

Abstract
An efficient route to a variety of carbazoles and dibenzofurans has been developed. It involves the reaction of o-iodoanilines or o-iodophenols with silylaryl triflates in the presence of CsF to afford the N- or O-arylated products, which are subsequently cyclized using a Pd catalyst to carbazoles and dibenzofurans in good to excellent yields. By using this methodology, the carbazole alkaloid mukonine has been synthesized in 76% overall yield in three steps.
Introduction
Heterocycles are a very important class of organic compounds, because of their wide application in medicine, agriculture, and technology.1 Among nitrogen heterocycles, carbazole alkaloids are a growing class of natural products, which display a wide variety of biological activities, such as antitumor, antibacterial, antimicrobial and anti-inflammatory activities.2 Recently, a number of carbazole derivatives have been used as organic materials, due to their well-known photoconducting3 and semiconducting properties,4 and their charge-transport and high thermal properties.5 Dibenzofurans and their derivatives have also attracted the attention of organic chemists for many years due to their occurrence in a wide variety of pharmaceuticals and natural products possessing useful biological activities.6 Considerable effort has been devoted to the development of efficient methods for the synthesis of a wide range of substituted carbazoles, including the reductive cyclization of 2-nitrobiphenyl derivatives by suitable organophosphorus reagents7 and the classical Fisher-Borsche synthesis starting with appropriate cyclohexanone arylhydrazones.8 Recently, transition metal catalysts have also been widely used to construct these ring systems. For example, the palladium-mediated oxidative cyclization of N,N-diarylamines to carbazoles9 and diaryl ethers to dibenzofurans,10 and the palladium-catalyzed double N-arylation of primary amines with 2,2-dihalobiphenyls11 have been employed to prepare carbazoles and dibenzofurans. Quite recently, Wu has utilized anionic cycloaromatization to synthesize 5-substituted dibenzofurans and carbazoles.12 Despite these significant recent improvements, there still are limitations in the present methods. For example, (a) stoichiometric amounts of palladium are needed in the oxidative cyclization reaction; (b) harsh reaction conditions are usually needed and some processes cannot tolerate many functional groups; and (c) the yields usually are not very good. One simple, new, efficient and general method to synthesize both the carbazole and dibenzofuran ring systems would be quite attractive because of the growing interest in these compounds.13
Although arynes have historically received much attention from organic chemists, their use as reagents in synthetic organic chemistry has been somewhat limited due to the harsh reaction conditions needed to generate arynes and the often uncontrolled reactivity exhibited by these species.14 Recently, silylaryl triflate 1a15 has been employed in the presence of CsF to generate benzyne under very mild reaction conditions. The resulting aryne readily undergoes a variety of electrophilic and nucleophilic reactions,16 several novel insertion reactions,17 and even Pd-catalyzed annulation reactions.18 We have reported that silylaryl triflates can react with a variety of nucleophiles, such as anilines and phenols to generate very high yields of the corresponding N- and O-arylated products and even more important is the fact that halides, such as iodides and bromides, are readily tolerated by the reaction conditions.16c,d We have taken advantage of this methodology to develop a simple, economical, and efficient one-pot, two-step procedure to synthesize the carbazole and dibenzofuran ring systems in good to excellent yields through the cross-coupling of o-iodoanilines or o-iodophenols with silylaryl triflates in the presence of CsF, followed by palladium-catalyzed intramolecular cyclization.19 Herein, we wish to provide a full account of the scope and limitations of this chemistry. We have also applied this chemistry to the high yield three-step synthesis of an interesting carbazole alkaloid, mukonine.
Results and Discussion
Preparation of the Aryne Precursors
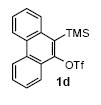
The arynes 1a-d were selected as substrates for our experiments. Aryne precursor 1a was selected as the simplest and most readily available aryne to study the scope of this chemistry, since it is commercially available. Aryne precursor 1b was selected to study the regioselectivity of the palladium-catalyzed cyclization step. The synthesis of silylaryl triflates 1a,20 1b,21 1c,22 and 1d23 have previously been reported.

Synthesis of Carbazoles and Analogues
The essential elements of our approach to the carbazole ring system are shown in Scheme 1. o-Iodoaniline is first allowed to react with the silylaryl triflate under very mild reaction conditions to afford the N-arylated o-iodoanilines A,16c which subsequently undergo intramolecular palladium-catalyzed arylation in the same pot to produce the carbazole derivatives B in good yields.
Scheme 1.
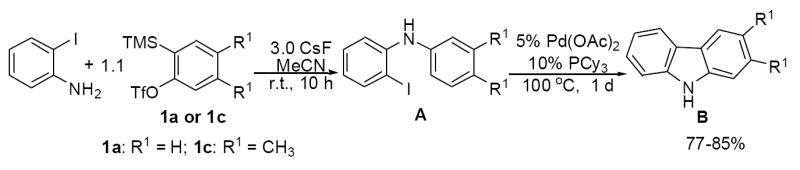
Our initial studies were directed towards achieving the optimal reaction conditions for the palladium-catalyzed intramolecular arylation step (A→B) (Scheme 1). A range of palladium catalysts were employed. We found that Pd(OAc)2 was the best catalyst. All other palladium catalysts [PdCl2(PPh3)2, Pd(PPh3)4, Pd(dba)2] examined afforded either comparable or lower yields. The ligand added to the reaction did not make much difference. Thus, the ligands dppe, dppm, and PCy3 (Cy = cyclohexyl) all worked well in our system. However, the solvent had a big effect on this cross-coupling reaction; DMF and toluene both gave very low yields of the carbazole products, while MeCN proved to be the best solvent for this process. The first step in this sequence had already been optimized during some of our earlier work on the N-arylation of amines.16c After our optimization work was complete, we settled on the following standard two step procedure. The iodoaniline (0.25 mmol), the silylaryl triflate (1.1 equiv) and CsF (3.0 equiv) were allowed to react at room temperature for 10 h in acetonitrile (4.0 mL) under air. Then Pd(OAc)2 (3.1 mg, 5 mol %) and PCy3 (7.0 mg, 10 mol %) were added and the reaction heated to 100 °C for 1 d under argon.
