription and translation are coupled in Archaea. Mol Biol Evol. 2007;24:893–895. doi: 10.1093/molbev/msm007. [DOI] [PubMed] [Google Scholar]
- Guerra S, Lopez-Fernandez LA, Pascual-Montano A, Munoz M, Harshman K, Esteban M. Cellular gene expression survey of vaccinia virus infection of human HeLa cells. J Virol. 2003;77:6493–6506. doi: 10.1128/JVI.77.11.6493-6506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harford C, Hamlin A, Riders E. Electron microscopic autoradiography of DNA synthesis in cell infected with vaccinia virus. Exp Cell Res. 1966;42:50–57. doi: 10.1016/0014-4827(66)90318-1. [DOI] [PubMed] [Google Scholar]
- Hershey JW. Translational control in mammalian cells. Ann Rev Bioch. 1991;60:717–755. doi: 10.1146/annurev.bi.60.070191.003441. [DOI] [PubMed] [Google Scholar]
- Kapp LD, Lorsch JR. The molecular mechanics of eukaryotic translation. Ann Rev Biochem. 2004;73:657–704. doi: 10.1146/annurev.biochem.73.030403.080419. [DOI] [PubMed] [Google Scholar]
- Katsafanas GC, Moss B. Histidine codons appended to the gene encoding the RP022 subunit of vaccinia virus RNA polymerase facilitate the isolation and purification of functional enzyme and associated proteins from virus-infected cells. Virology. 1999;258:469–479. doi: 10.1006/viro.1999.9744. [DOI] [PubMed] [Google Scholar]
- Katsafanas GC, Moss B. Vaccinia virus intermediate stage transcription is complemented by Ras-GTPase-activating protein SH3 domain-binding protein (G3BP) and cytoplasmic activation/proliferation-associated protein (p137) individually or as a heterodimer. J Biol Chem. 2004;279:52210–52217. doi: 10.1074/jbc.M411033200. [DOI] [PubMed] [Google Scholar]
- Maa JS, Esteban M. Structural and functional studies of a 39,000-Mr immunodominant protein of vaccinia virus. J Virol. 1987;61:3910–3919. doi: 10.1128/jvi.61.12.3910-3919.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallardo M, Leithe E, Schleich S, Roos N, Doglio L, Krijnse-Locker J. Relationship between vaccinia virus Intracellular cores, early mRNAs, and DNA replication sites. J Virol. 2002;76:5167–5183. doi: 10.1128/JVI.76.10.5167-5183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallardo M, Schleich S, Krijnse-Locker J. Microtubule-dependent organization of vaccinia virus core-derivd early mRNAs into distinct cytoplasmic structures. Mol Biol Cell. 2001;12:3875–3891. doi: 10.1091/mbc.12.12.3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller OL, Jr, Hamkalo BA, Thomas CA., Jr Visualization of bacterial genes in action. Science. 1970;169:392–395. doi: 10.1126/science.169.3943.392. [DOI] [PubMed] [Google Scholar]
- Moss B. Poxviridae: the viruses and their replicaton. In: Knipe DM, editor. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 2905–2946. [Google Scholar]
- Oh J, Broyles SS. Host cell nuclear proteins are recruited to cytoplasmic vaccinia virus replication complexes. J Virol. 2005;79:12852–12860. doi: 10.1128/JVI.79.20.12852-12860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S, Resch W, Moss B. Vaccinia virus D10 protein has mRNA decapping activity, providing a mechanism for control of host and viral gene expression. Proc Natl Acad Sci USA. 2007;104:2139–2144. doi: 10.1073/pnas.0611685104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politz JC, Taneja KL, Singer RH. Characterization of hybridization between synthetic oligodeoxynucleotides and RNA in living cells. Nucleic Acids Res. 1995;23:4946–4953. doi: 10.1093/nar/23.24.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice AP, Roberts BE. Vaccinia virus induces cellular mRNA degradation. J Virol. 1983;47:529–539. doi: 10.1128/jvi.47.3.529-539.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas C, Gil J, Melkova Z, Esteban M, Diaz-Guerra M. Vaccinia virus E3L protein is an inhibitor of the interferon (IFN)-induced 2–5A synthetase enzyme. Virology. 1998;243:406–414. doi: 10.1006/viro.1998.9072. [DOI] [PubMed] [Google Scholar]
- Sanz P, Moss B. A new vaccinia virus intermediate transcription factor. J Virol. 1998;72:6880–6883. doi: 10.1128/jvi.72.8.6880-6883.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm B, Locker JK. Cytoplasmic organization of poxvirus DNA replication. Traffic. 2005;6:839–846. doi: 10.1111/j.1600-0854.2005.00324.x. [DOI] [PubMed] [Google Scholar]
- Seet BT, Johnston JB, Brunetti CR, Barrett JW, Everett H, Cameron C, Sypula J, Nazarian SH, Lucas A, McFadden G. Poxviruses and immune evasion. Ann Rev Immunol. 2003;21:377–423. doi: 10.1146/annurev.immunol.21.120601.141049. [DOI] [PubMed] [Google Scholar]
- Shors T, Keck JG, Moss B. Down regulation of gene expression by the vaccinia virus D10 protein. J Virol. 1999;73:791–796. doi: 10.1128/jvi.73.1.791-796.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon S, Xu Y, Wang B, David MD, Schubert P, Kennedy D, Schrader JW. Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol Cell Biol. 2007;27:2324–2342. doi: 10.1128/MCB.02300-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Hoeve J, Morris C, Heisterkamp N, Groffen J. Isolation and chromosomal localization of CRKL, a human crk-like gene. Oncogene. 1993;8:2469–2474. [PubMed] [Google Scholar]
- Tolonen N, Doglio L, Schleich S, Locker JK. Vaccinia virus DNA replication occurs in endoplasmic reticulum- enclosed cytoplasmic mini-nuclei. Mol Biol Cell. 2001;12:2031–2046. doi: 10.1091/mbc.12.7.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CF, Oswald BW, Dellis S. Vaccinia virus late transcription is activated in vitro by cellular heterogeneous nuclear ribonucleoproteins. J Biol Chem. 2001;276:40680–40686. doi: 10.1074/jbc.M102399200. [DOI] [PubMed] [Google Scholar]
- Yuwen H, Cox JH, Yewdell JW, Bennink JR, Moss B. Nuclear localization of a double-stranded RNA-binding protein encoded by the vaccinia virus E3l gene. Virology. 1993;195:732–744. doi: 10.1006/viro.1993.1424. [DOI] [PubMed] [Google Scholar]
- Zelenin. Acridine orange as a probe for cell and molecular biology. In: Mason WT, editor. Fluorescent and luminescent probes for biological activity. San Diego: Academic Press; 1999. pp. 117–135. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.