

SUMMARY
Protein conformational stability is an important concern in many fields. Here, we describe a strategy for significantly increasing conformational stability by optimizing β-turn sequence. Proline and glycine residues are statistically preferred at several β-turn positions, presumably because their unique side chains contribute favorably to conformational stability in certain β-turn positions. However, β-turn sequences often deviate from preferred proline or preferred glycine. Therefore, our strategy involves replacing non-proline and non-glycine β-turn residues with preferred proline or preferred glycine residues. Here, we develop guidelines for selecting appropriate mutations, and present results for five mutations (S31P, S42G, S48P, T76P, Q77G) that significantly increase the conformational stability of Ribonuclease (RNase) Sa. The increases in stability ranged from 0.7 kcal/mol to 1.3 kcal/mol. The strategy was successful in overlapping or isolated β-turns, at buried (up to 50%) or completely exposed sites, and at relatively flexible or inflexible sites. Considering the significant number of β-turn residues in every globular protein and the frequent deviation of β-turn sequences from preferred proline and preferred glycine residues, this simple, efficient strategy will be useful for increasing the conformational stability of proteins.
Keywords: protein conformational stability, β-turn, proline, glycine
Introduction
Proteins carry out most of the important tasks in living cells, and they play an increasingly important role in the biotechnology and pharmaceutical industries. For proteins to perform these important tasks, they must fold to their proper, unique three-dimensional structure. This folding process depends on the protein having sufficient conformational stability. The conformational stability of a protein is defined as the free energy change (ΔG°) for the reaction: folded (or native, N) ⇔ unfolded (or denatured, D)1. This free energy change for most proteins is marginal – on the order of 5 to 15 kcal/mol. The marginal stability of proteins makes the use of certain proteins in industrial or pharmaceutical applications difficult especially when the protein is subjected to extremes of temperature and pH, or when the protein is subjected to destabilizing solvents such as organic solvents. Insufficient protein stability is also a common problem in general biochemical studies when an unstable recombinant protein of interest cannot be expressed at desirable levels due to aggregation and formation of inclusion bodies during expression.
Consequently, there is great interest in methods that can be used to increase the conformational stability of proteins. Many methods have been introduced2–10, but it is still difficult to efficiently and effectively stabilize proteins. Here, we have developed a strategy for significantly increasing protein stability by optimizing β-turn sequence. β-turns are the smallest type of secondary structure. They join other elements of secondary structure such as α-helices and β-strands and abruptly change the direction of the polypeptide chain. Due to their polar nature, β-turns occur most often at protein surfaces, but sometimes they do occur in the protein interior11. β-turns usually contain four residues designated as i (or −B1), i+1 (or L1), i+2 (or L2), i+3 (or +B1), and the distance between the α-carbons of the i and i+3 residues in a β-turn is less than 7 Å. They are sometimes, but not always, stabilized by a hydrogen bond. Two common types of β-turns are type I and type II β-turns, but there are several other β-turn types determined by the φ, ψ angles of the i+1 and i+2 residues12.
In statistical analyses of residues found in β-turns12; 13, protein structures display preferences for proline or glycine at certain β-turn positions. For example, in a type II turn, the O atom of the i+1 residue crowds the Cβ atom of the i+2 residue. Therefore, the i+2 residue of type II turns is usually glycine. Glycine is also often found at other positions of different β-turn types since its lack of a β-carbon sterically allows a wider range of φ, ψ angles than other amino acids. Proline is a residue that is also found often in turns presumably due to its unique restricted φ angle, which is entropically favorable at certain turn positions. For example, the pyrrolidene ring of proline residues restricts the φ angle of this residue to −60°. Furthermore, the φ angle of the i+1 residue of type I and II turns is −60° (see Hutchinson and Thornton12). Therefore, proline probably assumes the −60° φ angle that is required at certain β-turn positions with a reduced entropic cost relative to other, more intrinsically flexible amino acids.
Though proline and glycine are preferred at certain β-turn positions, turn regions in proteins show large sequence variation14–19. Therefore, our strategy involves replacing non-proline and non-glycine residues at β-turn positions where proline or glycine are preferred based on β-turn sequence statistics12; 13. Stabilization strategies based on sequence statistics are the easiest and most straightforward to use20, but it is not completely understood how these non-optimal β-turn sequences might be exploited to increase protein stability. Ohage et al.18 and others16; 21–23 have described approaches for optimizing β-turns. However, a strategy that focuses on both proline and glycine that tests different positions of different β-turn types has not been developed. Here, we present guidelines for using this strategy and results for several mutations that significantly increase the conformational stability of Ribonuclease (RNase) Sa.
Results and Discussion
Design strategy
To investigate whether stabilizing mutations to proline or glycine could be introduced at β-turn positions in RNase Sa, we used the program PROMOTIF24 and the 1.2 Å crystal structure of RNase Sa (PDB code: 1RGG25) to identify residues in β-turns. Then, we used β-turn sequence statistics collected by Guruprasad and Rajkumar13 to identify non-proline and non-glycine residues in positions where proline or glycine are preferred. The positional potentials of the wild-type residues and preferred proline or glycine residues are shown in Table 1. As an example, position 31 in RNase Sa is the i+1 position of a type I β-turn. A serine residue is found at position 31 in wild-type RNase Sa, but sequence statistics13 indicate that proline is preferred at the i+1 position of a type I β-turn (Table 1). Therefore, S31P is an example of a candidate for mutation. This process identified fourteen possible mutations out of thirty-two type I, II, I′, or II′ β-turn positions in RNase Sa. Mutations in type IV and VIII turns of RNase Sa were not made because the residues in these turns were mostly buried and therefore, probably not good targets for mutation.
