

Abstract
Midbrain and cerebellum development depends on an organizing center that is located at the midbrain–hindbrain junction of the vertebrate embryo. Expression of the two closely related transcription factors Pax2 and Pax5 overlaps spatially and temporally in this region of the developing central nervous system. To study a possible interaction of these transcription factors in midbrain and cerebellum patterning, we have generated Pax5, Krd double mutant mice. The transgene-induced Krd mutation corresponds to an ≈7-centimorgan chromosome 19 deletion that eliminates the entire Pax2 locus. The heterozygous Krd mutation deleting one Pax2 allele had no effect on midbrain and cerebellum development. Moreover, only minor developmental defects were previously observed at the midline of the inferior colliculus and anterior cerebellum in mice that were homozygous for a targeted Pax5 mutation. Similar morphological alterations were observed in 80% of all compound heterozygous Pax5 (+/−) Krd (+/−) mice. However, in the remaining 20% of compound heterozygotes, the inferior colliculi were missing, and the vermis of the cerebellum was severely disrupted due to the failure of the cerebellar primordia to fuse at the midline. Inactivation of the second Pax5 allele in Pax5 (−/−) Krd (+/−) mice resulted in complete loss of the posterior midbrain and cerebellum, as the tissue originating from the midbrain–hindbrain boundary region was deleted in the embryo as early as day 9.5. On the basis of these data, we propose that the cooperation of Pax2 and Pax5 is essential for normal functioning of the organizing center at the midbrain–hindbrain junction.
Keywords: Pax5 inactivation, Krd mutation
Early patterning of the central nervous system (CNS) results in subdivision of the neural tube along the anterior–posterior axis into the forebrain, midbrain, hindbrain, and spinal cord. Different strategies are subsequently used for the regional specialization of these CNS domains. Segmentation plays a prominent role in the compartmentalization and differentiation of the hindbrain (for review, see ref. 1). In contrast, the development of the midbrain and cerebellum depends on an organizing center that is located at the midbrain–hindbrain junction known as the isthmus (for review, see ref. 2). Isthmic tissue grafts were shown to induce an ectopic midbrain or cerebellum when transplanted into the chicken forebrain or hindbrain, respectively (for review, see ref. 3). Two secreted signaling molecules, Wnt1 and Fgf8, are specifically expressed in the organizing center at the isthmus and have thus been implicated in patterning of the midbrain region (4, 5). The Fgf8 protein was directly shown to have midbrain-inducing activity (6), whereas the midbrain and cerebellum failed to develop in mouse embryos homozygous for a Wnt1 null allele (7, 8). Four different transcription factors encoded by the homeobox genes En1 and En2 and the paired box genes Pax2 and Pax5 are transiently expressed in the midbrain–hindbrain junction of the mouse embryo (for review, see ref. 2). Targeted gene inactivation revealed a critical role for En1 in the early specification of the entire midbrain region (9), whereas the function of En2 is restricted to cerebellar patterning (10, 11). Herein we present evidence that the cooperation of Pax2 and Pax5 is indispensable for the function of the organizing center at the midbrain–hindbrain junction.
The Pax5 gene coding for the transcription factor BSAP is expressed in all B lymphoid tissues and testis in addition to the developing CNS (12). Consistent with this expression pattern, targeted gene disruption demonstrated an essential role for Pax5 in early pro-B cell development and in patterning of the midbrain and cerebellum (13, 14). The brain phenotype of homozygous Pax5 mutant mice was manifested as a change in the foliation pattern of the anterior cerebellum and as a reduction of the inferior colliculi near the midline. These morphological alterations did not impair the auditory function of the posterior midbrain (15) and were first visible only at embryonic day (E) 16.5 as a growth defect in the posterior region of the collicular neuroepithelium (13). The expression of the Pax5 gene is, however, already initiated at the three- to five-somite stage in the prospective midbrain of 8.25-day embryos (13, 16), indicating that a full week elapses between the onset of Pax5 expression and the first morphological manifestation of the Pax5 mutant phenotype. These observations combined with the mild brain phenotype suggest that the loss of Pax5 function is compensated for by another member of the Pax gene family that is expressed with a similar pattern in the developing midbrain–hindbrain boundary region.
A likely candidate for such a compensating gene is Pax2 because its expression pattern is temporally and spatially overlapping, yet not identical, with that of Pax5. The expression of Pax2 is highly dynamic in the embryonic midbrain region. It is already detectable in the late primitive streak embryo (day 7.5) and is subsequently observed in a relatively broad domain of the neural plate corresponding to the prospective midbrain and anterior hindbrain (16). At the one-somite stage, expression of the Wnt1 and En1 genes is initiated within the Pax2 expression domain followed by the onset of Pax5 and En2 transcription at the three- to five-somite stage (13, 16, 17). Upon neural tube closure, the expression of Pax2 becomes restricted to a narrow stripe at the isthmus, is thereafter down-regulated, and is no longer detectable at E11 (16, 17). At the same time, the Pax5 gene is transcribed in a broad domain centered around the midbrain–hindbrain boundary and is up-regulated to reach maximal expression at day 12.5 (12, 18). The Pax2 gene is expressed, in addition to the midbrain–hindbrain region, also in the developing kidney, eye, ear, and spinal cord (19, 20). Recently, a targeted null allele and a spontaneously occurring frameshift mutation of the mouse Pax2 gene have been described (21, 22). Analysis of homozygous mutant mice uncovered an essential role for Pax2 in midbrain, eye, ear, and kidney development consistent with Pax2 expression in these tissues (21–23). Curiously, the midbrain phenotype differed for the two Pax2 mutations analyzed. Whereas targeted inactivation of the Pax2 gene consistently resulted in exencephaly at the midbrain region (23), deletion of the posterior midbrain and cerebellum was frequently observed with the spontaneous Pax21Neu mutation (22).
