

Abstract
The Pebpb2 gene encodes a non-DNA binding subunit of the heterodimeric transcription factor, polyomavirus enhancer binding protein 2/core binding factor (PEBP2/CBF), and is rearranged in inversion of chromosome 16 associated with human acute myeloid leukemia. To investigate its physiological function, Pebpb2 was mutated by a targeting strategy to generate a null mutant. The homozygous mutation in mice proved lethal in embryos around embryonic day 12.5, apparently due to massive hemorrhaging in the central nervous system. In addition, definitive hematopoiesis in the liver was severely impaired. The observed phenotype was indistinguishable from that reported for homozygous disruption of AML1, which encodes a DNA binding subunit of PEBP2/CBF. Thus, the results indicate that the two subunits function together as a heterodimeric PEBP2/CBF in vivo and that PEBP2/CBF plays an essential role in the development of definitive hematopoiesis.
Growing evidence from targeting studies of mice suggests that a set of transcription factors are required for the development of primitive hematopoiesis in the yolk sac and definitive hematopoiesis in the fetal liver (1, 2). Null mutations in Scl-Tal1 (3) or Rbtn2 (4) cause a block of primitive erythropoiesis and homozygous mutant embryos die on embryonic days 8.5 (E8.5) to E10.5. Furthermore, Scl-Tal1 (−/−) embryonic stem (ES) cells do not contribute to adult type hematopoiesis on chimeric analysis (5). On the other hand, disruption of c-myb (6) or GATA-2 (7) mainly impairs definitive hematopoiesis, although primitive hematopoiesis is also hampered to some degree in the latter case. In c-myb and GATA-2 targeted embryos, no mature cells of any hematopoietic lineage, except for a megakaryocytic lineage for c-myb, are generated. Therefore, the observations strongly suggest that these transcription factors play an essential role in the growth and differentiation of hematopoietic progenitors and that the defects seen in null mutants are intrinsic to hematopoietic cells.
AML1 has also been shown by recent studies, including our own, to be essential for definitive hematopoiesis in the murine fetal liver (refs. 8 and 9 and unpublished observations). The product of AML1 is the DNA binding α subunit of the transcription factor polyomavirus enhancer binding protein 2 (PEBP2), also known as a core binding factor [(CBF); refs. 10–16]. AML1 is a mammalian homolog of the Drosophila genes, runt (17) and lozenge (18), and was originally identified at the breakpoint of the chromosome translocation, t(8;21), that is associated with human acute myeloid leukemia (19). Subsequently, AML1 was also found to be a frequent target of chromosome translocations associated with several other types of human leukemia (20–24).
The DNA binding domain of AML1, termed the Runt domain, carries an additional function of interacting with its partner protein, the β subunit (13, 15, 16). The β subunit of PEBP2/CBF is itself devoid of DNA binding activity, but on dimerizing with the α subunit it enhances its affinity for DNA (14, 25). The gene, Pebpb2, that encodes the β subunit is also the target of an inversion of chromosome 16, another chromosome anomaly associated with human acute myeloid leukemia (26). Gene targeting of Pebpb2 in the present study revealed that, as in the AML1 case, it functions to assure definitive hematopoiesis. This result firmly establishes that both PEBP2β and AML1 exert essential functions in hematopoiesis as a heterodimer.
MATERIALS AND METHODS
Targeting Vector and Homologous Recombination.
The targeting vector replaces a 1.2-kb region that encompasses the first exon of Pebpb2 with the neo cassette containing the neo gene regulated by the promoter and the poly(A) addition sequence of the murine phosphoglycerol kinase gene (Fig. 1A). The neo gene was flanked by a 1.1-kb PstI fragment and a 7.5-kb SmaI–SalI fragment, each corresponding to 5′ and 3′ genomic sequences of Pebpb2 surrounding the first exon. The gene for the diphtheria toxin A subunit was incorporated into the 3′ end of the vector. The vector was linearized with NotI and electroporated into J1 ES cells. Three hundred and twenty-six G418-resistant ES cell colonies were picked up, expanded as described previously (27), and screened for homologous recombination by Southern blotting using probe A depicted in Fig. 1A. One ES cell clone was identified as heterozygous for Pebpb2 disruption and injected into blastocysts derived from C57BL/6J mice. Chimeric mice were bred against C57BL/6J female mice. Southern blot analysis of tail DNA of F1 mice showed germ-line transmission of the mutant Pebpb2 allele. F1 mice heterozygous for the mutation were intercrossed to generate F2 offspring. For genotyping F2 mouse embryos and neonates, PCR was performed on genomic DNA as a template. The locations of primer 1, 5′-AGCTCTGCTGCAAACCCCGAGTCTG-3′, primer 2, 5′-ACGGGGTTGGCCTTAGGATTCCGTG-3′, and primer 3, 5′-AAGGGCCAGCTCATTCCTCCCACTC-3′, are depicted in Fig. 1A. PCR amplification (Perkin–Elmer) was performed for 35 cycles (94°C, 1 min; 72°C, 2 min; 72°C, 2 min), and the PCR products were confirmed by restriction enzyme digestion. All of the data presented below were obtained using F2 mouse embryos.