Table 1.
Positional potentials of wild-type and preferred proline or glycine residuesa.
| Residue Number | Position | Turn Type | Positional Potential of Wild-Type Residue | Positional Potential of Preferred Glycine or Prolineb |
|---|---|---|---|---|
| 31 | i+1 | I | Ser (1.61) | Pro (4.29) |
| 42 | i+3 | I | Ser (0.94) | Gly (2.57) |
| 48 | i | II | Ser (0.65) | Pro (1.91) |
| 76 | i+1 | II | Thr (0.71) | Pro (4.92) |
| 77 | i+2 | II | Gln (0.17) | Gly (9.39) |
Of the fourteen possible mutations, candidates were eliminated where the wild-type side chain was in the active site of the protein as determined by Yakovlev et al.26 or where the side chain was involved in hydrogen bonding as determined from analysis using the program pfis27 and the crystal structure of RNase Sa25. Also, for mutations to proline, candidates were eliminated where the amide nitrogen of the residue was a hydrogen bond donor since proline residues lack an amide hydrogen. Mutation of ionizable residues was also not considered. This is because the effect of mutating ionizable residues on overall stability has been shown to be difficult to predict in terms of electrostatic interaction energy28.
Additional guidelines for candidate selection were discovered based on results for the Y49P, Y86G, and Q94P variants. The stability changes associated with these three variants as determined by thermal denaturations monitored by circular dichroism (CD) spectroscopy are shown in Table 2. The Y86G mutation increased the conformational stability of RNase Sa by only 0.4 kcal/mol. The Y49P mutation did not change the stability of the protein significantly as shown by its near-zero ΔΔG value, and the Q94P mutation destabilized the protein significantly by 0.8 kcal/mol.
Table 2.
Parameters characterizing the thermal unfolding of RNase Sa and β-turn variantsa.
| Protein | Position | Turn Type | ΔHmb | ΔΔHmc | ΔSmd | ΔΔSme | Tmf | ΔTmg | ΔΔGoh |
|---|---|---|---|---|---|---|---|---|---|
| Wild-type | 91 | 283 | 48.4 | ||||||
| S31P | i+1 | I | 92 | 1 | 284 | 1 | 50.9 | 2.5 | 0.7 |
| S42G | i+3 | I | 91 | 0 | 281 | −2 | 51.0 | 2.6 | 0.7 |
| S48P | i | II | 87 | −4 | 267 | −16 | 52.9 | 4.5 | 1.3 |
| Y49P | i+1 | II | 85 | −6 | 265 | −18 | 47.7 | −0.7 | −0.2 |
| T76P | i+1 | II | 96 | 5 | 295 | 12 | 51.9 | 3.5 | 1.0 |
| Q77G | i+2 | II | 88 | −3 | 271 | −12 | 51.3 | 2.9 | 0.8 |
| Y86G | i+2 | I′ | 90 | −1 | 279 | −4 | 49.9 | 1.5 | 0.4 |
| Q94P | i+1 | I | 84 | −7 | 263 | −20 | 45.7 | −2.7 | −0.8 |
| T76P-Q77G | 84 | −7 | 257 | −26 | 54.2 | 5.8 | 1.6 | ||
| S31P-S42G-Q77G | 81 | −10 | 247 | −36 | 55.2 | 6.8 | 1.9 |
The experiments were done in 30 mM Mops at pH 7.0.
ΔHm, enthalpy of unfolding at Tm (in kcal mol−1). The error is ± 5 kcal mol−1.
ΔΔHm = ΔHm(mutant) − ΔHm(wild-type). The error is ± 7 kcal mol−1.
ΔSm = ΔHm/Tm (in cal mol−1 K−1). The error is ± 15 cal mol−1 K−1.
ΔΔSm = ΔSm(mutant) − ΔSm(wild-type). The error is ± 21 cal mol−1 K−1.
Tm = T (in °C), where ΔG° = 0. The error is ± 0.3 °C.
ΔTm = Tm(mutant) − Tm(wild-type) (in °C). The error is 0.4 °C.
ΔΔG° = 0.283 (ΔSm for wild-type in kcal mol−1 K−1) * Δ;Tm. The error is ≤ 0.2 kcal mol−1. A positive value indicates an increase in stability.
Substitution of aromatic tyrosine residues in the Y49P and Y86G variants did not significantly increase the stability of RNase Sa. This is probably because these aromatic residues provide stabilizing interactions in β-turns. A study by Gibbs et al.29 suggested that aromatic residues provide stabilizing interactions in a type II′ β-turn. There is also evidence in RNase Sa that suggests that aromatic residues in β-turns provide stabilizing interactions. For example, the Y49N mutation (position 49 is the i+1 position of a type II β-turn) in RNase Sa decreases the stability of the protein by 1 kcal/mol (S.T., unpublished observations). The hydroxyl group of this tyrosine is not involved in hydrogen bonding interactions, so it seems that the decrease in stability in the Y49N variant is caused by the loss of stabilizing interactions provided by the aromatic portion of the tyrosine. This loss of 1 kcal/mol is similar in magnitude to the stability gained in the T76P variant (position 76 is also the i+1 position of a type II β-turn) (Table 2). Therefore, in the Y49P variant, the gain in stability upon introduction of preferred proline is probably counterbalanced by the loss of stabilizing interactions provided by the aromatic tyrosine resulting in a near-zero change in conformational stability. Further evidence that aromatic residues can provide stabilizing interactions in β-turns is shown in our results from a previous study where we compared solubility values of 20 variants of RNase Sa at position 76 (i+1 position of a type II β-turn). We also measured the stability for each of these variants, and the results showed that the T76Y, T76F, and T76W variants all result in stability increases similar in magnitude to the T76P variant suggesting that the stabilizing contribution of aromatic residues in this case is similar to that of a preferred proline in a β-turn. Combining these RNase Sa results with the results of Gibbs et al.29 suggests that significant stability cannot be gained by replacing aromatic residues in turns with glycine or proline. Indeed, this is what is observed for the Y49P and Y86G variants (Table 2).