A characteristic and unusual feature of mammalian Pax genes is the association of disease syndromes with heterozygosity for null alleles, known as haploinsufficiency (for review, see refs. 24 and 25). The function of these transcription factors is, therefore, thought to be particularly sensitive to gene dosage, since mutation of one allele already results in developmental abnormalities. Haploinsufficiency of PAX2 has recently been shown to be responsible for the dominantly inherited renal-coloboma syndrome in two human families (26, 27). Heterozygous patients exhibit bilateral optic nerve colobomas and various degrees of renal hypoplasia ranging from progressive kidney failure to the absence of any defect (26, 27). A phenotype closely resembling the syndrome of these patients (26) was previously described for the dominant mouse mutation Krd (kidney and retinal defects) (28). This mutation was generated by a transgene insertion on chromosome 19 that resulted in deletion of an approximately 7-centimorgan DNA segment that includes the Pax2 locus (28). Homozygous Krd embryos die prior to implantation due to the deletion of several recessive genes in addition to Pax2. Heterozygous Krd mice show a high incidence of kidney defects, including small, cystic, and hypoplastic kidneys, and abnormalities of eye development manifested as optic nerve defects and hypocellularity of the retinal cell layers resulting in altered electroretinograms (28, 29). The kidney and eye defects of heterozygous Krd mice not only closely resemble those of human patients with heterozygous point mutations in PAX2 (26, 27) but also are identical with the phenotype of heterozygous Pax2 mutant mice (21, 22). Hence, deletion of Pax2 is sufficient to account for the haploinsufficient phenotype of the Krd mutation.
Herein we have studied the interaction between Krd and Pax5 in patterning of the midbrain and cerebellum region by crossing the targeted Pax5 mutation (13) into the Krd (+/−) genetic background. No morphological abnormalities were observed in the midbrain and cerebellum of single mutant Krd (+/−) mice, indicating that deletion of only one Pax2 allele has no effect on midbrain development. However, progressive inactivation of one and both Pax5 alleles on the Krd (+/−) background resulted in increasing loss of the posterior midbrain and cerebellum. In Pax5 (−/−) Krd (+/−) embryos, most, if not all, tissue originating from the midbrain–hindbrain boundary is missing as early as E9.5. These data suggest that the cooperation of Pax2 and Pax5 is essential for normal development of the midbrain and cerebellum.
MATERIALS AND METHODS
Mice.
Krd (+/−) mutant mice of the C3H strain (28) were crossed with Pax5 (+/−) mutant mice (13) bred on the C57BL/6J and 129/Sv hybrid background. The genotype of the mutant mice was determined by PCR assay using oligonucleotides specific for either the Pax5 locus (14) or the chloramphenicol acetyltransferase gene insertion associated with the Krd mutation (28, 30).
Histological Analysis.
Brains of newborn and adult mice were dissected, fixed overnight at 4°C in PBS containing 4% paraformaldehyde, washed in PBS, and then processed for paraffin sectioning. Sections (6–8 μm) were stained with hematoxylin and eosin.
β-Galactosidase Staining.
Dissected embryos were briefly washed in PBS; fixed for 5–10 min on ice in a buffer containing 100 mM sodium phosphate (pH 7.3), 0.2% glutaraldehyde, 2 mM MgCl2, and 5 mM EGTA; and then stained with 5-bromo-4-chloro-3-indolyl β-d-galactoside as described (31).
Whole-Mount in Situ Hybridization Analysis.
Whole mount in situ hybridization was performed essentially as described (32). Single-stranded RNA probes were labeled with digoxigenin-UTP (Boehringer Mannheim). After hybridization, the labeled RNA probes were detected with anti-digoxigenin antibodies coupled to alkaline phosphatase. The Krox20 probe contained 750 bp of C-terminal coding sequences (33). The En1 and En2 probes contained 600 and 800 bp of 3′ untranslated sequences, respectively (34). The Wnt1 and Fgf8 probes consisted of a 900-bp and 400-bp cDNA fragment from the respective 3′ untranslated region (4).
RESULTS
One Functional Pax2 Allele Is Sufficient for Normal Development of the Midbrain and Cerebellum in Heterozygous Krd Mutant Mice.
Mutation of one Pax2 allele leads to a semidominant kidney and eye phenotype both in humans and mice (21, 22, 26, 27). Heterozygous Krd mutant mice also exhibit similar kidney and retinal defects, thus indicating that the Krd deletion can be regarded as a null mutation of the Pax2 locus in heterozygotes (28). During embryogenesis, the Pax2 gene is also transiently expressed in the midbrain–hindbrain junction in addition to the developing kidney, eye, and ear (19, 20). Recently, a defect in neural tube closure at the midbrain region was reported in embryos heterozygous for a targeted Pax2 mutation. In these heterozygous embryos, exencephaly was observed at a low frequency and in a genetic background-dependent manner (23). During our analysis of heterozygous Krd (+/−) embryos, we did not observe any case of exencephaly among all midgestation embryos (E9.5–E12.5) analyzed, suggesting that the Krd mutation does not interfere with neural tube closure on the mixed genetic background contributed by the inbred strains C57BL/6J, 129/Sv, and C3H (data not shown). Moreover, detailed inspection and histological analysis did not uncover any developmental abnormalities of the midbrain and cerebellum in adult Krd (+/−) mice (Fig. 1 D–F) compared with wild-type animals (Fig. 1 A–C). The inferior and superior colliculi of the midbrain were fully developed, and the foliation of the cerebellum was entirely normal. We conclude therefore that elimination of one Pax2 allele by the Krd deletion does not affect the development of CNS structures that originate from the mid–hindbrain boundary region. The absence of a semidominant midbrain phenotype, therefore, makes the heterozygous Krd mutation well suited for investigating its potential interaction with Pax5 in midbrain and cerebellum development by crossing the targeted Pax5 mutation (13) into the Krd (+/−) genetic background.