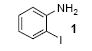
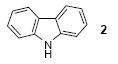
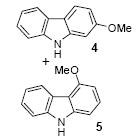
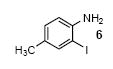
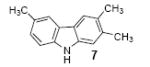
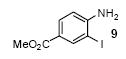
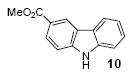
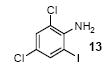
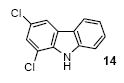
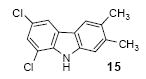
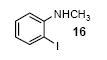
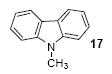
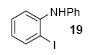
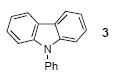
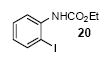
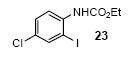
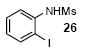
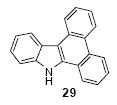
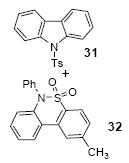
We next studied the scope and limitations of this two-step cross-coupling process by allowing a wide variety of iodoanilines and their derivatives to react with the silylaryl triflates 1a-d. The results are summarized in Table 1. A variety of o-iodoanilines react with silylaryl triflates 1a, 1b or 1c to afford, after Pd-catalyzed cyclization, high yields of the desired carbazoles (Table 1, entries 1-9). When a slight excess (1.1 equiv) of the silylaryl triflate 1a was allowed to react with o-iodoaniline (entry 1), the desired carbazole was obtained in a 77% yield under our standard reaction conditions. When 2.4 equiv of the silylaryl triflate 1a were allowed to react with o-iodoaniline, the N-phenylcarbazole was isolated in only a 66% yield (entry 2). o-Iodoaniline also reacts with the methoxy-substituted silylaryl triflate 1b, followed by palladium-catalyzed cyclization, to afford two carbazole derivatives 4 and 5 in a 5:1 ratio (entry 3). The fact that carbazole 4 is the major product can be easily explained by a steric effect during the Pd-catalyzed cyclization. The Pd-catalyzed cyclization is occurring at the less hindered position away from the methoxy group. When silylaryl triflate 1c was employed with 2-iodo-4-methylaniline, we obtained the desired product in a 68% yield (entry 4). Approximately a 4% yield of the isomeric product 1,2,6-trimethylcarbazole was also observed as detected by GC/MS. Again the Pd-catalyzed cyclization is occurring with high regioselectivity for the less hindered position away from the methyl group. When substituted iodoanilines were allowed to react with silylaryl triflate 1a, the corresponding carbazole derivatives could be obtained in good to excellent yields (entries 5-8). Similarly, 2,4-dichloro-6-iodoaniline reacts with 1.1 equiv of silylaryl triflate 1c to afford the desired product in an 85% yield. Approximately a 5% yield of the isomeric product 6,8-dichloro-1,2-dimethylcarbazole was also observed as detected by GC/MS (entry 9). The presence of a chlorine in the starting anilines does not appear to interfere with the overall process (entries 7-9). N-Methylcarbazole is also readily obtained in an 82% yield when N-methyl-2-iodoaniline was employed as the substrate (entry 10). N-Phenylcarbazole was obtained in a slightly higher yield by this cross-coupling procedure, when N-phenyl-2-iodoaniline was employed, instead of 2-iodoaniline, in the presence of an excess of the aryne (compare entries 2 and 12). N-(2-Iodophenyl)methanesulfonamide and ethyl 2-iodophenylcarbamate and their derivatives also react well with the silylaryl triflate 1a or 1c to afford high yields of the corresponding products (entries 13-18); again, only about 5% of a product isomeric with the products shown in entries 16 and 18 was observed by GC/MS analysis. Interestingly, when silylaryl triflate 1d was allowed to react with N-(2-iodophenyl)methanesulfonamide, we did not obtain the expected product, but rather the deprotected dibenzo[a,c]carbazole in a 62% yield (entry 19). Apparently the extended π-conjugation of the dibenzo[a,c]carbazole facilitates loss of the methanesulfonyl group. It is interesting that the reaction of N-tosyl-2-iodoaniline and silylaryl triflate 1a afforded a 1:1 ratio of compounds 31 and 32 in an 83% overall yield (entry 20). It seems rather surprising that the Pd-catalyzed cyclization onto the tosyl group competes so effectively with cyclization onto the phenyl ring. One can also start with N-(2-iodobenzyl)methanesulfonamide or N-benzyl-2-iodobenzenesulfonamide and silylaryl triflate 1a and produce the corresponding six membered ring products in good yields (entries 21 and 22). None of the product from cyclization onto the benzyl group in entry 22 is observed, possibly because this would involve formation of a seven membered ring.
Table 1.