The Q94P mutation destabilized RNase Sa despite mutation to a preferred proline. One reason might be that Gln94 is close in sequence to Cys96, which forms a disulfide bond with Cys7 in RNase Sa. A mutation to proline close to this bond might strain the bond and destabilize the protein – especially since this disulfide bond contributes dramatically to the stability of RNase Sa (~ 7 kcal/mol)30. Another possible reason the Q94P variant destabilizes RNase Sa is that Gln94 is close in space to Asp79 which is a buried carboxyl group that does not form any intramolecular hydrogen bonds and hence, contributes unfavorably to the conformational stability of RNase Sa31. Gln94 is the only polar residue near Asp79, and mutating it to the more hydrophobic proline might make Asp79 contribute even more unfavorably to the conformational stability of RNase Sa.
Therefore, based on the results for the Y49P, Y86G, and Q94P variants, the following additional guidelines for using the strategy were established: mutation of aromatic residues and mutation of residues that were close in sequence to residues involved in the disulfide bond (Cys7 and Cys96) were avoided. Overall, this elimination process resulted in seven candidates for mutation. Five of these variants (S31P, S42G, S48P, T76P, and Q77G) were made. The other two (Q32P and T59P) were not made, because they are similar to the S48P mutation, i.e., a mutation at the i position of a type II β-turn. Note that Ser31 is at position i+1 in a type I β-turn, but Gln32 is at position i in a type II β-turn. This is a consequence of overlapping turns as discussed further below. Figure 1 shows the five candidates for mutation in RNase Sa (Ser31, Ser42, Ser48, Thr76, and Gln77).
Figure 1.
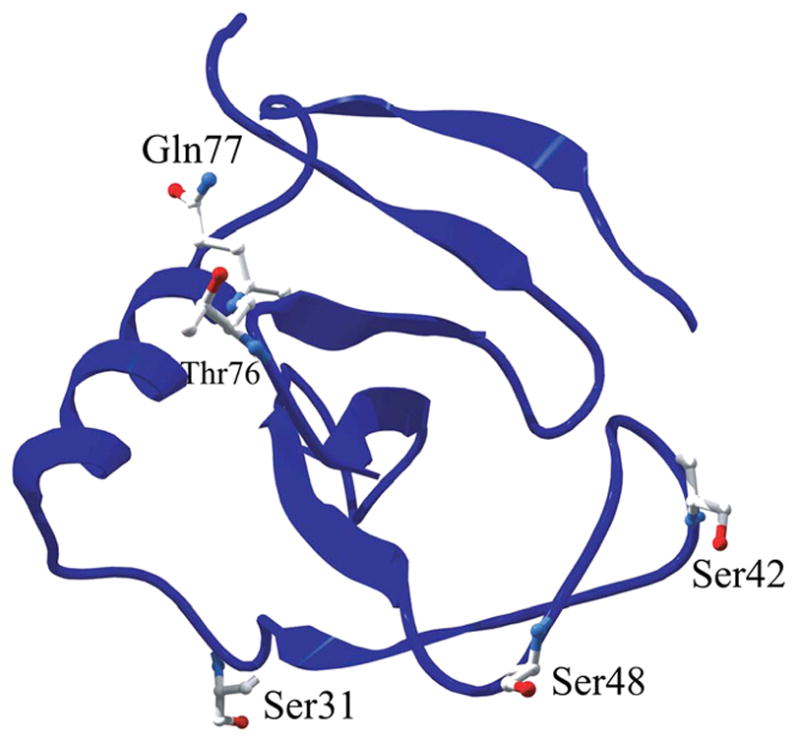
Ribbon diagram showing candidates for optimizing β-turn sequence in RNase Sa. Ser31, Ser42, and Gln77 are the candidates shown. The figure was generated using the Swiss-Pdb Viewer program55 and the 1.2 Å crystal structure of RNase Sa (PDB code: 1RGG25).
None of the mutations significantly affected the protein structure
To confirm that these mutations in RNase Sa did not change the solution structure of the protein, CD spectra of the wild-type protein and several of the β-turn variants were compared (data not shown). The results show similar CD spectra for wild-type and the β-turn variants suggesting that the variants retained structure similar to wild-type RNase Sa. In addition, all of the variants retained enzymatic activity (data not shown). In general, mutations to proline or glycine do not appear to significantly affect protein structure as determined by X-ray crystallography. For example, several studies have reported crystal structures of mutants involving proline and glycine substitutions in β-turns or other locations in protein structures16; 17; 23; 32, and each study found no significant change in structure upon mutation to proline or glycine. Therefore, results from X-ray crystallography and our results suggest that these types of substitutions do not cause significant structural changes relative to the wild-type proteins.