Figure 1.
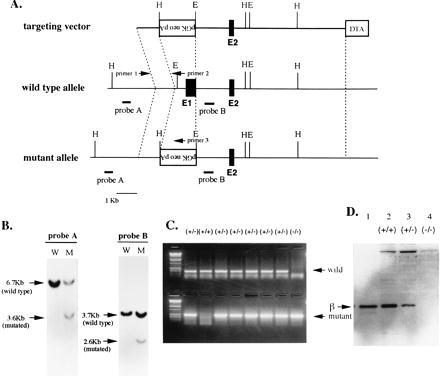
Targeting of the Pebpb2 gene by homologous recombination. (A) The targeting strategy used to delete the first exon of Pebpb2. In the targeting vector the transcriptional direction of neo is opposite those of the Pebpb2 and DTA genes. Probe A was used to screen homologous recombination in ES cells, whereas probe B was used to check the single integration of the vector. Sets of sense primer 1 and antisense primers 2 or 3 were used for PCR to genotype F2 embryos. E1 and E2 indicate the first and second exons of Pebpb2. H and E represent HindIII and EcoRI sites, respectively. (B) Southern blot analysis of ES cell clones. Genomic DNAs of ES cell clones were digested by HindIII (left two lanes) or EcoRI (right two lanes) and hybridized with probe A or probe B, respectively. (C) Genotyping of F2 E12.5 embryos by PCR. Genomic DNAs from F2 embryos were used as templates for PCR. The primers used are indicated in A. (D) Immunoblot analysis of the PEBP2β protein. Proteins extracted from wild-type, heterozygous, and homozygous mutant F2 embryos were run on a gel, and the blotted filter was probed with anti-PEBP2β antiserum. In lane 1, 200-pg of PEBP2β protein purified from an E. coli lysate was run as a positive control.
Reverse Transcription–PCR (RT-PCR) Analysis.
E12.5 live embryos were selected on the basis of the presence of a heart beat. Total RNA samples were prepared from the livers of embryos using Isogen (Nippon Gene, Toyama, Japan) and treated with RQ1 RNase-free DNase (Promega). RNA (2.5 μg) was reverse transcribed at 42°C for 40 min using 200 units of SuperScript II (GIBCO/BRL) and an oligo(dT) primer. PCR amplification of the cDNAs was performed for 25, 30, or 35 cycles, and the amplified products were found to be in linear ranges for the cycle number quantitatively. PCR products from hypoxanthine phosphoribosyltransferase transcripts were used as standards. The primer sets for βH1 globin, ɛ globin, ζ globin, c-myb, GATA-2, flk-2/flt-3, and hypoxanthine phosphoribosyltransferase were as described previously (28–30). The primers used for AML1 were 5′-CCTGCTTGGGTGTGAGGCCG-3′ (nt 1366∼1385) and 5′-GCCTCGCTCATCTTGCCGGG-3′ (nt 1638∼1657) of Pebpa2b (31).
Histological Examination.
Embryos were fixed in Bouin’s fluid for 18 hr and embedded in paraffin. Transverse sections with a 5-μm thickness were mounted on slide glasses and stained with hematoxylin and eosin as described previously (27). Peripheral blood cells collected from the cord vessels and the cells liberated from the fetal liver by touch preparations were smeared on slide glasses and stained with May-Giemsa (Merck).
In Vitro Colony Forming Assays.
Single cell suspensions were prepared from the yolk sac and fetal liver of E11.5 embryos by passing the tissue through 18-gauge needles 20 times. The cells were washed twice with α-minimum essential medium containing 10% (vol/vol) fetal calf serum (HyClone) and plated in medium containing 1.2% (wt/vol) methylcellulose (Wako Pure Chemical, Osaka), 30% (vol/vol) fetal calf serum, 1% (wt/vol) BSA (Sigma), 0.1 mM 2-mercaptoethanol, recombinant erythropoietin (2 units/ml), granulocyte colony-stimulating factor (10 ng/ml) (Chugai Pharmaceutical, Tokyo), IL-3 (10 ng/ml), and stem cell factor (10 ng/ml) (Amgen). Colony numbers were scored after 7 days of incubation at 37°C in humidified air containing 5% CO2.
Immunoblot Analysis.