Synthesis of carbazoles and analoguesa
| entry | substrate | aryl triflate | CsF
(equiv) |
product | % isolated
yield |
|---|---|---|---|---|---|
| 1 |
|
1a | 3.0 |
|
77 |
| 2 | 1 | 1a | 5.0 |
|
66b |
| 3 | 1 | 1b | 3.0 |
|
61
(5:1) |
| 4 |
|
1c | 3.0 |
|
68 |
| 5 | 6 | 1a | 3.0 |
|
69 |
| 6 |
|
1a | 3.0 |
|
68 |
| 7 |
|
1a | 3.0 |
|
72 |
| 8 |
|
1a | 3.0 |
|
87 |
| 9 | 13 | 1c | 3.0 |
|
85 |
| 10 |
|
1a | 3.0 |
|
82 |
| 11 | 16 | 1c | 3.0 |
|
71 |
| 12 |
|
1a | 3.0 |
|
76c |
| 13 |
|
1a | 3.0 |
|
85 |
| 14 | 20 | 1c | 3.0 |
|
85 |
| 15 |
|
1a | 3.0 |
|
86 |
| 16 | 23 | 1c | 3.0 |
|
85 |
| 17 |
|
1a | 3.0 |
|
85 |
| 18 | 26 | 1c | 3.0 |
|
85 |
| 19 | 26 |
|
3.0 |
|
62 |
| 20 |
|
1a | 3.0 |
|
83
(1:1) |
| 21 |
|
1a | 3.0 |
|
66 |
| 22 |
|
1a | 3.0 |
|
62 |
Reaction conditions: 0.25 mmol of aryl iodide are allowed to react with 1.1 equiv of the aryl triflate and the number of equiv of CsF shown in the table in 4.0 mL of MeCN as the solvent at room temperature for 10 h, followed by the addition of 5 mol % Pd(OAc)2 and 10 mol % PCy3 and heating for 1 d at 100 °C .
2.4 Equiv of aryl triflate were emp loyed and the reaction was run at room temperature for 2 d, followed by the addition of 5 mol % Pd(OAc)2 and 10 mol % PCy3 and heating for 1 d at 100 °C.
The reaction was run at room temperature for 1.5 d, followed by the addition of 5 mol % Pd(OAc)2 and 10 mol % PCy3 and heating for 1 d at 100 °C.
Although there are numerous examples in the literature of analogous palladium-catalyzed intramolecular cyclizations, the exact mechanism for this process is not well established at this time. Nevertheless, it seems clear that oxidative addition of the aryl halide to Pd(0) generates an arylpalladium species, which cyclizes to form a palladium(II)cycle, which in turn undergoes reductive elimination to the heterocycle while regenerating the Pd(0) catalyst.
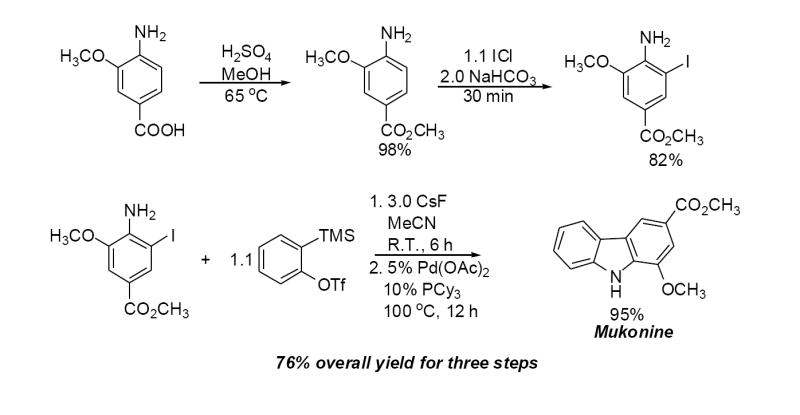
Synthesis of Mukonine
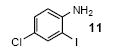
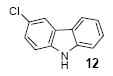
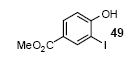
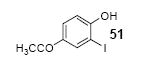
Mukonine [1-methoxy-3-(methoxycarbonyl)carbazole] is an alkaloid obtained from the Indian curry-leaf tree (Murraya koenigii).25 Mukonine has been synthesized by several different methods.26 By employing our procedure, mukonine can be obtained in a 76% overall yield in three steps from commercially available 4-amino-3-methoxybenzoic acid (Scheme 2). 3-Methoxy-4-aminobenzoic acid reacts with methanol to afford methyl 3-methoxy-4-aminobenzoate in a 98% yield. Iodination using ICl in dichloromethane affords methyl 4-amino-3-iodo-5-methoxybenzoate in an 82% yield. Employing our standard reaction conditions on methyl 4-amino-3-iodo-5-methoxybenzoate, mukonine can be obtained in a 95% yield in only one additional step.
Scheme 2.
Synthesis of Dibenzofurans and Analogues
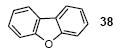
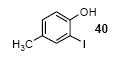
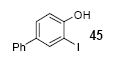
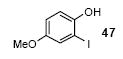
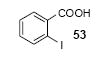
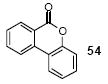
Since some effort has been devoted to the synthesis of carbazoles and related compounds, it seemed natural for us to extend this chemistry to the synthesis of dibenzofurans and related compounds by using o-iodophenol and derivatives. Indeed, we have also investigated the use of o-iodophenols in this process. Here, we needed to use 1.2 equiv of silylaryl triflates 1a or 1c, and 3.5 equiv of CsF to get the yields specified. The results are summarized in Table 2. o-Iodophenol reacts with silylaryl triflate 1a or 1c to afford, after Pd-catalyzed cyclization, the corresponding dibenzofurans in 70% and 67% yields respectively (entries 1 and 2). A variety of substituted iodophenols have also been employed in this process; most of them work quite well and afford the desired compounds in modest to good yields (entries 3-10). Similar to our carbazole results, when silylaryl triflate 1c was used in this cross-coupling process, approximately 4-5% yields of the expected isomeric products were observed as detected by GC/MS (entries 2, 4-6, and 8). It is noteworthy that the presence of an electron-withdrawing ester or ketone moiety on the phenol presents no difficulties and, in fact, these substrates gave slightly higher yields of the desired products (entries 9 and 10). In this process, one can also start with 2-iodobenzoic acid and allow it to react with silylaryl triflate 1a. Cyclization produces the desired six membered ring product benzo[c]coumarin in a 46% yield (entry 11).
Table 2.