Stability increases for the β-turn variants in RNase Sa
To determine the conformational stability changes caused by the β-turn mutations, thermal denaturations monitored by circular dichroism spectroscopy at pH 7 were performed. Previously, our lab has characterized the folding and stability of RNase Sa and several variants26; 28; 30; 31; 33–39. The unfolding of RNase Sa and its variants is reversible and follows a 2-state mechanism. The thermodynamic parameters characterizing the thermal unfolding of these variants are shown in Table 2. The S31P, S42G, S48P, T76P, and Q77G mutations resulted in significant increases in conformational stability as shown by the positive ΔΔG values greater than 0.5 kcal/mol and the increased Tm values. The double mutant, T76P-Q77G, containing two mutations in the same turn, stabilized the protein by 1.6 kcal/mol. A triple mutant, S31P-S42G-Q77G, was also constructed, and it stabilized RNase Sa by 1.9 kcal/mol.
Thermodynamic effects of mutations to proline
In general, mutations to proline are expected to stabilize proteins by decreasing conform ational entropy in the denatured state. Matthews et al.40 estimated this stabilization to be on the order of 0.8 kcal/mol based on their A82P mutation in T4 lysozyme. The change in conformational entropy, or ΔΔS value, for A82P was negative (−10 cal mol−1 K−1) suggesting a decrease in conformational entropy in the denatured state, but did not appear significant based on the error40. Prajapati et al.32 did another study of general mutations to proline that involved thirteen mutations to proline in four different proteins. They did observe significant ΔΔS values, but the ΔΔS values varied widely including negative and positive values (−55 to 47 cal mol−1 K−1). For our stabilizing mutations to proline in β-turns (S31P, S48P, and T76P), we also observed both positive and negative ΔΔS values (−16 to 12 cal mol−1 K−1), but these values were not significant based on the error associated with the measurements (Table 2).
Other studies have investigated the effects of mutations to proline in β-turns. Suzuki and co-workers41; 42 have investigated how mutations to proline at second sites (i.e., i+1 residues) of type I and II β-turns increase the thermal stability of oligo-1,6-glucosidase from Bacillus cereus, though they did not measure Tm and ΔΔG° values. Furthermore, even though Choi and Mayo43 introduced prolines at second sites of β-turns in protein G (K10P and A48P), they observed slight decreases in stability presumably because Lys10 formed favorable electrostatic interactions in the folded state and because the amide nitrogen of Ala48 was a hydrogen bond donor. In agreement with the results of Suzuki and co-workers41; 42, the mutations to proline at second sites of type I and type II β-turns in RNase Sa (S31P and T76P) significantly increased protein stability (Table 2). In addition, the mutation to preferred proline at the first site of a type II turn in RNase Sa (S48P), resulted in the largest increase in conformational stability (1.3 kcal/mol) of all the mutations to preferred proline or preferred glycine (Table 2).
Thermodynamic effects of mutations to glycine
Mutations to glycine in certain positions (not necessarily in β-turns) can relieve steric strain in the folded state caused by residues that contain a β-carbon. For example, certain residues are located in the left-handed α-helical (L-α) region of the Ramachandran plot (0°<φ<90°, 0°<ψ<90°)44. Non-glycine residues in this region may cause steric strain involving the β-carbon of the residue and the peptide backbone. Indeed, Takano et al.45 found that Gly to Ala substitutions at rigid L-α positions led to significant destabilization (1–2 kcal/mol) of human lysozyme. Masumoto et al.46 found similar results in hen egg white lysozyme. Consequently, there have been several studies involving non-Gly to Gly mutations in L-α positions resulting in stability increases of 1 to 2 kcal/mol21; 22; 47. In contrast, Nicholson et al.48 found that this strategy worked neither for the N55G variant nor the K124G variant in T4 lysozyme. However, Asn55 in T4 lysozyme is at the i+1 position of a type I′ β-turn, and Asn is highly preferred even relative to Gly at this position (the positional potentials are 6.07 and 1.73 for Asn and Gly, respectively13). Therefore, this might be a possible reason for the slight decrease in stability observed for the N55G mutation in T4 lysozyme. As for the K124G variant, Nicholson et al.48 noted that Lys124 is involved in favorable electrostatic interactions in wild-type T4 lysozyme. The stabilizing results at positions 42 and 77 in RNase Sa (Table 2), whose φ, ψ angles are in the L-α region as determined by the program pfis27, agree with those of Kimura et al.21, Stites et al.47, and Kim et al.22, and they suggest that non-glycine residues in the L-α region do cause strain which, by mutation to glycine, can be relieved to significantly increase protein stability. However, care must be taken to avoid mutating highly preferred Asn residues at i+1 positions of type I′ β-turns or residues that participate in favorable electrostatic interactions. In terms of changes in conformational entropy, mutations to glycine might be expected to have positive ΔΔS values40. However, our two stabilizing mutations to glycine (S42G and Q77G) did not have significant ΔΔS values (Table 2).
Stabilizing mutations occurred in both isolated and overlapping β-turns
Some of the stabilizing mutations occurred in isolated β-turns while others were in β-turns that shared one or more residues with other β-turns. Such overlapping turns appear to be very common in proteins. In their survey of 205 proteins, Hutchinson and Thornton12 found that 58% of β-turns share one or more residues with other β-turns. In RNase Sa, 75% of the β-turns overlap with other β-turns as determined by PROMOTIF24. Therefore, it is not surprising that some of the candidates for mutation in RNase Sa occurred in overlapping β-turns. According to the PROMOTIF24 analysis of RNase Sa structure, position 48 is in a type II turn that overlaps with a type VIII turn. Positions 31 and 42 are also in turns that overlap with other turns. On the other hand, positions 76 and 77 are in an isolated turn. All of these mutations significantly increased the stability of RNase Sa, so it seems that significant increases in stability can be obtained by optimizing either isolated or overlapping β-turns.