Figure 1.
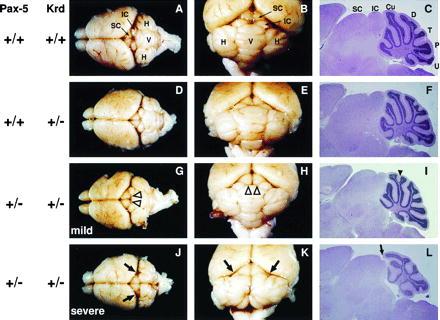
Brain phenotype of compound heterozygous Pax5 (+/−) Krd (+/−) mice. The entire brain (dorsal view in A, D, G, and J), a close-up view of the cerebellum and colliculi (B, E, H, and K), and histological sections of the midbrain and cerebellum (C, F, I, and L) are shown for 4-week-old mice of the genotypes indicated to the left. The sagittal plane of sectioning was close to the midline. Open arrowheads highlight the lateral displacement of the inferior colliculi in Pax5 (+/−) Krd (+/−) mice (G and H); a solid arrowhead indicates the intraculminate fissure present in the cerebella of these mice (I). Arrows point to the absence of the inferior colliculus in the lateral midbrain (J and K) and at the midline (L) of severely affected Pax5 (+/−) Krd (+/−) mice. SC, superior colliculus; IC, inferior colliculus; V, vermis; H, hemisphere; Cu, culmen; D, declive; T, tuber vermis; P, pyramis; U, uvula.
Absence of the Inferior Colliculi and Disruption of the Vermis in a Subset of Compound Heterozygous Pax5, Krd Mutant Mice.
Mice that were heterozygous for both the Pax5 and Krd mutations were born at the expected Mendelian frequency (18 of 76) in crosses between Pax5 (+/−) and Krd (+/−) mutant mice. Morphological analysis revealed two different phenotypes in the adult brains of these compound heterozygous mice (Fig. 1 and Table 1). The majority (80%) of these mice displayed only mild phenotypic abnormalities in the midbrain and cerebellum (Fig. 1 G–I). The inferior colliculi were laterally displaced and thus underdeveloped near the midline (Fig. 1 G and H), but the development of the superior colliculi was normal. Moreover, the cerebellum of these mice showed a minor alteration of the foliation pattern, as the lobule culmen was bifurcated due to the presence of an intraculminate fissure near the midline (Fig. 1I, arrowhead). This mild brain phenotype of compound heterozygous Pax5 (+/−) Krd (+/−) mice closely resembles that of homozygous Pax5 mutant mice (13). Hence, the Krd and Pax5 mutations affect midbrain patterning in a dosage-dependent manner as inactivation of one Pax5 allele combined with the heterozygous Krd deletion can result in a similar brain phenotype as mutation of both Pax5 alleles.
Table 1.
Variability of the brain phenotype in compound heterozygous Pax5, Krd mutant mice
| Pax5 | Krd | Phenotype | E16–18 | Newborn | 1–3 weeks | Total |
|---|---|---|---|---|---|---|
| +/− | +/− | Mild | 18 | 14 | 36 | 68 (80%) |
| +/− | +/− | Severe | 5 | 4 | 8 | 17 (20%) |
A more severe phenotype in the midbrain and cerebellum was observed in 20% of compound heterozygous mice (Fig. 1 J–L and Table 1). In these animals, even the lateral aspect of the inferior colliculi was lost. Moreover, the structure of the cerebellum was severely disrupted near the midline due to the presence of a deep furrow that divided the central part, known as the vermis, into two parts (Fig. 1 J and K). As a consequence, the foliation pattern in the two separated halves of the vermis was completely abnormal (Fig. 1L). One of the main functions of the vermis is to control the fine coordination of movement (35). Consistent with this notion, all compound heterozygous mice with a disrupted vermis displayed ataxia that was manifested as classical swaying behavior. These mice could not move straight on a plane surface but instead pivoted in circles. Interestingly, the swaying mice and the mice displaying the mild brain phenotype were fertile and thus could be mated with Pax5 (+/−) mice. Among the compound heterozygous progeny of these crosses, the mild and severe midbrain phenotype segregated again at a ratio of about 4:1 (data not shown), indicating that the observed phenotypic variability is inherent to the Pax5 (+/−) Krd (+/−) genotype (see Discussion).
The cerebellum develops during embryogenesis from the cerebellar anlage that becomes morphologically visible between E11 and E12 as two bilateral thickenings on both sides of the roof of the fourth ventricle (36, 37). As development proceeds, these bilateral thickenings expand, grow together, and fuse in the midline at approximately day 15 of gestation. After birth the cerebellum is subdivided into different folia by a complex folding process, whereby eight folds are derived from the fused central part of the cerebellum, thus constituting the vermis. A failure of the cerebellar primordia to fuse at the midline could, therefore, explain the disruption of the vermis in 20% of all Pax5 (+/−) Krd (+/−) mice. To test this hypothesis, we have analyzed the brain of these mice at birth. Like in adult mice, two distinct brain phenotypes were observed with newborn mice of the Pax5 (+/−) Krd (+/−) genotype. In 14 cases, the brains of compound heterozygous mutants were morphologically very similar to those of wild-type mice (Table 1 and Fig. 2 A and C). The cerebella of four mice were, however, not fused at the midline (Fig. 2D), which thus explains the disruption of the vermis in a subset of compound heterozygous mice. In this context it is interesting to note that we have not yet found an adult compound heterozygous mouse with developmental abnormalities in the vermis and inferior colliculi that were intermediate between the mild and severe phenotypes shown in Fig. 1 (data not shown). It appears therefore that either the completion or failure of midline fusion determines the severity of the brain phenotype in compound heterozygous mice.