Synthesis of dibenzofurans and analoguesa
| entry | substrate | aryl triflate | CsF
(equiv) |
product | % isolated
yield |
|---|---|---|---|---|---|
| 1 |
|
1a | 3.5 |
|
70 |
| 2 | 37 | 1c | 3.5 |
|
67b |
| 3 |
|
1a | 3.5 |
|
68 |
| 4 | 40 | 1c | 3.5 |
|
68b |
| 5 |
|
1a | 3.5 |
|
63 |
| 6 | 43 | 1c | 3.5 |
|
61b |
| 7 |
|
1a | 3.5 |
|
63 |
| 8 |
|
1c | 3.5 |
|
77b,c |
| 9 |
|
1a | 3.5 |
|
80 |
| 10 |
|
1a | 3.5 |
|
76 |
| 11 |
|
1a | 4.0 |
|
46d |
Reaction conditions: 0.25 mmol of aryl iodide are allowed to react with 1.2 equiv of aryl triflate and the number of equiv of CsF shown in the table in 4.0 mL of MeCN as the solvent at room temperature for 10 h, followed by the addition of 5 mol % Pd(OAc)2 and 10 mol % PCy3 and heating for 1 d at 100 °C.
About 4% of the isomeric product was detected by GC/MS.
In this reaction, 10 mo l % Pd(OAc)2 and 20 mol % PCy3 were used.
1.5 Equiv of aryl triflate were used.
Conclusions
In summary, we have developed a simple and efficient one-pot, two-step procedure to synthesize the carbazole and dibenzofuran ring systems and related compounds. This process involves the reaction of o-iodoanilines or o-iodophenols with silylaryl triflates in the presence of CsF to afford the N- or O-arylated products, which are subsequently cyclized to carbazoles and dibenzofurans in situ using a Pd catalyst. The starting materials are commercially available or can be easily prepared using known chemistry. The yields are good to excellent. Several new and multisubstituted carbazoles and dibenzofurans have been synthesized using this chemistry. By using this methodology, the carbazole alkaloid mukonine has been synthesized in 76% overall yield in three steps from a commercially available starting material.
Experimental Section
General
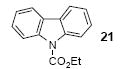
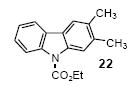
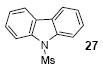
The 1H and 13C NMR spectra were recorded at 300 and 75.5 MHz or 400 and 100 MHz respectively. All melting points are uncorrected. High resolution mass spectra were recorded on a Kratos MS50TC double focusing magnetic sector mass spectrometer using EI at 70 eV. All reagents were used directly as obtained commercially unless otherwise noted. All yields reported represent an average of at least two independent runs. Silylaryl triflate 1a is commercially available. The product characterization data, and 1H and 13C NMR spectra for compounds 2, 14, 15, 17, 21, 22, 27, 33, 34, 38, and 50 can be found in the supporting information for our previous communication.19
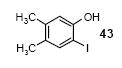
Preparation of methyl 4-amino-3-iodo-5-methoxybenzoate
To a solution of 0.905 g of methyl 4-amino-3-methoxybenzoate (5 mmol) and 0.84 g of NaHCO3 (10 mmol) in 15 mL of CH2Cl2 at room temperature was added 0.893 g of ICl (5.5 mmol) in 5 mL of CH2Cl2. The reaction mixture was stirred at room temperature for 30 min. The resulting solution was washed with saturated NaHCO3 (20 mL) solution and extracted with CH2Cl2 (20 mL). The combined CH2Cl2 fractions were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to afford 1.25 g of the desired product (82% yield) as a white solid: mp 93-94 °C; 1H NMR (300 MHz, CDCl3) δ 3.86 (s, 3H), 3.89 (s, 3H), 4.69 (s, 2H), 7.39 (d, J = 1.8 Hz, 1H), 8.00 (d, J = 1.8 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 52.2, 56.2, 80.7, 110.7, 120.5, 133.3, 142.0, 145.3, 166.3; IR (CDCl3, cm-1) 3495, 3391, 3001, 2949, 2838, 1694, 1268; HRMS m/z 306.9701 (calcd C9H10INO3, 306.9705).
General Procedure for the Synthesis of Carbazoles and Related Compounds
In a 4 dram vial, the silylaryl triflate (0.275 mmol) and CsF (0.75 mmol) were added to a solution of the o-iodoaniline (0.25 mmol) in acetonitrile (4 mL). The reaction mixture was allowed to stir at room temperature for 10 h under air. The vial was then flushed with argon and Pd(OAc)2 (5 mol %, 3.1 mg) and PCy3 (10 mol %, 7.0 mg) were added to the reaction, which was heated to 100 °C for 1 d. The resulting solution was washed with brine (20 mL) and extracted with diethyl ether (20 mL). The combined ether fractions were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to afford the desired product. Schlenk tube techniques are not necessary.
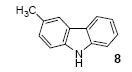
2,3,6-Trimethylcarbazole (7)
The indicated compound was obtained in a 68% yield as a white solid: mp 208-209 °C (lit.27 211-211.5 °C); 1H NMR (300 MHz, acetone-d6) δ 2.38 (s, 6H), 2.47 (s, 3H), 7.15 (dd, J = 8.4, 1.5 Hz, 1H), 7.25 (s, 1H), 7.33 (d, J = 8.4 Hz, 1H), 7.81 (s, 1H), 9.89 (s, 1H); 13C NMR (75 MHz, acetone-d6) δ 19.4, 29.1, 20.8, 110.6, 111.6, 119.7, 120.5, 121.4, 123.5, 126.3, 126.9, 127.5, 134.4, 138.6, 139.6; IR (CDCl3, cm-1) 3393, 2963, 2934, 2852, 1464; HRMS m/z 209.1209 (calcd C15H15N, 209.1204).