Protein was extracted from E12.5 live embryos using Isogen. One hundred-microgram aliquots of protein per lane were separated by electrophoresis on 0.1% (wt/vol) SDS-15% (wt/vol) polyacrylamide gels. Proteins were then transferred electrophoretically onto poly(vinylidene difluoride) membranes (Bio-Rad). After blocking, the membranes were first incubated with an 8000-fold-diluted anti-PEBP2β rabbit serum (32) and then a 3000-fold-diluted biotin-conjugated anti-rabbit IgG solution (Kirkegaard & Perry Laboratories). Streptavidin was coupled to biotin using an ABC kit (Vector Laboratories). Proteins were visualized by immersing the membranes in ECL detection reagent (Amersham).
RESULTS
Generation of Mouse Embryos Homozygous for Pebpb2 Disruption.
The first exon of Pebpb2 encodes the amino-terminal 26 amino acids including the initiating methionine of the PEBP2β polypeptide (25). This was replaced by the neo gene derived from the targeting vector (Fig. 1A). Because both the cap site of the transcripts and the initiating methionine of the open reading frame were deleted, there should be theoretically no authentic transcripts or polypeptide generated from the mutant allele. This was indeed the case as shown below. By screening the ES cells transfected with the targeting vector, one homologous recombinant was obtained. The results of Southern blot analysis are shown in Fig. 1B. Probe A was derived from the genomic sequence and located 5′ to the homologous sequence used in the vector. A 3.6-kb mutated band was detected in addition to a 6.7-kb wild-type band in HindIII-digested genomic DNA. Probe B was derived from the first intron and was present in the vector. This probe revealed a 2.6-kb mutated band in addition to a 3.7-kb wild-type band in EcoRI-digested genomic DNA. Therefore, this clone was considered to harbor the expected mutation on one allele of Pebpb2.
F1 mice heterozygous for the mutation were obtained as described in Materials and Methods. They developed normally and were mated with each other. No live F2 offspring carrying the homozygous mutation was born (Table 1), indicating that the mutation in both Pebpb2 alleles is embryonically lethal. Therefore, we genotyped the F2 embryos by PCR using genomic DNA and different sets of primers. An example of the results obtained for one littermate is shown in Fig. 1C. We found that the Pebpb2 (−/−) embryos lived until E11.5 but that half were dead at E12.5 as diagnosed by cessation of heart beat (Table 1). At E13.5, all of the homozygous embryos were dead. Thus, the homozygous mutation appears to cause embryonic death between E11.5 and E13.5.
Table 1.
Genotypes of F2 embryos and offsprings obtained by intercross of Pebpb2/Cbfb +/−
| Stages | Genotype
|
Total | |||
|---|---|---|---|---|---|
| −/−
|
|||||
| +/+ | +/− | Alive | Dead | ||
| E10.5 | 0 | 7 | 5 | 0 | 12 |
| E11.5 | 25 | 50 (2) | 14 | 1 | 90 |
| E12.5 | 31 (1) | 52 | 12 | 12 | 107 |
| E13.5 | 0 | 3 | 0 | 2 | 5 |
| PO | 17 | 37 | 0 | 54 | |
Numbers in parenthesis represent degenerated dead embryos. They did not show the phenotypes observed in the −/− dead embryos. PO are offsprings at postnatal day 0.
To examine the expression of Pebpb2, RT-PCR was performed on E12.5 embryonic RNAs. Two primers, for the second and fifth exons, were employed, but no bands were generated from cDNAs prepared from homozygous mutant embryos (data not shown). Synthesis of the PEBP2β protein in the embryos was also examined by immunoblot analysis (Fig. 1D). Protein extracts were prepared and run on SDS-polyacrylamide gels, and the blotted filter was probed with anti-PEBP2β antibodies (32). Compared with the wild-type embryo, approximately half or none of the normal amount of PEBP2β was detected for heterozygous or homozygous mutant embryos, respectively. Thus, the homozygous mutant embryos expressed neither Pebpb2 transcripts nor polypeptides.
Defect of Definitive Hematopoiesis in the Pebpb2 (−/−) Embryos.
Until E11.5 no immediately apparent abnormalities were observed in Pebpb2 (−/−) embryos by gross anatomical and histological examinations. In contrast, severe hemorrhage in cerebral ventricles and spinal canals was seen in 14 of 24 E12.5 Pebpb2 (−/−) embryos (Fig. 2B). Fig. 2C is an enlarged view of hemorrhage in a lateral ventricle. This type of hemorrhage was considered the most probable principal cause of embryonic death. Although it was difficult to assign exactly the primary sites of intraventricular hemorrhage, the junction of the brainstem ventricle and the spinal canal is shown as an example (Fig. 2D). Hemorrhage was observed in the nerve cell layer as foci of various sizes, some appearing to extend into and lift the ependymal epithelium (indicated by an arrow). Blood cells detected in the cavity (arrowhead) could have flowed out through rupture sites in the ependyma and their nucleated nature indicated a primitive hematopoietic origin.
Figure 2.