Figure 2.
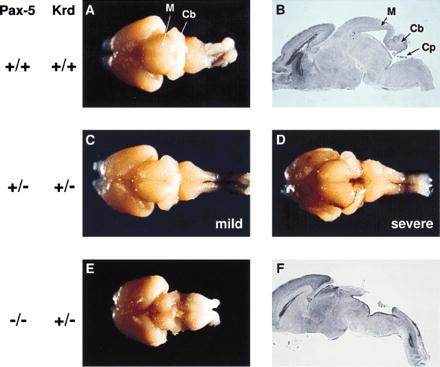
Absence of the cerebellum and posterior midbrain in Pax5 (−/−) Krd (+/−) mutant mice. The brains of newborn mice of the genotypes indicated to the left are shown as dorsal view (A and C–E). Midsagittal sections through the brains shown in A and E are displayed in B and F, respectively. M, midbrain; Cb, cerebellum; Cp, choroid plexus.
Early Deletion of the Cerebellum and Posterior Midbrain in Pax5 (−/−) Krd (+/−) Embryos.
Mice of the Pax5 (−/−) Krd (+/−) genotype were obtained by crossing compound heterozygous Pax5 (+/−) Krd (+/−) mice with Pax5 (+/−) mice. Pax5 (−/−) Krd (+/−) progeny were present at about the expected frequency of 12.5% during embryogenesis and at birth, indicating that these mutants could develop to term (Table 2). These mice were, however, unable to feed after birth since no milk could be found in their stomach. Consequently, they died within 24 h of birth, demonstrating that inactivation of the second Pax5 allele on the Krd (+/−) background is incompatible with postnatal life (Table 2). Superficial examination of the brains of newborn mutants revealed a large deletion of collicular and cerebellar tissues encompassing the posterior midbrain and cerebellum (Fig. 2E). As shown by the midsagittal section in Fig. 2F, the cerebellum and posterior midbrain were entirely missing, and the choroid plexus was fused to the remnants of the anterior midbrain. Interestingly, the analysis of 13 newborn Pax5 (−/−) Krd (+/−) mice revealed that the variability of the brain phenotype associated with this genotype was less pronounced than that of compound heterozygous mutant mice. In a few cases, some rudimentary tissue of the cerebellum was found on both lateral sides of the brain (data not shown).
Table 2.
Perinatal lethality of Pax5 (−/−) Krd (+/−) mutant mice
| Pax5 | Krd | E8.5–12.5 | E16–18 | Newborn | 1–3 weeks |
|---|---|---|---|---|---|
| −/− | +/+ | 33/321 | 4/73 | 5/76 | 7/105 |
| −/− | +/− | 44/321 | 9/73 | 13/76 | 0/105 |
The progeny of crosses between Pax5 (+/−) Pax2 (+/+) females and Pax5 (+/−) Krd (+/−) males were genotyped at different developmental stages, and the numbers of offspring with the two genotypes indicated to the left are shown as a fraction of the total number of mice analyzed. Progeny of both genotypes are expected at a frequency of 12.5% in these crosses.
To investigate the onset of the tissue deletion in the developing CNS of Pax5 (−/−) Krd (+/−) embryos, we have taken advantage of the fact that the inactivated Pax5 allele expresses an in-frame lacZ fusion gene under the control of the endogenous Pax5 locus (13). LacZ gene expression in Pax5 mutant embryos can, therefore, be used to trace cells originating from the midbrain–hindbrain boundary region. Strong β-galactosidase staining was observed in the corresponding brain region of Pax5 (−/−) embryos at day 9.5 (Fig. 3A) in agreement with our previous conclusion that Pax5 is not involved in autoregulation of its own transcription (13). Weaker, but consistent, lacZ expression was detected at the midbrain–hindbrain boundary in heterozygous Pax5 (+/−) mutant embryos regardless of whether one copy of the Pax2 locus was in addition deleted by the Krd mutation (Fig. 3 D and E). Importantly, lacZ expression was entirely lost at the 20-somite stage in Pax5 (−/−) Krd (+/−) embryos (Fig. 3B), whereas some β-galactosidase activity was still detectable in the 15-somite embryo (Fig. 3C). These data therefore demonstrate that complete deletion of the Pax5-expressing CNS tissue is achieved between the 15 and 20 somite stages in Pax5 (−/−) Krd (+/−) embryos. This finding was confirmed by whole-mount in situ hybridization analysis of gene transcripts that are specifically expressed at the midbrain–hindbrain junction. As illustrated for the Fgf8 gene, no expression could be detected at day 9.5 in the corresponding CNS region of Pax5 (−/−) Krd (+/−) embryos (Fig. 3I) in contrast to wild-type (Fig. 3G), compound heterozygous (Fig. 3H), and Pax5 (−/−) embryos (Fig. 3F). The Fgf8 gene was, however, transcribed in other regions of the Pax5 (−/−) Krd (+/−) embryo, including the previously described expression domains in the surface ectoderm of the forebrain and in the tail bud (4, 38, 39). Furthermore, transcripts of the En1, En2, and Wnt1 genes could also not be detected at day 9.5 in the midbrain–hindbrain junction of Pax5 (−/−) Krd (+/−) embryos (data not shown). Thus these data demonstrate that inactivation of both Pax5 alleles combined with the heterozygous Krd deletion eliminating one Pax2 allele results in an early loss of CNS tissue that normally gives rise to the posterior midbrain and cerebellum.