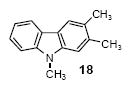
2,3,9-Trimethylcarbazole (18)
The indicated compound was obtained in a 71% yield as a light yellow solid: mp 88-89 °C; 1H NMR (300 MHz, acetone-d6) δ 2.43 (s, 3H), 2.46 (s, 3H), 3.78 (s, 3H), 7.15-7.23 (m, 2H), 7.33 (d, J = 8.1 Hz, 1H), 7.41 (td, J = 6.9, 0.9 Hz, 1H), 7.83 (s, 1H), 8.02 (d, J = 7.8 Hz, 1H); 13C NMR (75 MHz, acetone-d6) δ 19.3, 20.3, 38.3, 114.6, 115.2, 120.2, 120.9, 124.1, 124.3, 126.5, 127.0, 132.9, 136.9, 137.4, 138.7; IR (CDCl3, cm-1) 3016, 2961, 2935, 2899, 1601; HRMS m/z 209.1209 (calcd C15H15N, 209.1204).
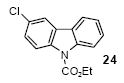
Ethyl 3-chlorocarbazole-9-carboxylate (24)
The indicated compound was obtained in an 85% yield as a white solid: mp 102-104 °C; 1H NMR (300 MHz, CDCl3) δ 1.54 (t, J = 7.2 Hz, 3H), 4.57 (q, J = 7.2 Hz, 2H), 7.24-7.50 (m, 3H), 7.84-7.88 (m, 2H), 8.18 (d, J = 9.0, 1H), 8.24 (d, J = 9.0, 1H); 13C NMR (75 MHz, CDCl3) δ 14.7, 63.5, 116.6, 117.5, 119.6, 120.0, 123.7, 124.9, 127.3, 127.4, 128.1, 129.1, 136.8, 138.8, 152.3; IR (CDCl3, cm-1) 3128, 3064, 2981, 2919, 1723, 1441; HRMS m/z 273.0560 (calcd C15H12ClNO2, 273.0556).
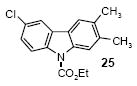
Ethyl 6-chloro-2,3-dimethylcarbazole-9-carboxylate (25)
The indicated compound was obtained in an 85% yield as a yellow solid: mp 125-126 °C; 1H NMR (300 MHz, CDCl3) δ 1.54 (t, J = 7.2 Hz, 3H), 2.37 (s, 3H), 2.40 (s, 3H), 4.56 (q, J = 7.2 Hz, 2H), 7.33 (dd, J = 8.7, 2.1 Hz, 1H), 7.59 (s, 1H), 7.80 (d, J = 2.1 Hz, 1H), 8.02 (s, 1H), 8.13 (d, J = 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 14.7, 20.2, 21.2, 63.4, 117.2, 117.5, 119.2, 120.4, 122.9, 126.5, 127.6, 128.9, 132.4, 136.7, 137.3, 137.6, 152.4; IR (CDCl3, cm-1) 2980, 2920, 2860, 1727, 1476; HRMS m/z 301.0873 (calcd C17H16ClNO2, 301.0869).
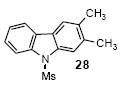
2,3-Dimethyl-9-(methanesulfonyl)carbazole (28)
The indicated compound was obtained in an 85% yield as white solid: mp 123-125 °C; 1H NMR (300 MHz, CDCl3) δ 2.41 (s, 3H), 2.43 (s, 3H), 2.93 (s, 3H), 7.38-7.45 (m, 2H), 7.74 (s, 1H), 7.91-7.94 (m, 2H), 8.12 (dd, J = 7.2, 1.2 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 20.2, 21.1, 38.4, 115.0, 115.5, 120.0, 120.8, 124.3, 124.5, 126.8, 127.1, 133.2, 137.2, 137.3, 138.6; IR (CDCl3, cm-1) 3061, 3032, 3012, 2930, 2856, 1471; HRMS m/z 273.0829 (calcd C15H15NO2S, 273.0823).
9-Tosylcarbazole (31)
The indicated compound was obtained in a 41% yield as a white solid: mp 129-130 °C (lit.28 128.5-130 °C); 1H NMR (300 MHz, CDCl3) δ 2.34 (s, 3H), 7.22 (dd, J = 8.7, 0.6 Hz, 2H), 7.42 (td, J = 7.5, 0.9 Hz, 2H), 7.57 (td, J = 7.5, 1.2 Hz, 2H), 7.77 (dt, J = 8.4, 1.8 Hz, 2H), 8.05-8.08 (m, 2H), 8.36 (dt, J = 8.4, 0.6 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 20.7, 115.2, 120.5, 124.4, 126.6, 126.7, 127.8, 130.1, 134.9, 138.5, 145.8; IR (CDCl3, cm-1) 3109, 3067, 2902, 1595, 1452; HRMS m/z 321.0827 (calcd C19H15NO2S, 321.0823).
2-Methyl-5,5-dioxide-6-phenyldibenzo[c,e][1,2]thiazine (32)
The indicated compound was obtained in a 42% yield as a white solid: mp 208-210 °C; 1H NMR (400 MHz, CDCl3) δ 2.55 (s, 3H), 6.98-7.01 (m, 1H), 7.22-7.25 (m, 2H), 7.33-7.40 (m, 6H), 7.83 (s, 1H), 7.84 (d, J = 8.0 Hz, 1H), 8.03-8.05 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 22.3, 123.4, 123.8, 125.4, 125.5, 125.7, 126.4, 128.3, 128.6, 129.5, 129.7, 132.4, 132.7, 138.4, 140.4, 143.5; IR (CDCl3, cm-1) 3067, 2925, 2853, 2253, 1600, 1492; HRMS m/z 321.0827 (calcd C19H15NO2S, 321.0823).