Stability correlation with side chain burial and backbone flexibility
The extent to which side chains are buried has been suggested to play a role in the stability changes observed for β-turn mutations. Takano et al.16 investigated the role of turn residues in the structure and stability of human lysozyme. They observed that the increase in stability upon mutations to preferred glycine was dependent upon whether the side chain was buried or exposed to solvent. For example, substitutions on the surface hardly changed the conformational stability whereas mutation at a buried site (Gln58) significantly increased the stability of human lysozyme. Their other mutations to glycine in β-turns (R50G and H78G) did not stabilize the protein, but this could have been due to the disruption of favorable electrostatic interactions. The stabilizing mutations to glycine in our study (S42G and Q77G) were at partially buried sites (52% and 39%, respectively) (Table 3). In contrast, the stabilizing mutations to proline in this study (S31P, S48P, and T76P) were at completely exposed sites, yet they yielded significant stability increases. Therefore, our results suggest that this strategy works at buried sites (up to 50%) as well as completely exposed sites.
Table 3.
Percent side chain burial and B-factors for the stabilizing β-turn variants in RNase Sa.
| Protein | % Side-chain buriala | B-factor (Å2)b | ΔΔG° (kcal/mol) |
|---|---|---|---|
| S31P | 0.7 | 12 | 0.7 |
| S42G | 52.0 | 27 | 0.7 |
| S48P | 2.1 | 16 | 1.3 |
| T76P | −2.5 | 30 | 1.0 |
| Q77G | 39.4 | 24 | 0.8 |
Values for the wild-type residue.
Average B-factors for backbone atoms obtained from the 1.2Å RNase Sa crystal structure (PDB code: 1RGG25).
The flexibility of the protein structure at the site of mutation has also been suggested to correlate with stability changes of mutations involving glycine and proline. Takano et al.45 replaced glycine residues with alanine in human lysozyme to gauge the strain caused by having non-glycine residues in sites with left-handed helical φ, ψ angles (i.e., sites where a β-carbon might cause steric strain). They showed that mutations at relatively flexible positions of the protein (B-factors ≥ 13 Å2) did not show large stability changes. Similarly, Yutani et al49 replaced conserved proline residues in the tryptophan synthase alpha subunit with Ala or Gly and only saw significant stability changes in less mobile regions. Matthews and co-workers have also shown that mutations that significantly affect protein stability tend to be at rigid sites50; 51. In contrast, the S42G, S48P, T76P, and Q77G mutations in this study were at sites with B-factors > 15 Å2 (Table 3), yet they still yielded significant increases in stability. Therefore, it seems that significant increases in stability can be achieved by optimization of relatively flexible or inflexible β-turn sites.
Additivity of multiple β-turn mutations
To investigate the possibility of cooperative stability increases in multiple β-turn variants, a double mutant containing mutations in the same turn (T76P-Q77G) and a triple mutant containing mutations in three different turns (S31P-S42G-Q77G) were made. The results in Table 2 show that the stability increases for these multiple mutants were essentially additive. For example, the stability increase for the T76P-Q77G variant was 1.6 kcal/mol while the sum for the individual mutations is 1.8 kcal/mol. Therefore, it appears that multiple β-turn mutants yield increases in stability that are additive even when multiple mutations are made in the same turn.
Conclusions and general guidelines
β-turn sequence statistics for large sets of proteins have been collected and show that proline and glycine residues are preferred at several positions of several β-turn types12; 13. However, β-turn sequences in RNase Sa and other proteins12–19 often deviate from preferred proline or preferred glycine residues. Here we show that mutating non-proline and non-glycine residues to preferred proline or preferred glycine residues in β-turns results in significant increases in the conformational stability of RNase Sa. Also, there were a large number of candidates (seven) for stabilizing β-turn mutations in the relatively small RNase Sa, and five of these mutations representing a wide range of β-turn positions result in significant increases in conformational stability of RNase Sa. Other studies have also shown that optimizing β-turn sequence can yield large increases in protein stability16; 18; 21–23. A large percentage of protein residues are found in β-turns. For example, it has been estimated that a quarter of all protein residues are in β-turns52, and in RNase Sa, about 31% of the residues are in β-turns (not including residues in type IV or type VIII turns). Therefore, the variability of β-turn sequences (i.e., deviation from preferred residues) and the large percentage of turn residues found in proteins should provide ample opportunities for using this strategy to stabilize any protein of interest.
Based on this study as well as others, the following guidelines should be useful when attempting to increase protein stability by mutating non-proline and non-glycine residues to preferred proline or preferred glycine: 1) The wild-type residue should not be involved in stabilizing interactions such as hydrogen bonds or favorable electrostatic interactions. Also, since proline lacks an amide hydrogen, mutations to proline should be avoided at positions where the amide nitrogen is a hydrogen bond donor. 2) Bulky hydrophobic or aromatic residues such as Val, Leu, Ile, Phe, Tyr, and Trp should not be targeted as they might be partially buried and provide favorable hydrophobic interactions. 3) Mutations to proline near in sequence to Cys residues involved in disulfide bonds should be avoided. 4) Asparagine residues in the i+1 position of type I′ β-turns should not be targeted as they are highly preferred in these positions13. 5) Mutations in type IV or type VIII turns are less desirable as these turns are more often buried than other β-turn types. Following these guidelines, we have been successful in increasing the stability of RNase Sa by 0.7 to 1.9 kcal/mol. The strategy was effective in overlapping or isolated β-turns, at buried (up to 50%) or completely exposed sites, and at relatively flexible or inflexible sites. Also, the method is simple to use in that it doesn’t require complex calculations or sequence/structural information of homologous proteins. Further development of this strategy is necessary as data for several positions in type I (the i position), type I′ (the i+2 position), and type II′ (the i+1 position) β-turns is still lacking. The strategy presented here has not been included in lists of popularly considered strategies for increasing protein stability2; 7; 10, but our results demonstrate the efficiency and effectiveness of increasing protein stability by optimizing β-turn sequence and should be useful for those interested in increasing protein stability.