Figure 3.
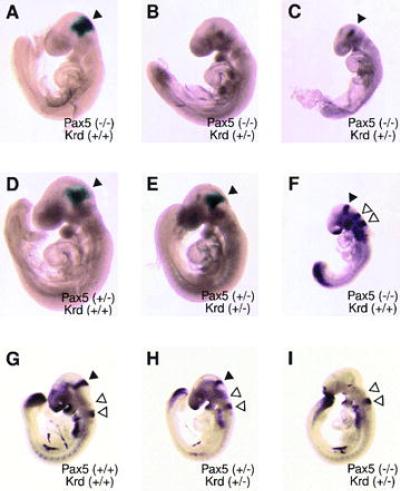
Early deletion of midbrain–hindbrain boundary tissue in Pax5 (−/−) Krd (+/−) embryos. (A–E) β-Galactosidase staining of E9.5 embryos of the indicated genotypes. An arrowhead denotes the expression domain of the Pax5–lacZ allele in the mid–hindbrain boundary region. (F–I) Whole-mount in situ hybridization of E9.5 embryos with Fgf8 and Krox20 probes. A solid arrowhead points to the Fgf8 expression domain in the mid–hindbrain region; open arrowheads denote expression of the Krox20 control gene in rhombomeres 3 and 5 of the hindbrain (33). All embryos were analyzed at the 20- to 21-somite stage except for the embryos in C and F, which contained only 14 or 15 somites. Note that the expression of Krox20 is down-regulated in rhombomere 3 during the transition from the 15- to 20-somite stage (33) and that intensive β-galactosidase staining is already seen at the 7-somite stage in the midbrain region of Pax5 mutant embryos (13).
DISCUSSION
Pax2, Pax5, and Pax8 constitute a subgroup within the mammalian Pax gene family whose protein products are highly related in sequence (12, 40). The three transcription factors also recognize DNA with identical sequence specificity (41) and are expressed in a partially overlapping manner in the developing CNS of the mouse embryo (12, 16, 18). Pax2 expression is initiated in the primordia of the midbrain and anterior hindbrain at E7.5 (16, 17) followed by expression of the Pax5 gene in the midbrain–hindbrain boundary region at E8.25 (13). Starting at E9, Pax8 is also weakly expressed in this region of the CNS (12, 42). To gain insight into the function and possible redundancy of these Pax transcription factors in midbrain and cerebellum development, we have crossed the targeted Pax5 mutation (13) into mice heterozygous for the Krd deletion, which eliminates the entire Pax2 locus (28). Deletion of one Pax2 allele did not affect midbrain development in Krd (+/−) mice. Likewise, targeted disruption of both Pax5 alleles in Pax5 (−/−) mice had only a minimal effect on morphogenesis of the inferior colliculus and cerebellum close to the midline (13). However, progressive inactivation of one and both Pax5 alleles on the Krd (+/−) background resulted in increasing deletion of the posterior midbrain and cerebellum. All tissue at the midbrain–hindbrain boundary was completely lost in Pax5 (−/−) Krd (+/−) embryos at the 20-somite stage (E9.5), as shown by the absence of midbrain-specific expression of the lacZ (Pax5), Fgf8, Wnt1, En1, and En2 genes. These experiments identify the period from E8.25 (onset of Pax5 expression) to E9.5 (tissue loss) as the critical time window for the interaction of Krd and Pax5 in midbrain and cerebellum development.
The Krd deletion eliminates several genes in addition to the Pax2 locus (28). Hence, another gene within the Krd deletion region could cooperate with Pax5 in midbrain and cerebellum patterning. However, we regard this as unlikely for the following two reasons. (i) The mammalian genome contains only a few genes that exhibit truly haploinsufficient phenotypes upon heterozygous mutation (43). Pax2 is one of these genes as shown by the haploinsufficiency of Pax2 mutations in humans (26, 27) and mice (21, 22) (see also Introduction). (ii) The specificity of the Krd (+/−) Pax5 (−/−) phenotype strongly argues that the cooperating gene located in the Krd deletion would have to be expressed in the developing midbrain–hindbrain boundary region. Thus it is highly improbable that the Krd deletion contains, apart from Pax2, a second gene that is both haploinsufficient and important for midbrain and cerebellum development. We therefore interpret the brain phenotype of Krd, Pax5 double mutant mice to indicate that the Pax2 gene cooperates with Pax5 in midbrain and cerebellum patterning. In this view, the gene dosage of Pax2 and Pax5 is critical for normal functioning of the organizing center at the midbrain–hindbrain boundary, suggesting that the two transcription factors may fulfill similar partially redundant functions in this region of the CNS. The wild-type Pax8 gene is, however, unable to compensate for the loss of Pax2 and Pax5 function and thus appears to be dispensable for early patterning of the midbrain and cerebellum in agreement with the late onset of its expression in this brain region (42).