6-Benzyl-5,5-dioxidedibenzo[c,e][1,2]thiazine (36)
The indicated compound was obtained in a 62% yield as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 5.05 (s, 2H), 7.18 (s, 5H), 7.23 (dd, J = 8.0, 1.6 Hz, 1H), 7.28 (td, J = 8.0, 1.2 Hz, 1H), 7.33 (td, J = 8.0, 1.2 Hz, 1H), 7.54 (td, J = 7.6, 1.2 Hz, 1H), 7.65 (td, J = 8.0, 1.2 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.93 (dd, J = 7.6, 1.6 Hz, 1H), 7.99 (dd, J = 7.6, 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 52.9, 122.2, 122.7, 125.6, 125.7, 125.7, 125.8, 127.7, 127.9, 128.4, 128.8, 130.4, 132.5, 132.7, 135.4, 135.9, 138.8; IR (CDCl3, cm-1) 3064, 3030, 2923, 2851, 1601, 1477; HRMS m/z 321.0827 (calcd C19H15NO2S, 321.0823).
General Procedure for the Synthesis of Dibenzofurans and Related Compounds
In a 4 dram vial, the silylaryl triflate (0.3 mmol) and CsF (0.875 mmol) were added to a solution of the o-iodophenol (0.25 mmol) in acetonitrile (4 mL). The rest of the procedure is the same as that used in the synthesis of the carbazoles. Schlenk tube techniques are not necessary.
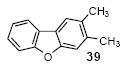
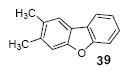
2,3-Dimethyldibenzofuran (39)
The indicated compound was obtained in a 67% yield as a white solid: mp 90-91 °C; 1H NMR (300 MHz, CDCl3) δ 2.39 (s, 3H), 2.40 (s, 3H), 7.34-7.42 (m, 3H), 7.52 (d, J = 7.5 Hz, 1H), 7.68 (s, 1H), 7.87 (dd, J = 7.5, 0.9 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 20.2, 20.9, 111.7, 112.4, 120.4, 121.1, 122.1, 122.6, 124.6, 126.6, 131.4, 136.6, 155.3, 156.4; IR (CDCl3, cm-1) 3046, 2968, 2941, 2922, 1448; HRMS m/z 196.0890 (calcd C14H12O, 196.0888).
3-Methyldibenzofuran (41)
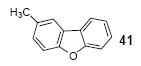
The indicated compound was obtained in a 68% yield as a white solid: mp 42-43 °C; 1H NMR (400 MHz, CDCl3) δ 2.49 (s, 3H), 7.22-7.25 (m, 1H), 7.31 (td, J = 7.6, 1.2 Hz, 1H), 7.40-7.45 (m, 2H), 7.54 (d, J = 8.0 Hz, 1H), 7.73 (t, J = 0.8 Hz, 1H), 7.90 (dd, J = 8.0, 0.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.6, 111.4, 111.9, 120.8, 120.9, 122.7, 124.4, 124.5, 127.2, 128.4, 132.4, 154.7, 156.7; IR (CDCl3, cm-1) 3026, 2978, 2941, 2921, 1446; HRMS m/z 182.0734 (calcd C13H10O, 182.0731).
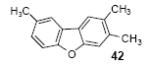
2,3,6-Trimethyldibenzofuran (42)
The indicated compound was obtained in a 68% yield as a white solid: mp 97-98 °C; 1H NMR (400 MHz, CDCl3) δ 2.35 (s, 3H), 2.36 (s, 3H), 2.46 (s, 3H), 7.17 (dd, J = 8.4, 1.2 Hz, 1H), 7.28 (s, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.61 (s, 1H), 7.63 (d, J = 0.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 20.2, 21.0, 21.6, 111.2, 112.4, 120.5, 121.0, 122.1, 124.6, 127.6, 131.2, 132.1, 136.4, 154.7, 155.6; IR (CDCl3, cm-1) 3014, 2970, 2940, 2921, 1454; HRMS m/z 210.1048 (calcd C15H14O, 210.1044).
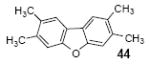
2,3,6,7-Tetramethyldibenzofuran (44)
The indicated compound was obtained in a 61% yield as a white solid: mp 183-184 °C; 1H NMR (400 MHz, CDCl3) δ 2.40 (s, 6H), 2.41 (s, 6H), 7.32 (s, 2H), 7.64 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 20.2, 20.9, 112.3, 120.8, 122.3, 131.0, 135.7, 155.3; IR (CDCl3, cm-1) 3026, 2971, 2919, 2856, 1454; HRMS m/z 224.1204 (calcd C16H16O, 224.1201).
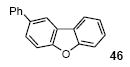
3-Phenyldibenzofuran (46)
The indicated compound was obtained in a 63% yield as a white solid: mp 94-96 °C; 1H NMR (400 MHz, CDCl3) δ 7.31-7.37 (m, 2H), 7.43-7.48 (m, 3H), 7.55-7.66 (m, 5H), 7.96 (dd, J = 7.6, 0.8 Hz, 1H), 8.11 (d, J = 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 112.0, 119.4, 120.9, 123.1, 124.5, 124.9, 126.9, 127.3, 127.6, 127.7, 129.1, 136.6, 141.6, 156.0, 156.9; IR (CDCl3, cm-1) 3057, 3032, 2920, 1601, 1470; HRMS m/z 280.0892 (calcd C18H12O, 280.0888).
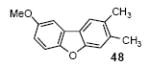
6-Methoxy-2,3-dimethyldibenzofuran (48)
The indicated compound was obtained in a 67% yield as a white solid: mp 97-98 °C; 1H NMR (400 MHz, CDCl3) δ 2.38 (s, 3H), 2.39 (s, 3H), 3.88 (s, 3H), 6.90 (dd, J = 8.4, 2.4 Hz, 1H), 7.05 (d, J = 2.4 Hz, 1H), 7.29 (s, 1H), 7.59 (s, 1H), 7.73 (d, J = 8.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 20.2, 20.8, 55.9, 96.8, 110.7, 112.2, 117.8, 120.4, 120.7, 122.2, 131.3, 135.0, 155.4, 157.7, 159.6; IR (CDCl3, cm-1) 3015, 2999, 2979, 2939, 1590; HRMS m/z 226.0996 (calcd C15H14O2, 226.0993).