Materials and Methods
Preparation of mutant plasmids and proteins
Primers were ordered from Integrated DNA Technologies at www.idtdna.com. Site-directed mutagenesis was performed using the QuikChange® Site-Directed Mutagenesis Kit from Stratagene. Mini-preps of the mutant plasmids were performed using the QIAprep® Spin Miniprep Kit from Qiagen. Sequencing of the mutant plasmids was done at the Gene Technologies Laboratory, Department of Biology, Texas A&M University. All proteins were expressed and purified as described previously53.
Thermal denaturation experiments
Mops buffer at 30 mM concentration was used at pH 7.0. Thermal denaturation experiments were done using an AVIV Circular Dichroism Spectrometer Model 62DS as previously described27; 30. The data were analyzed using KaleidaGraph version 3.6.4 (Synergy Software). The analysis of thermal denaturation curves has been previously described54.
Acknowledgments
We thank Dr. Abbas Razvi and Yun Wei for their helpful suggestions and comments.
This work has been funded by National Institutes of Health Grant T32 GM065088 (to S.R.T.), by the NSF REU Site Program in Biochemistry at Texas A&M University (DBI-0552822) (to S.S.), by grants GM-37039 and GM-52483 from the National Institutes of Health (USA), and grants BE-1060 and BE-1281 from the Robert A. Welch Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pace CN. Measuring and increasing protein stability. Trends Biotechnol. 1990;8:93–8. doi: 10.1016/0167-7799(90)90146-o. [DOI] [PubMed] [Google Scholar]
- 2.Imoto T. Stabilization of protein. Cell Mol Life Sci. 1997;53:215–23. doi: 10.1007/PL00000593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shortle D. Mutational studies of protein structures and their stabilities. Q Rev Biophys. 1992;25:205–50. doi: 10.1017/s0033583500004674. [DOI] [PubMed] [Google Scholar]
- 4.Matthews BW. Studies on protein stability with T4 lysozyme. Adv Protein Chem. 1995;46:249–78. doi: 10.1016/s0065-3233(08)60337-x. [DOI] [PubMed] [Google Scholar]
- 5.Terwilliger TC. Engineering the stability and function of gene V protein. Adv Protein Chem. 1995;46:177–215. doi: 10.1016/s0065-3233(08)60335-6. [DOI] [PubMed] [Google Scholar]
- 6.Serrano L, Kellis JT, Jr, Cann P, Matouschek A, Fersht AR. The folding of an enzyme. II. Substructure of barnase and the contribution of different interactions to protein stability. J Mol Biol. 1992;224:783–804. doi: 10.1016/0022-2836(92)90562-x. [DOI] [PubMed] [Google Scholar]
- 7.Shaw A, Bott R. Engineering enzymes for stability. Curr Opin Struct Biol. 1996;6:546–50. doi: 10.1016/s0959-440x(96)80122-9. [DOI] [PubMed] [Google Scholar]
- 8.Pace CN, Shirley BA, McNutt M, Gajiwala K. Forces contributing to the conformational stability of proteins. Faseb J. 1996;10:75–83. doi: 10.1096/fasebj.10.1.8566551. [DOI] [PubMed] [Google Scholar]
- 9.Pace CN. Conformational stability of globular proteins. Trends Biochem Sci. 1990;15:14–7. doi: 10.1016/0968-0004(90)90124-t. [DOI] [PubMed] [Google Scholar]
- 10.Lehmann M, Wyss M. Engineering proteins for thermostability: the use of sequence alignments versus rational design and directed evolution. Curr Opin Biotechnol. 2001;12:371–5. doi: 10.1016/s0958-1669(00)00229-9. [DOI] [PubMed] [Google Scholar]
- 11.Rose GD, Young WB, Gierasch LM. Interior turns in globular proteins. Nature. 1983;304:654–7. doi: 10.1038/304654a0. [DOI] [PubMed] [Google Scholar]
- 12.Hutchinson EG, Thornton JM. A revised set of potentials for beta-turn formation in proteins. Protein Sci. 1994;3:2207–16. doi: 10.1002/pro.5560031206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guruprasad K, Rajkumar S. Beta-and gamma-turns in proteins revisited: a new set of amino acid turn-type dependent positional preferences and potentials. J Biosci. 2000;25:143–56. [PubMed] [Google Scholar]
- 14.Castagnoli L, Vetriani C, Cesareni G. Linking an easily detectable phenotype to the folding of a common structural motif. Selection of rare turn mutations that prevent the folding of Rop. J Mol Biol. 1994;237:378–87. doi: 10.1006/jmbi.1994.1241. [DOI] [PubMed] [Google Scholar]
- 15.Brunet AP, Huang ES, Huffine ME, Loeb JE, Weltman RJ, Hecht MH. The role of turns in the structure of an alpha-helical protein. Nature. 1993;364:355–8. doi: 10.1038/364355a0. [DOI] [PubMed] [Google Scholar]
- 16.Takano K, Yamagata Y, Yutani K. Role of amino acid residues at turns in the conformational stability and folding of human lysozyme. Biochemistry. 2000;39:8655–65. doi: 10.1021/bi9928694. [DOI] [PubMed] [Google Scholar]