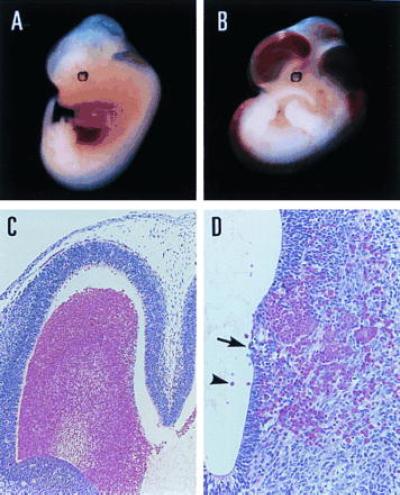
Gross and microscopic views of hemorrhagic lesions in E12.5 Pebpb2 (−/−) embryos. (A) is a Pebpb2 (+/−) embryo, demonstrating a normal appearance. Severe hemorrhage is evident in (B) of a Pebpb2 (−/−) embryo. (C) Hemorrhage in the lateral ventricle of a (−/−) embryo. (D) Histological view of the isthmus of a (−/−) embryo. See text for an explanation of the arrow and arrowhead in D.
The second major abnormality in the Pebpb2 (−/−) embryos was seen in the livers that were smaller than those of Pebpb2 (+/−) embryos (data not shown). The histology of the liver sections from Pebpb2 (+/−) embryos showed that the sinusoidal cavities were filled with numerous large and relatively mature hematopoietic cells (Fig. 3A, arrow). Many small cells, possessing densely stained nuclei and found to be scattered in the entire field (arrowheads), were judged to be hematopoietic cells at less differentiated stages. In contrast, the livers of Pebpb2 (−/−) embryos were almost completely devoid of hematopoietic cells, and their sinusoidal cavities were empty (Fig. 3B). Because the E12.5 liver is the site of definitive hematopoiesis, the results indicated this process to be severely impaired in Pebpb2 (−/−) embryos. The presence or absence of hematopoietic cells in the livers was also confirmed by preparing tissue smears. From Pebpb2 (−/−) embryos, only large cells of stromal origin with lightly stained nuclei were observed (Fig. 3D). In touch preparations from Pebpb2 (+/−) embryonic livers, on the other hand, numerous hematopoietic progenitors representing erythroblasts and myeloblasts could be identified (see Fig. 3C legend).
Figure 3.
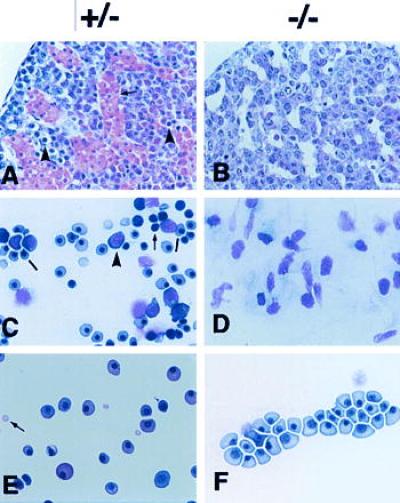
Histological examination of livers and hematopoietic cells from E12.5 Pebpb2 (+/−) and (−/−) embryos. (A and B) Sections of liver were stained with hematoxylin and eosin. (C and D) Touch preparations of the liver tissue were stained with May-Giemsa. In (C), the arrow indicates an early hematopoietic progenitor, the arrowhead, a more differentiated myeloid progenitor and the bar, an erythroid cell. (E and F) Smears of peripheral blood were stained with May-Giemsa. See text for an explanation of the arrow and arrowheads in A and F.
The defect in definitive hematopoiesis in the Pebpb2 (−/−) embryos was confirmed by examining the gene transcripts of transcription factors known to be essential for the development of definitive hematopoiesis. A semiquantitative RT-PCR analysis of the c-myb and GATA-2 transcripts revealed that their expression in the E12.5 fetal liver was markedly (c-myb) or significantly (GATA-2) reduced in the Pebpb2 (−/−) mutants (Fig. 4). In contrast, comparable levels of AML1 transcripts were detected in both (+/−) and (−/−) fetal livers.
Figure 4.
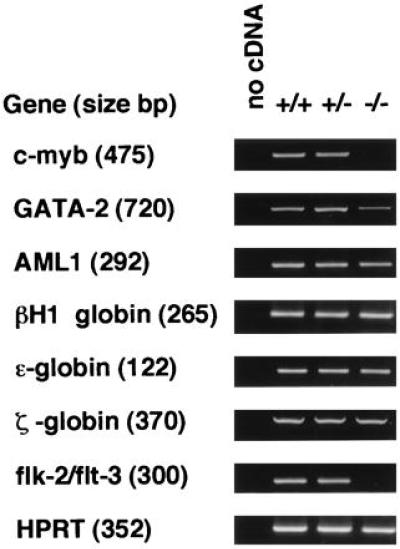
Semiquantitative RT-PCR analysis of the transcripts. Results for samples after 30 cycles of PCR in the linear range for amplification are shown as representative. In the case of flk-2/flt-3, PCR was repeated twice because of the very low level of transcripts in the sample RNA. Bands in agarose gels were ethidium bromide stained. In the lanes of no cDNA, PCR was performed without addition of cDNA to the reaction mixture.