In zebrafish, the formation of the midbrain–hindbrain boundary was previously shown to depend on the function of the Pax[b] gene, because neutralizing antibodies raised against the Pax[b] protein prevented morphogenesis of this CNS region in injected embryos (44). Recently, several Pax[b] alleles with intragenic mutations have been isolated in a large-scale genetic screen for zebrafish developmental mutants (45). These noi (no isthmus) mutations interfered with the development of the midbrain–hindbrain boundary region in homozygous embryos (45), thus genetically confirming the result of the antibody injection experiments. We have recently cloned all three members of the zebrafish Pax2/5/8 subfamily, which unequivocally identified Pax[b] as the zebrafish orthologue of the mouse Pax2 gene based on sequence similarity (P. Pfeffer and M.B., unpublished data). Thus in fish, the loss of Pax2 is sufficient to interfere with normal functioning of the organizing center at the midbrain–hindbrain boundary. A similar phenotype has recently been reported for the spontaneous mutation Pax21Neu of the mouse that resulted in frequent deletion of the posterior midbrain and cerebellum (22). The midbrain phenotype of homozygous Pax21Neu mice is, therefore, similar to that of Pax5 (−/−) Krd (+/−) mice, suggesting that the inactivation of the second Pax2 allele may be functionally equivalent to disruption of two Pax5 alleles. However, exencephaly rather than deletion of the midbrain region was observed in embryos that were homozygous for a targeted Pax2 gene mutation (23). The reason for this discrepancy is at present unclear, particularly since both mutations appear to be null alleles of the Pax2 gene (22, 23). Interestingly, the exencephalic embryos lacking Pax2 still express the Pax5 gene in a region corresponding to the midbrain–hindbrain junction (23). Moreover, the lacZ gene at the inactivated Pax5 locus is also normally expressed in homozygous Pax5 mutant embryos (ref. 13; see Fig. 3A). Hence, Pax5 is neither subject to autoregulation by its own product nor to cross-regulation by Pax2. Likewise, Pax2 expression appears to be normal in Pax5 (−/−) embryos (data not shown). It is therefore highly likely that the severe midbrain phenotype of Pax5 (−/−) Krd (+/−) embryos results from the failure to activate common target genes rather than from disruption of auto- or cross-regulatory loops of Pax2 and Pax5.
The haploinsufficient nature of mammalian Pax genes was revealed by the discovery that mutation of one allele of either Pax3 or Pax6 causes Splotch or Small eye in the mouse and Waardenburg syndrome or aniridia in humans, respectively (for review, see refs. 46–48). Heterozygous mutation at the Pax3 or Pax6 locus results in variable phenotypes in humans and mice (46–48). However, this phenotypic variability does not correlate with specific mutations, since large chromosomal deletions cause the same spectrum of developmental abnormalities as single point mutations (for review, see ref. 25). Phenotypic variability is also a hallmark of the renal-coloboma syndrome, which has recently been associated with single nucleotide mutations of the human PAX2 gene (26, 27). Herein we have reported considerable variation of the midbrain phenotype in compound heterozygous Pax5 (+/−) Krd (+/−) mice. The severity of the brain phenotype in these mice appears to be determined by stochastic events that either allow or prevent midline fusion of the cerebellar primordia during embryogenesis. In analogy to other Pax gene mutations, we propose that the fluctuation of the Pax5 (+/−) Krd (+/−) phenotype is caused by a critical threshold requirement of Pax2 and Pax5 in midbrain and cerebellum development.
During Drosophila development, the Pax genes paired (prd) and gooseberry (gsb) are involved together with the segment polarity genes engrailed (en) and wingless (wg) in a regulatory network controlling the segmentation process in the ectoderm (for review, see refs. 2 and 49). Interestingly, the murine homologues of these Drosophila genes are not only expressed in the midbrain–hindbrain boundary region of the mouse embryo but also play an important role in the development of this region of the CNS as shown by gene targeting experiments. Inactivation of the Wnt1 gene resulted in the deletion of the entire midbrain and cerebellum (7, 8), whereas the function of the En1 gene was required for morphogenesis of the posterior midbrain and cerebellum (9). Recently, the expression of the En1 transgene in the midbrain–hindbrain junction of Wnt1 null embryos was shown to rescue midbrain and cerebellum development, indicating that En1 is a target of the Wnt1 signaling pathway (50). Pax2 and Pax5 are also part of the regulatory network controlling midbrain and cerebellum patterning (refs. 13 and 22 and this study). Interestingly, the brain phenotype of Pax5 (−/−) Krd (+/−) embryos resembles that of En1 mutant mice (9) at the morphological level, although it may be slightly more extreme than the En1 (−/−) phenotype based on gene expression data (9). Moreover, the expression of the En2 gene depends on a midbrain-specific enhancer containing functionally important binding sites for Pax2 and Pax5 (42). Thus these findings indicate that the regulatory interactions of Pax, Wnt, and En genes have been conserved from Drosophila to mammals similar to the highly conserved role of Pax6 in eye development (51).
Acknowledgments
We are grateful to J. M. Jones for maintenance of the Krd mouse colony; to P. Charnay, G. Martin, A. Joyner, and H. Varmus for providing Krox20, Fgf8, En1, En2, and Wnt1 probes; and to P. Pfeffer and T. Jenuwein for critical reading of the manuscript. This work was in part supported by an European Molecular Biology Organization short term fellowship (P.U.), by a grant from the Austrian Industrial Research Promotion Fund and by National Institutes of Health Grant GM24872.