- 17.Predki PF, Agrawal V, Brunger AT, Regan L. Amino-acid substitutions in a surface turn modulate protein stability. Nat Struct Biol. 1996;3:54–8. doi: 10.1038/nsb0196-54. [DOI] [PubMed] [Google Scholar]
- 18.Ohage EC, Graml W, Walter MM, Steinbacher S, Steipe B. Beta-turn propensities as paradigms for the analysis of structural motifs to engineer protein stability. Protein Sci. 1997;6:233–41. doi: 10.1002/pro.5560060125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou HX, Hoess RH, DeGrado WF. In vitro evolution of thermodynamically stable turns. Nat Struct Biol. 1996;3:446–51. doi: 10.1038/nsb0596-446. [DOI] [PubMed] [Google Scholar]
- 20.Steipe B, Schiller B, Pluckthun A, Steinbacher S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J Mol Biol. 1994;240:188–92. doi: 10.1006/jmbi.1994.1434. [DOI] [PubMed] [Google Scholar]
- 21.Kimura S, Kanaya S, Nakamura H. Thermostabilization of Escherichia coli ribonuclease HI by replacing left-handed helical Lys95 with Gly or Asn. J Biol Chem. 1992;267:22014–7. [PubMed] [Google Scholar]
- 22.Kim J, Brych SR, Lee J, Logan TM, Blaber M. Identification of a key structural element for protein folding within beta-hairpin turns. J Mol Biol. 2003;328:951–61. doi: 10.1016/s0022-2836(03)00321-8. [DOI] [PubMed] [Google Scholar]
- 23.Vega MC, Martinez JC, Serrano L. Thermodynamic and structural characterization of Asn and Ala residues in the disallowed II′ region of the Ramachandran plot. Protein Sci. 2000;9:2322–8. doi: 10.1110/ps.9.12.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hutchinson EG, Thornton JM. PROMOTIF--a program to identify and analyze structural motifs in proteins. Protein Sci. 1996;5:212–20. doi: 10.1002/pro.5560050204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sevcik J, Dauter Z, Lamzin VS, Wilson KS. Ribonuclease from Streptomyces aureofaciens at atomic resolution. Acta Crystallogr D Biol Crystallogr. 1996;52:327–344. doi: 10.1107/S0907444995007669. [DOI] [PubMed] [Google Scholar]
- 26.Yakovlev GI, Mitkevich VA, Shaw KL, Trevino S, Newsom S, Pace CN, Makarov AA. Contribution of active site residues to the activity and thermal stability of ribonuclease Sa. Protein Sci. 2003;12:2367–73. doi: 10.1110/ps.03176803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hebert EJ, Giletto A, Sevcik J, Urbanikova L, Wilson KS, Dauter Z, Pace CN. Contribution of a conserved asparagine to the conformational stability of ribonucleases Sa, Ba, and T1. Biochemistry. 1998;37:16192–200. doi: 10.1021/bi9815243. [DOI] [PubMed] [Google Scholar]
- 28.Pace CN, Alston RW, Shaw KL. Charge-charge interactions influence the denatured state ensemble and contribute to protein stability. Protein Sci. 2000;9:1395–8. doi: 10.1110/ps.9.7.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gibbs AC, Bjorndahl TC, Hodges RS, Wishart DS. Probing the structural determinants of type II′ beta-turn formation in peptides and proteins. J Am Chem Soc. 2002;124:1203–13. doi: 10.1021/ja011005e. [DOI] [PubMed] [Google Scholar]
- 30.Pace CN, Hebert EJ, Shaw KL, Schell D, Both V, Krajcikova D, Sevcik J, Wilson KS, Dauter Z, Hartley RW, Grimsley GR. Conformational stability and thermodynamics of folding of ribonucleases Sa, Sa2 and Sa3. J Mol Biol. 1998;279:271–86. doi: 10.1006/jmbi.1998.1760. [DOI] [PubMed] [Google Scholar]
- 31.Trevino SR, Gokulan K, Newsom S, Thurlkill RL, Shaw KL, Mitkevich VA, Makarov AA, Sacchettini JC, Scholtz JM, Pace CN. Asp79 makes a large, unfavorable contribution to the stability of RNase Sa. J Mol Biol. 2005;354:967–978. doi: 10.1016/j.jmb.2005.09.091. [DOI] [PubMed] [Google Scholar]
- 32.Prajapati RS, Das M, Sreeramulu S, Sirajuddin M, Srinivasan S, Krishnamurthy V, Ranjani R, Ramakrishnan C, Varadarajan R. Thermodynamic effects of proline introduction on protein stability. Proteins. 2007;66:480–91. doi: 10.1002/prot.21215. [DOI] [PubMed] [Google Scholar]
- 33.Grimsley GR, Shaw KL, Fee LR, Alston RW, Huyghues-Despointes BM, Thurlkill RL, Scholtz JM, Pace CN. Increasing protein stability by altering long-range coulombic interactions. Protein Sci. 1999;8:1843–9. doi: 10.1110/ps.8.9.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shaw KL, Grimsley GR, Yakovlev GI, Makarov AA, Pace CN. The effect of net charge on the solubility, activity, and stability of ribonuclease Sa. Protein Sci. 2001;10:1206–15. doi: 10.1110/ps.440101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pace CN, Horn G, Hebert EJ, Bechert J, Shaw K, Urbanikova L, Scholtz JM, Sevcik J. Tyrosine hydrogen bonds make a large contribution to protein stability. J Mol Biol. 2001;312:393–404. doi: 10.1006/jmbi.2001.4956. [DOI] [PubMed] [Google Scholar]