Primitive hematopoiesis starts at E7.5 in the yolk sac. This did not appear to be affected by the homozygous mutation of Pebpb2, because a number of erythrocytes could be identified in the hearts of E11.5 (−/−) embryos (data not shown). Stained smears of peripheral blood from E12.5 embryos (Fig. 3E and 3F) demonstrated nucleated large erythrocytes, diagnosed as primitive erythrocytes, and no apparent morphological differences could be detected between these in heterozygous and homozygous mutants. Mature erythrocytes that appeared small and enucleated (Fig. 3E, arrow) were the products of terminal differentiation in definitive erythropoiesis. They were detected in smears from heterozygous but not from homozygous mutants. The presence of primitive erythropoiesis was also examined by RT-PCR of RNAs prepared from E12.5 livers (Fig. 4). Transcripts of embryonic type hemoglobin (βH1, ɛ, and ζ globin) were detected to similar degrees in both the heterozygous and homozygous mutants.
We then examined whether the Pebpb2 (−/−) embryos harbor hematopoietic progenitor cells that can grow and differentiate into erythroid and myeloid lineages. Cells from E11.5 yolk sacs and livers were cultured in semisolid medium under assay conditions permitting the growth of definitive hematopoietic progenitors. Similar numbers of erythroid, myeloid, and erythroid-myeloid mixed colonies could be counted in the case of wild-type and heterozygous mutants (Table 2). Homozygous mutants, however, generated no colonies at all in either lineage, suggesting that the defect in definitive hematopoiesis in the (−/−) embryos lies at the stage of hematopoietic progenitor cells, although the possibility of microenvironmental alteration in hematopoiesis could not be excluded. flk-2/flt-3 is known to be preferentially expressed in the fetal liver during definitive hematopoiesis and is used as a marker for early hematopoietic progenitors (33). Transcripts of flk-2/flt-3 were found to be below detectable levels as measured by RT-PCR in liver RNA samples from E12.5 homozygous mutants (Fig. 4). The lack of hematopoietic cells of any lineage as observed by histology and in touch preparations (Fig. 3) is in agreement with the results of colony formation in vitro and RT-PCR of flk-2/flt-3.
Table 2.
Hematopoietic progenitors in yolk sac and fetal liver
| Genotype (n) | Yolk sac (E11.5)
|
Genotype (n) | Fetal liver (E11.5)
|
||||
|---|---|---|---|---|---|---|---|
| E | M | Mix | E | M | Mix | ||
| +/+ (n = 12) | 10 ± 16 | 94 ± 60 | 15 ± 18 | +/+ (n = 12) | 14 ± 11 | 87 ± 29 | 18 ± 13 |
| +/− (n = 16) | 5 ± 5 | 86 ± 40 | 11 ± 10 | +/− (n = 16) | 14 ± 9 | 96 ± 32 | 23 ± 15 |
| −/− (n = 5) | 0 | 0 | 0 | −/− (n = 5) | 0 | 0 | 0 |
Abbreviations: n, numbers of embryos analyzed; E, erythroid colonies; M, myeloid colonies; Mix, erythroid/myeloid mixed colonies. Numbers represent colonies per 1 × 105 yolk sac cells or 2 × 104 fetal liver cells, respectively, mean ± SD.
DISCUSSION
The phenotype observed for Pebpb2 (−/−) mutants is indistinguishable from that reported for AML1-targeted embryos (refs. 8 and 9 and unpublished observations). In both cases, F2 homozygous (−/−) embryos die between E11.5 and E12.5. The direct cause of death appears to be severe hemorrhaging, mainly in the cerebral ventricles and spinal canals. The underlying mechanism is not considered to involve any deficiency of platelet formation, because GATA-2 (−/−) embryos, which lack all hematopoietic lineages, do not show signs of bleeding (7). Furthermore, NF-E2 (−/−) embryos devoid of platelet formation can survive to the neonatal stage (34). Instead, necrosis of the endothelial cells in the capillaries that perforate into the nerve layers has been suggested as the mechanism of hemorrhage in AML1 targeted embryos (9). The small hemorrhagic foci along the capillaries that were observed in E11.5 Pebpb2 (−/−) embryos to precede massive bleeding into the cerebral cavity (data not shown) are in line with this hypothesis. With both the Aml1 and Pebpb2 targeting, the impairment of definitive hematopoiesis in the fetal liver, a second major finding, is likely to be intrinsic to hematopoietic cells with the defect probably lying at the stage of early progenitors. Primitive erythropoiesis in the yolk sac, on the other hand, did not appear to be affected in either case.