ABBREVIATIONS
- CNS
central nervous system
- E
embryonic day(s)
References
- 1.Lumsden A, Krumlauf R. Science. 1996;274:1109–1115. doi: 10.1126/science.274.5290.1109. [DOI] [PubMed] [Google Scholar]
- 2.Joyner A L. Trends Genet. 1996;12:15–20. doi: 10.1016/0168-9525(96)81383-7. [DOI] [PubMed] [Google Scholar]
- 3.Bally-Cuif L, Wassef M. Curr Opin Genet Dev. 1995;5:450–458. doi: 10.1016/0959-437x(95)90048-l. [DOI] [PubMed] [Google Scholar]
- 4.Crossley P H, Martin G R. Development (Cambridge, UK) 1995;121:439–451. doi: 10.1242/dev.121.2.439. [DOI] [PubMed] [Google Scholar]
- 5.Wilkinson D G, Bailes J A, McMahon A P. Cell. 1987;50:79–88. doi: 10.1016/0092-8674(87)90664-7. [DOI] [PubMed] [Google Scholar]
- 6.Crossley P H, Martinez S, Martin G R. Nature (London) 1996;380:66–68. doi: 10.1038/380066a0. [DOI] [PubMed] [Google Scholar]
- 7.McMahon A P, Bradley A. Cell. 1990;62:1073–1085. doi: 10.1016/0092-8674(90)90385-r. [DOI] [PubMed] [Google Scholar]
- 8.Thomas K R, Capecchi M R. Nature (London) 1990;346:847–850. doi: 10.1038/346847a0. [DOI] [PubMed] [Google Scholar]
- 9.Wurst W, Auerbach A B, Joyner A L. Development (Cambridge, UK) 1994;120:2065–2075. doi: 10.1242/dev.120.7.2065. [DOI] [PubMed] [Google Scholar]
- 10.Joyner A L, Herrup K, Auerbach B A, Davis C A, Rossant J. Science. 1991;251:1239–1243. doi: 10.1126/science.1672471. [DOI] [PubMed] [Google Scholar]
- 11.Millen K J, Wurst W, Herrup K, Joyner A L. Development (Cambridge, UK) 1994;120:695–706. doi: 10.1242/dev.120.3.695. [DOI] [PubMed] [Google Scholar]
- 12.Adams B, Dörfler P, Aguzzi A, Kozmik Z, Urbánek P, Maurer-Fogy I, Busslinger M. Genes Dev. 1992;6:1589–1607. doi: 10.1101/gad.6.9.1589. [DOI] [PubMed] [Google Scholar]
- 13.Urbánek P, Wang Z-Q, Fetka I, Wagner E F, Busslinger M. Cell. 1994;79:901–912. doi: 10.1016/0092-8674(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 14.Nutt S L, Urbánek P, Rolink A, Busslinger M. Genes Dev. 1997;11:476–491. doi: 10.1101/gad.11.4.476. [DOI] [PubMed] [Google Scholar]
- 15.Reimer K, Urbánek P, Busslinger M, Ehret G. Audiology. 1996;35:55–61. doi: 10.3109/00206099609071930. [DOI] [PubMed] [Google Scholar]
- 16.Rowitch D H, McMahon A P. Mech Dev. 1995;52:3–8. doi: 10.1016/0925-4773(95)00380-j. [DOI] [PubMed] [Google Scholar]
- 17.Püschel A W, Westerfield M, Dressler G R. Mech Dev. 1992;38:197–208. doi: 10.1016/0925-4773(92)90053-m. [DOI] [PubMed] [Google Scholar]
- 18.Asano M, Gruss P. Mech Dev. 1992;39:29–39. doi: 10.1016/0925-4773(92)90023-d. [DOI] [PubMed] [Google Scholar]
- 19.Nornes H O, Dressler G R, Knapik E W, Deutsch U, Gruss P. Development (Cambridge, UK) 1990;109:797–809. doi: 10.1242/dev.109.4.797. [DOI] [PubMed] [Google Scholar]
- 20.Dressler G R, Deutsch U, Chowdhury K, Nornes H O, Gruss P. Development (Cambridge, UK) 1990;109:787–795. doi: 10.1242/dev.109.4.787. [DOI] [PubMed] [Google Scholar]
- 21.Torres M, Gómez-Pardo E, Dressler G R, Gruss P. Development (Cambridge, UK) 1995;121:4057–4065. doi: 10.1242/dev.121.12.4057. [DOI] [PubMed] [Google Scholar]
- 22.Favor J, Sandulache R, Neuhäuser-Klaus A, Pretsch W, Chatterjee B, Senft E, Wurst W, Blanquet V, Grimes P, Spörle R, Schughart K. Proc Natl Acad Sci USA. 1996;93:13870–13875. doi: 10.1073/pnas.93.24.13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torres M, Gómez-Pardo E, Gruss P. Development (Cambridge, UK) 1996;122:3381–3391. doi: 10.1242/dev.122.11.3381. [DOI] [PubMed] [Google Scholar]
- 24.Stuart E T, Kioussi C, Gruss P. Annu Rev Genet. 1994;28:219–236. doi: 10.1146/annurev.ge.28.120194.001251. [DOI] [PubMed] [Google Scholar]
- 25.Strachan T, Read A P. Curr Opin Genet Dev. 1994;4:427–438. doi: 10.1016/0959-437x(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 26.Sanyanusin P, Schimmenti L A, McNoe L A, Ward T A, Pierpont M E M, Sullivan M J, Dobyns W B, Eccles M R. Nat Genet. 1995;9:358–364. doi: 10.1038/ng0495-358. [DOI] [PubMed] [Google Scholar]