- 36.Pace CN. Polar group burial contributes more to protein stability than nonpolar group burial. Biochemistry. 2001;40:310–3. doi: 10.1021/bi001574j. [DOI] [PubMed] [Google Scholar]
- 37.Takano K, Scholtz JM, Sacchettini JC, Pace CN. The Contribution of Polar Group Burial to Protein Stability Is Strongly Context-dependent. J Biol Chem. 2003;278:31790–31795. doi: 10.1074/jbc.M304177200. [DOI] [PubMed] [Google Scholar]
- 38.Trefethen JM, Pace CN, Scholtz JM, Brems DN. Charge-charge interactions in the denatured state influence the folding kinetics of ribonuclease Sa. Protein Sci. 2005;14:1934–8. doi: 10.1110/ps.051401905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trevino SR, Scholtz JM, Pace CN. Amino Acid Contribution to Protein Solubility: Asp, Glu, and Ser Contribute more Favorably than the other Hydrophilic Amino Acids in RNase Sa. J Mol Biol. 2007;366:449–60. doi: 10.1016/j.jmb.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matthews BW, Nicholson H, Becktel WJ. Enhanced protein thermostability from site-directed mutations that decrease the entropy of unfolding. Proc Natl Acad Sci U S A. 1987;84:6663–7. doi: 10.1073/pnas.84.19.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watanabe K, Suzuki Y. Protein thermostabilization by proline substitutions. J Mol Catal B Enz. 1998;4:167–180. [Google Scholar]
- 42.Watanabe K, Masuda T, Ohashi H, Mihara H, Suzuki Y. Multiple proline substitutions cumulatively thermostabilize Bacillus cereus ATCC7064 oligo-1,6-glucosidase. Irrefragable proof supporting the proline rule. Eur J Biochem. 1994;226:277–83. doi: 10.1111/j.1432-1033.1994.tb20051.x. [DOI] [PubMed] [Google Scholar]
- 43.Choi EJ, Mayo SL. Generation and analysis of proline mutants in protein G. Protein Eng Des Sel. 2006;19:285–9. doi: 10.1093/protein/gzl007. [DOI] [PubMed] [Google Scholar]
- 44.Ramachandran GN, Sasisekharan V. Conformation of polypeptides and proteins. Adv Protein Chem. 1968;23:283–438. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 45.Takano K, Yamagata Y, Yutani K. Role of amino acid residues in left-handed helical conformation for the conformational stability of a protein. Proteins. 2001;45:274–80. doi: 10.1002/prot.1147. [DOI] [PubMed] [Google Scholar]
- 46.Masumoto K, Ueda T, Motoshima H, Imoto T. Relationship between local structure and stability in hen egg white lysozyme mutant with alanine substituted for glycine. Protein Eng. 2000;13:691–5. doi: 10.1093/protein/13.10.691. [DOI] [PubMed] [Google Scholar]
- 47.Stites WE, Meeker AK, Shortle D. Evidence for strained interactions between side-chains and the polypeptide backbone. J Mol Biol. 1994;235:27–32. doi: 10.1016/s0022-2836(05)80008-7. [DOI] [PubMed] [Google Scholar]
- 48.Nicholson H, Soderlind E, Tronrud DE, Matthews BW. Contributions of left-handed helical residues to the structure and stability of bacteriophage T4 lysozyme. J Mol Biol. 1989;210:181–93. doi: 10.1016/0022-2836(89)90299-4. [DOI] [PubMed] [Google Scholar]
- 49.Yutani K, Hayashi S, Sugisaki Y, Ogasahara K. Role of conserved proline residues in stabilizing tryptophan synthase alpha subunit: analysis by mutants with alanine or glycine. Proteins. 1991;9:90–8. doi: 10.1002/prot.340090203. [DOI] [PubMed] [Google Scholar]
- 50.Alber T, Sun DP, Nye JA, Muchmore DC, Matthews BW. Temperature-sensitive mutations of bacteriophage T4 lysozyme occur at sites with low mobility and low solvent accessibility in the folded protein. Biochemistry. 1987;26:3754–8. doi: 10.1021/bi00387a002. [DOI] [PubMed] [Google Scholar]
- 51.Matthews BW. Structural and genetic analysis of protein stability. Annu Rev Biochem. 1993;62:139–60. doi: 10.1146/annurev.bi.62.070193.001035. [DOI] [PubMed] [Google Scholar]
- 52.Shepherd AJ, Gorse D, Thornton JM. Prediction of the location and type of beta-turns in proteins using neural networks. Protein Sci. 1999;8:1045–55. doi: 10.1110/ps.8.5.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hebert EJ, Grimsley GR, Hartley RW, Horn G, Schell D, Garcia S, Both V, Sevcik J, Pace CN. Purification of ribonucleases Sa, Sa2, and Sa3 after expression in Escherichia coli. Protein Expr Purif. 1997;11:162–8. doi: 10.1006/prep.1997.0776. [DOI] [PubMed] [Google Scholar]
- 54.Pace CN, Scholtz JM. Measuring the conformational stability of a protein. In: Creighton TE, editor. Protein structure: A practical approach. IRL Press; Oxford: 1997. pp. 299–321. [Google Scholar]
- 55.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–23. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]