The present study together with previous reports (8, 9) strongly suggest that the DNA binding subunit of PEBP2/CBF cannot function in vivo in the absence of the non-DNA binding subunit and that the two proteins exert their functions as one entity: the PEBP2/CBF heterodimer, in vivo. We previously reported that AML1 is expressed in hematopoietic cells in the fetal liver as revealed by in situ hybridization (35). Expression of Pebpb2 was also confirmed in the E12.5 fetal liver by RT-PCR (data not shown). It remains to be seen whether expression of AML1 and Pebpb2 is coordinated in hematopoietic progenitors. An analogous situation has been reported for a Scl-Tal1/Rbtn2 protein complex detected in cells of the erythroid lineage (36, 37). Scl-Tal1 is a DNA binding helix–loop–helix protein, whereas Rbtn2 contains a LIM domain that is considered to mediate protein–protein interactions. Targeting of each gene results in an identical phenotype, namely a defect in primitive erythropoiesis (3, 4).
In addition to Pebpa2b/Cbfa2/AML1, two more genes, Pebpa2a/Cbfa1/AML3 (13) and Pebpa2c/Cbfa3/AML2 (38–40), are known to encode a PEBP2/CBF α subunit. The polypeptides from all three genes can form heterodimers with the β subunit which therefore is presumed to have effects on expression of genes regulated by Pebpa2a and Pebpa2c as well as AML1. The phenotype seen in the Pebpb2 homozygous mutant suggests that Pebpa2a and Pebpa2c begin to exert their critical function after E12.5 in mouse development.
Recently, homozygous mutants of Pebpb2/Cbfb were generated by disrupting exon 5 that allowed the synthesis of only one of the three alternatively spliced transcripts: β3, but not the β1 or β2 isoforms (41, 42). The β3 transcript skips exon 5, and its product differs from the other two isoforms in carboxyl-terminal short stretches of amino acid residues (14, 25). The β3 protein was originally thought not to heterodimerize with the α protein (25). This observation was the basis of the targeting strategy employed by Sasaki et al. and Wang et al. (41, 42). More recently, it was found that the DNA binding ability of the α protein depends on the reduced state of the two cysteine residues in the Runt domain and that the β3 protein protects this reduction and significantly stimulates DNA binding (43). Furthermore, the β3 protein stimulated transcription as efficiently as the β1 and β2 proteins in a transient reporter assay with which the contribution of both the α and β subunits to the transactivation could be evaluated (T. Kanno, Y. Kanno, and Y.I., unpublished observations). The homozygous embryos reported by Sasaki et al. and Wang et al. synthesized the β3 transcript and polypeptide (41, 42), whereas neither transcripts nor polypeptides of any β isoform could be synthesized in our experiments. The small number of colonies that formed in vitro with Pebpb2/Cbfb (−/−) embryos (42) may be attributed to remaining β3 activity. However, the major phenotypic characters exhibited by the homozygous embryos of Sasaki et al. (41), Wang et al. (42), and our own were nearly identical. There is a possibility that the homozygous mutant embryos of Sasaki et al.(41) and Wang et al. (42) would have died earlier but survived up to E12.5 due to the presence of the β3 protein. Our results proved that the embryos died at E12.5 in the absence of any β activity. The main question remaining to be answered is what is the additional function of the β1/β2 isoforms that is absent in the β3.
PEBP2/CBF is presumed to interact with other essential transcription factors within the regulatory network, such as c-myb and GATA-2, to control the growth and differentiation of hematopoietic cells. Chimeric polypeptides generated by rearrangement of AML1 or Pebpb2 should interfere with their normal function, thus leading to deregulation of lineage and stage-specific differentiation of hematopoietic cells. The unraveling of the molecular mechanisms of hematopoietic cell development and leukemogenesis constitutes a major challenge for future research.
Acknowledgments
We thank N. Yanai for useful suggestions, H. Yamanaka and Y. Sugitani for technical assistance, and I. Imamura for secretarial assistance. This work was supported in part by research grants from the Ministry of Education, Science, and Culture of Japan (Y.I., M.S., and T.N.) and the Proposal-Based Advanced Industrial Technology R & D Program (Y.I. and M.S.).