- 27.Sanyanusin P, McNoe L A, Sullivan M J, Weaver R G, Eccles M R. Hum Mol Genet. 1995;4:2183–2184. doi: 10.1093/hmg/4.11.2183. [DOI] [PubMed] [Google Scholar]
- 28.Keller S A, Jones J M, Boyle A, Barrow L L, Killen P D, Green D G, Kapousta N V, Hitchcock P F, Swank R T, Meisler M H. Genomics. 1994;23:309–320. doi: 10.1006/geno.1994.1506. [DOI] [PubMed] [Google Scholar]
- 29.Green, D. C., Kapousta-Bruneau, N. V., Hitchcock, P. F. & Keller, S. A. (1997) Invest. Opthal. Visual Sci., in press. [PubMed]
- 30.Keller S A, Rosenberg M P, Johnson T M, Howard G, Meisler M H. Genes Dev. 1990;4:1316–1321. doi: 10.1101/gad.4.8.1316. [DOI] [PubMed] [Google Scholar]
- 31.Sanes J R, Rubenstein J L R, Nicolas J-F. EMBO J. 1986;5:3133–3142. doi: 10.1002/j.1460-2075.1986.tb04620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilkinson D G, Nieto M A. Methods Enzymol. 1993;225:361–373. doi: 10.1016/0076-6879(93)25025-w. [DOI] [PubMed] [Google Scholar]
- 33.Wilkinson D G, Bhatt S, Chavrier P, Bravo R, Charnay P. Nature (London) 1989;337:461–464. doi: 10.1038/337461a0. [DOI] [PubMed] [Google Scholar]
- 34.Millen K J, Hui C-C, Joyner A L. Development (Cambridge, UK) 1995;121:3935–3945. doi: 10.1242/dev.121.12.3935. [DOI] [PubMed] [Google Scholar]
- 35.Ghez C. In: Principles of Neural Science. Kandel E R, Schwartz J H, Jessell T M, editors. London: Pentice–Hill International; 1991. pp. 626–646. [Google Scholar]
- 36.Miale I L, Sidman R L. Exp Neurol. 1961;4:277–296. doi: 10.1016/0014-4886(61)90055-3. [DOI] [PubMed] [Google Scholar]
- 37.Hatten M E, Heintz N. Annu Rev Neurosci. 1995;18:385–408. doi: 10.1146/annurev.ne.18.030195.002125. [DOI] [PubMed] [Google Scholar]
- 38.Heikinheimo M, Lawshé A, Shackleford G M, Wilson D B, MacArthur C A. Mech Dev. 1994;48:129–138. doi: 10.1016/0925-4773(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 39.Ohuchi H, Yoshioka H, Tanaka A, Kawakami Y, Nohno T, Noji S. Biochem Biophys Res Commun. 1994;204:882–888. doi: 10.1006/bbrc.1994.2542. [DOI] [PubMed] [Google Scholar]
- 40.Walther C, Guenet J L, Simon D, Deutsch U, Jostes B, Goulding M D, Plachov D, Balling R, Gruss P. Genomics. 1991;11:424–434. doi: 10.1016/0888-7543(91)90151-4. [DOI] [PubMed] [Google Scholar]
- 41.Kozmik Z, Kurzbauer R, Dörfler P, Busslinger M. Mol Cell Biol. 1993;13:6024–6035. doi: 10.1128/mcb.13.10.6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song D-L, Chalepakis G, Gruss P, Joyner A L. Development (Cambridge, UK) 1996;122:627–635. doi: 10.1242/dev.122.2.627. [DOI] [PubMed] [Google Scholar]
- 43.Fisher E, Scambler P. Nat Genet. 1994;7:5–7. doi: 10.1038/ng0594-5. [DOI] [PubMed] [Google Scholar]
- 44.Krauss S, Maden M, Holder N, Wilson S W. Nature (London) 1992;360:87–89. doi: 10.1038/360087a0. [DOI] [PubMed] [Google Scholar]
- 45.Brand M, Heisenberg C-P, Jiang Y-J, Beuchle D, Lun K, Furutani-Seiki M, Granato M, Haffter P, Hammerschmidt M, Kane D A, Kelsh R N, Mullins M C, Odenthal J, van Eeden F J M, Nüsslein-Volhard C. Development (Cambridge, UK) 1996;123:179–190. doi: 10.1242/dev.123.1.179. [DOI] [PubMed] [Google Scholar]
- 46.Hanson I, van Heyningen V. Trends Genet. 1995;11:268–272. doi: 10.1016/s0168-9525(00)89073-3. [DOI] [PubMed] [Google Scholar]
- 47.Glaser T, Walton D S, Cai J, Epstein J A, Jepeal L, Maas R L. In: Molecular Genetics of Ocular Disease. Wiggs J L, editor. New York: Wiley–Liss; 1995. pp. 51–82. [Google Scholar]
- 48.Tassabehji M, Newton V E, Liu X-Z, Brady A, Donnai D, Krajewska-Walasek M, Murday V, Norman A, Obersztyn E, Reardon W, Rice J C, Trembath R, Wieacker R, Whiteford M, Winter R, Read A P. Hum Mol Genet. 1995;4:2131–2137. doi: 10.1093/hmg/4.11.2131. [DOI] [PubMed] [Google Scholar]
- 49.Noll M. Curr Opin Genet Dev. 1993;3:595–605. doi: 10.1016/0959-437x(93)90095-7. [DOI] [PubMed] [Google Scholar]
- 50.Danielian P S, McMahon A P. Nature (London) 1996;383:332–334. doi: 10.1038/383332a0. [DOI] [PubMed] [Google Scholar]
- 51.Halder G, Callaerts P, Gehring W. Science. 1995;267:1788–1792. doi: 10.1126/science.7892602. [DOI] [PubMed] [Google Scholar]