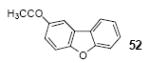
3-Acetyldibenzofuran (52)
The indicated compound was obtained in a 76% yield as a white solid: mp 69-70 °C; 1H NMR (300 MHz, CDCl3) δ 2.71 (s, 3H), 7.38 (td, J = 7.5, 1.2 Hz, 1H), 7.70 (td, J = 7.5, 1.2 Hz, 1H), 7.57 (s, 1H), 7.60 (s, 1H), 7.99 (dd, J = 7.5, 0.6 Hz, 1H), 8.10 (dd, J = 8.7, 1.8 Hz, 1H), 8.57 (d, J = 1.8 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 27.0, 111.8, 112.2, 121.2, 121.8, 123.6, 123.9, 124.8, 128.2, 128.3, 132.7, 157.1, 159.1, 197.5; IR (CDCl3, cm-1) 3063, 3002, 2922, 2849, 1678, 1598; HRMS m/z 210.0683 (calcd C14H10O2, 210.0680).
Acknowledgments
We are grateful to the National Institutes of Health Kansas University Center of Excellence in Chemical Methodologies and Library Development (P50 GM069663) for their generous financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pozharskii AF, Soldatenkov AT, Katritzky AR. Heterocycles in Life and Society-An Introduction to Heterocyclic Chemsitry and Biochemistry and the Role of Heterocycles in Science, Technology, Medicine and Agriculture. John Wiley & Sons; Chichester, England: 1997. [Google Scholar]
- 2.For reviews, see: Knölker H-J, Reddy KR. Chem Rev. 2002;102:4303–4427. doi: 10.1021/cr020059j.Knölker H-J. Top Curr Chem. 2005;244:115–148.Chakraborty DP. In: The Alkaloids. Bossi A, editor. Vol. 44. Academic Press; New York: 1993. p. 257.Gallagher PT. Science of Synthesis. Vol. 10. Thieme; Stuttgart: 2000. p. 693.Omura S, Sasaki Y, Iwai Y, Takeshima H. J Antibiot. 1995;48:535–548. doi: 10.7164/antibiotics.48.535.Knölker HJ. In: Advances in Nitrogen Heterocycles. Moody CJ, editor. Vol. 1. JAI; Greenwich: 1995. p. 173.Moody CJ. Synlett. 1994:681–688.Bergman J, Pelcman B. Pure Appl Chem. 1990;62:1967–1976.Sakano K, Ishimaru K, Nakamura S. J Antibiot. 1980;33:683–689. doi: 10.7164/antibiotics.33.683.Das KC, Chakraborty DP, Bose PK. Experientia. 1965;21:340. doi: 10.1007/BF02144703.
- 3.a Ganguly T, Farmer L, Li W, Bergeron JY, Gravel D, Durocher G. Macromolecules. 1993;26:2315–2322. [Google Scholar]; b Ganguly T, Bergeron JY, Farmer L, Gravel D, Durocher G. J Lumin. 1994;59:247–256. [Google Scholar]
- 4.Bouchard J, Wakim S, Leclerc M. J Org Chem. 2004;69:5705–5711. doi: 10.1021/jo049419o. [DOI] [PubMed] [Google Scholar]
- 5.a Justin Thomas KR, Lin JT, Tao Y-T, Ko C-W. J Am Chem Soc. 2001;123:9404–9411. doi: 10.1021/ja010819s. [DOI] [PubMed] [Google Scholar]; b Biswas M, Das SK. Polymer. 1982;23:1713–1725. [Google Scholar]; c Biswas M, Mishra GC. Makromol Chem. 1981;182:261–264. [Google Scholar]; d Biswas M, Das SK. Eur Polym J. 1981;17:1245–1251. [Google Scholar]
- 6.a Abe H, Uchiyama M, Tanaka Y, Saitô H. Tetrahedron Lett. 1976;17:3807–3810. [Google Scholar]; b Morris HR, Taylor GW, Masento MS, Jermyn KA, Kay RR. Nature. 1987;328:811–814. doi: 10.1038/328811a0. [DOI] [PubMed] [Google Scholar]; c Hogberg H-E, Hjalmarsson M. Tetrahedron Lett. 1978;19:5215–5218. [Google Scholar]; d Kokubun T, Harborne JB, Eagles J, Waterman PG. Phytochemistry. 1995;39:1039–1042. [Google Scholar]
- 7.a Cadogan JIG. Quart Rev. 1962;16:208–239. [Google Scholar]; b Cadogan JIG, Cameron-Wood M. Proc Chem Soc. 1962;361 [Google Scholar]; c Cadogan JIG, Cameron-Wood M, Mackie RK, Searle RJG. J Chem Soc. 1965:4831–4837. [Google Scholar]; d Cadogan JIG. Synthesis. 1969:11–17. [Google Scholar]
- 8.Gilchrist TL. Heterocyclic Chemistry. Pitman Publishing Ltd; London: 1985. [Google Scholar]
- 9.Åkermark B, Eberson L, Jonsson E, Pettersson E. J Org Chem. 1975;40:1365–1367. [Google Scholar]
- 10.a Davidson JM, Triggs C. J Org Chem Soc A. 1968:1331–1334. [Google Scholar]; b Itatani H, Yoshimoto H. J Org Chem. 1973;38:76–79. [Google Scholar]; c Clarke FRS, Norman RPC, Thomas CB, Wilson SJ. J Chem Soc Perkin Trans 1. 1974;38:1289–1294. [Google Scholar]; d Akermark B, Eberson L, Jonsson E, Petterson E. J Org Chem. 1975;40:1365–1367. [Google Scholar]; e De Lombaert S, Blanchard L, Stamford LB, Tan J, Wallace EM, Satoh Y, Fitt J, Hoyer D, Simonsbergen D, Moliterni J, Marcopoulos N, Savage P, Chou M, Trapani AJ, Jeng AY. J Med Chem. 2000;43:488–504. doi: 10.1021/jm990507o. [DOI] [PubMed] [Google Scholar]