ABBREVIATIONS
- E
embryonic day
- PEBP2
polyomavirus enhancer binding protein 2
- CBF
core binding factor
- ES cell
embryonic stem cell
- RT-PCR
reverse transcription–PCR
References
- 1.Orkin S H. J Biol Chem. 1995;270:4955–4958. doi: 10.1074/jbc.270.10.4955. [DOI] [PubMed] [Google Scholar]
- 2.Shivdasani R A, Orkin S H. Blood. 1996;87:4025–4039. [PubMed] [Google Scholar]
- 3.Shivdasani R A, Mayer E L, Orkin S H. Nature (London) 1995;373:432–434. doi: 10.1038/373432a0. [DOI] [PubMed] [Google Scholar]
- 4.Warren A J, Colledge W H, Carlton M B L, Evans M J, Smith A J H, Rabbitts T H. Cell. 1994;78:45–57. doi: 10.1016/0092-8674(94)90571-1. [DOI] [PubMed] [Google Scholar]
- 5.Porcher C, Swat W, Rockwell K, Fujiwara Y, Alt F W, Orkin S H. Cell. 1996;86:47–57. doi: 10.1016/s0092-8674(00)80076-8. [DOI] [PubMed] [Google Scholar]
- 6.Mucenski M L, McLain K, Kier A B, Swerdlow S H, Schreiner C M, Miller T A, Pietryga D W, Scott W J, Potter S S. Cell. 1991;65:677–689. doi: 10.1016/0092-8674(91)90099-k. [DOI] [PubMed] [Google Scholar]
- 7.Tsai F-Y, Keller G, Kuo F C, Weiss M, Chen J, Rosenblatt M, Alt F W, Orkin S H. Nature (London) 1994;371:221–226. doi: 10.1038/371221a0. [DOI] [PubMed] [Google Scholar]
- 8.Okuda T, van Deursen J, Hiebert S W, Grosveld G, Downing J R. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 9.Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe A H, Speck N A. Proc Natl Acad Sci USA. 1996;93:3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamachi Y, Ogawa E, Asano M, Ishida S, Murakami Y, Satake M, Ito Y, Shigesada K. J Virol. 1990;64:4808–4819. doi: 10.1128/jvi.64.10.4808-4819.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang S, Speck N A. Mol Cell Biol. 1992;12:89–102. doi: 10.1128/mcb.12.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bae S C, Yamaguchi-Iwai Y, Ogawa E, Maruyama M, Inuzaka M, Kagoshima H, Shigesada K, Satake M, Ito Y. Oncogene. 1993;8:809–814. [PubMed] [Google Scholar]
- 13.Ogawa E, Maruyama M, Kagoshima H, Inuzuka M, Lu J, Satake M, Shigesada K, Ito Y. Proc Natl Acad Sci USA. 1993;90:6859–6863. doi: 10.1073/pnas.90.14.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang S, Wang Q, Crute B E, Melnikova I N, Keller S R, Speck N A. Mol Cell Biol. 1993;13:3324–3339. doi: 10.1128/mcb.13.6.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kagoshima H, Shigesada K, Satake M, Ito Y, Miyoshi H, Ohki M, Pepling M, Gergen P. Trends Genet. 1993;9:338–341. doi: 10.1016/0168-9525(93)90026-e. [DOI] [PubMed] [Google Scholar]
- 16.Meyers S, Downing J R, Hiebert S W. Mol Cell Biol. 1993;13:6336–6345. doi: 10.1128/mcb.13.10.6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kania M A, Bonner A S, Duffy J B, Gergen J P. Genes Dev. 1990;4:1701–1713. doi: 10.1101/gad.4.10.1701. [DOI] [PubMed] [Google Scholar]
- 18.Daga A, Karlovich C A, Dumstrei K, Banerjee U. Genes Dev. 1996;10:1194–1205. doi: 10.1101/gad.10.10.1194. [DOI] [PubMed] [Google Scholar]
- 19.Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. Proc Natl Acad Sci USA. 1991;88:10431–10434. doi: 10.1073/pnas.88.23.10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nucifora G, Begy C R, Erickson P, Drabkin H A, Rowley J D. Proc Natl Acad Sci USA. 1993;90:7784–7788. doi: 10.1073/pnas.90.16.7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitani K, Ogawa S, Tanaka T, Miyoshi H, Kurokawa M, Mano H, Yazaki Y, Ohki M, Hirai H. EMBO J. 1994;13:504–510. doi: 10.1002/j.1460-2075.1994.tb06288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romana S P, Mauchauffe M, Le Coniat M, Chumakov I, Le Paslier D, Berger R, Bernard O A. Blood. 1995;85:3662–3670. [PubMed] [Google Scholar]
- 23.Golub T R, Barker G F, Bohlander S K, Hiebert S W, Ward D C, Bray-Ward P, Morgan E, Raimondi S C, Rowley J D, Gilliland D G. Proc Natl Acad Sci USA. 1995;92:4917–4921. doi: 10.1073/pnas.92.11.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shurtleff S A, Buijs A, Behm F G, Rubnitz J E, Raimondi S C, Hancock M L, Chan G C-F, Pui C-H, Grosveld G, Downing J R. Leukemia. 1995;9:1985–1989. [PubMed] [Google Scholar]
- 25.Ogawa E, Inuzuka M, Maruyama M, Satake M, Naito-Fujimoto M, Ito Y, Shigesada K. Virology. 1993;194:314–331. doi: 10.1006/viro.1993.1262. [DOI] [PubMed] [Google Scholar]
- 26.Liu P, Tarle S A, Hajra A, Claxton D F, Marlton P, Freedman M, Siciliano M J, Collins F S. Science. 1993;261:1041–1044. doi: 10.1126/science.8351518. [DOI] [PubMed] [Google Scholar]
- 27.Nakai S, Kawano H, Yudate T, Nishi M, Kuno J, Nagata A, Jishage K, Hamada H, Fujii H, Kawamura K, Shiba K, Noda T. Genes Dev. 1995;9:3109–3121. doi: 10.1101/gad.9.24.3109. [DOI] [PubMed] [Google Scholar]
- 28.Koopman P, Gubbay J, Collignon J, Lovell-Badge R. Nature (London) 1989;342:940–942. doi: 10.1038/342940a0. [DOI] [PubMed] [Google Scholar]
- 29.McClanahan T, Dalrymple S, Barkett M, Lee F. Blood. 1993;81:2903–2915. [PubMed] [Google Scholar]
- 30.Weiss M J, Keller G, Orkin S H. Genes Dev. 1994;8:1184–1197. doi: 10.1101/gad.8.10.1184. [DOI] [PubMed] [Google Scholar]
- 31.Bae S-C, Ogawa E, Maruyama M, Oka H, Satake M, Shigesada K, Jenkins N A, Gilbert D J, Copeland N G, Ito Y. Mol Cell Biol. 1994;14:3242–3252. doi: 10.1128/mcb.14.5.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu J, Maruyama M, Satake M, Bae S C, Ogawa E, Kagoshima H, Shigesada K, Ito Y. Mol Cell Biol. 1995;15:1651–1661. doi: 10.1128/mcb.15.3.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matthews W, Jordan C T, Wiegand G W, Pardoll D, Lemischka I R. Cell. 1991;65:1143–1152. doi: 10.1016/0092-8674(91)90010-v. [DOI] [PubMed] [Google Scholar]
- 34.Shivdasani R A, Rosenblatt M F, Zucker-Franklin D, Jackson C W, Hunt P, Saris C J M, Orkin S H. Cell. 1995;81:695–704. doi: 10.1016/0092-8674(95)90531-6. [DOI] [PubMed] [Google Scholar]
- 35.Satake M, Nomura S, Yamaguchi-Iwai Y, Takahama Y, Hashimoto Y, Niki M, Kitamura Y, Ito Y. Mol Cell Biol. 1995;15:1662–1670. doi: 10.1128/mcb.15.3.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valge-Archer V E, Osada H, Warren A J, Forster A, Li J, Baer R, Rabbitts T H. Proc Natl Acad Sci USA. 1994;91:8617–8621. doi: 10.1073/pnas.91.18.8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wadman I, Li J, Bash R O, Forster A, Osada H, Rabbitts T H, Baer R. EMBO J. 1994;13:4831–4839. doi: 10.1002/j.1460-2075.1994.tb06809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levanon D, Negreanu V, Bernstein Y, Bar-Am I, Avivi L, Groner Y. Genomics. 1994;23:425–432. doi: 10.1006/geno.1994.1519. [DOI] [PubMed] [Google Scholar]
- 39.Wijmenga C, Speck N A, Draopoli N C, Hofker M H, Liu P, Collins F S. Genomics. 1995;26:611–614. doi: 10.1016/0888-7543(95)80185-o. [DOI] [PubMed] [Google Scholar]
- 40.Bae S C, Takahashi E, Zhang Y W, Ogawa E, Shigesada K, Namba Y, Satake M, Ito Y. Gene. 1995;159:245–248. doi: 10.1016/0378-1119(95)00060-j. [DOI] [PubMed] [Google Scholar]
- 41.Sasaki K, Yagi H, Bronson R T, Tominaga K, Matsunashi T, Deguchi K, Tani Y, Kishimoto T, Komori T. Proc Natl Acad Sci USA. 1996;93:12359–12363. doi: 10.1073/pnas.93.22.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Stacy T, Miller J D, Lewis A F, Gu T-L, Huang X, Bushweller J H, Bories J-C, Alt F W, Ryan G, Liu P P, Wynshaw-Boris A, Binder M, Marin-Padilla M, Sharpe A H, Speck N A. Cell. 1996;87:697–708. doi: 10.1016/s0092-8674(00)81389-6. [DOI] [PubMed] [Google Scholar]
- 43.Kagoshima H, Akamatsu Y, Ito Y, Shigesada K. J Biol Chem. 1996;271:33074–33082. doi: 10.1074/jbc.271.51.33074. [DOI] [PubMed] [Google Scholar]