- 11.Nozaki K, Takahashi K, Nakano K, Hiyama T, Tang H-Z, Fujiki M, Yamagushi S, Tamao K. Angew Chem Int Ed Engl. 2003;42:2051–2053. doi: 10.1002/anie.200250648. [DOI] [PubMed] [Google Scholar]
- 12.a Lee C-Y, Lin C-F, Lee J-L, Chiu C-C, Lu W-D, Wu M-J. J Org Chem. 2004;69:2106–2110. doi: 10.1021/jo0303158. [DOI] [PubMed] [Google Scholar]; b Wu M-J, Lee C-Y, Lin C-F. Angew Chem Int Ed Engl. 2002;41:4077–4079. doi: 10.1002/1521-3773(20021104)41:21<4077::AID-ANIE4077>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 13.a Istvan EJ, Ling Y, Kassoum N, Tork T, Wu X, Cao Y, Guo R, Li B, Zhu X, Huang Y, Long YQ. J Med Chem. 2001;44:4313–4324. doi: 10.1021/jm010016f. [DOI] [PubMed] [Google Scholar]; b Yasuo K, Yutaka A, Takeshi S.J Org Chem 2001668612–8615.11735545 [Google Scholar]; c Silvere A, Edwige N, Mireille L, Cecile M, Cao W, Kiefer DW, Sheila C, Gerard L, Patrice F.Bioorg Med Chem Lett 200212209–212.11755356 [Google Scholar]
- 14.For reviews on the use of arynes in organic synthesis, see: Kessar SV. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 4. Pergamon Press; New York: 1991. pp. 483–515.Pellissier H, Santelli M. Tetrahedron. 2003;59:701–730.
- 15.Himeshima Y, Sonoda T, Kobayashi H. Chem Lett. 1983:1211–1214. [Google Scholar]
- 16.a Yoshida H, Honda Y, Shirakawa E, Hiyama T. Chem Commun. 2001:1880–1881. doi: 10.1039/b103745p. [DOI] [PubMed] [Google Scholar]; b Yoshida H, Shirakawa E, Honda Y, Hiyama T. Angew Chem Int Ed Engl. 2002;41:3247–3249. doi: 10.1002/1521-3773(20020902)41:17<3247::AID-ANIE3247>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; c Liu Z, Larock RC. Org Lett. 2003;5:4673–4675. doi: 10.1021/ol0358612. [DOI] [PubMed] [Google Scholar]; d Liu Z, Larock RC. Org Lett. 2004;6:99–102. doi: 10.1021/ol0361406. [DOI] [PubMed] [Google Scholar]
- 17.a Liu Z, Larock RC. J Am Chem Soc. 2005;127:13112–13113. doi: 10.1021/ja054079p. [DOI] [PubMed] [Google Scholar]; b Tambar UK, Stoltz BM. J Am Chem Soc. 2005;127:5340–5341. doi: 10.1021/ja050859m. [DOI] [PubMed] [Google Scholar]
- 18.a Liu Z, Zhang X, Larock RC. J Am Chem Soc. 2005;127:15716–15717. doi: 10.1021/ja055781o. [DOI] [PubMed] [Google Scholar]; b Zhang X, Larock RC. Org Lett. 2005;7:3973–3976. doi: 10.1021/ol0514597. [DOI] [PubMed] [Google Scholar]
- 19.Liu Z, Larock RC. Org Lett. 2004;6:3739–3741. doi: 10.1021/ol048564l. [DOI] [PubMed] [Google Scholar]
- 20.Himeshima Y, Sonoda T, Kobayashi H. Chem Lett. 1983:1211–1214. [Google Scholar]
- 21.Peña D, Pérez D, Guitián E, Castedo L. J Am Chem Soc. 1999;121:5827–5828. [Google Scholar]
- 22.Yoshida H, Sugiura S, Kunai A. Org Lett. 2002;4:2767–2769. doi: 10.1021/ol0262845. [DOI] [PubMed] [Google Scholar]
- 23.Peña D, Pérez D, Guitián E, Castedo L. J Org Chem. 2000;65:6944–6950. doi: 10.1021/jo000535a. [DOI] [PubMed] [Google Scholar]
- 24.a Campeau L-C, Parisien M, Jean A, Fagnou K. J Am Chem Soc. 2006;128:581–590. doi: 10.1021/ja055819x. [DOI] [PubMed] [Google Scholar]; b Parisien M, Damien V, Fagnou K. J Org Chem. 2005;70:7578–7584. doi: 10.1021/jo051039v. [DOI] [PubMed] [Google Scholar]
- 25.Chakraborty DP. In: Prog Chem Org Nat Prod. Herz W, Grisebach H, Kirby GW, editors. Vol. 34. Springer; Wien: 1977. p. 299. [Google Scholar]
- 26.a Knölker H-J, Wolpert M. Tetrahedron. 2003;59:5317–5322. [Google Scholar]; b Kuwahara A, Nakano K, Nozaki K. J Org Chem. 2005;70:413–419. doi: 10.1021/jo048472+. [DOI] [PubMed] [Google Scholar]
- 27.Kuroki M, Tsunashima Y. J Heterocyclic Chem. 1981;18:709–714. [Google Scholar]
- 28.Wassmundt FW, Babic GT. J Org Chem. 1982;47:3585–3587. [Google Scholar]