

Abstract
The ability of commercially available amino acid derivatives, especially Fmoc-Trp(Boc)-OH, to differentiate enantiomers of chiral phosphonates, phosphinates, phosphates, phosphine oxides, and phosphonamidates is demonstrated with 31P, 13C, and 1H NMR spectroscopy. The chiral differentiation provided a rapid and convenient method for measuring the enantiomeric purity of these phosphorus compounds.
1. Introduction
Chiral phosphorus oxide derivatives 1 are a class of compound that have been widely utilized in both chemistry and biology. Chiral phosphine oxides and phosphinate monoesters are important precursors to the corresponding phosphines that are used as chiral ligands for metal catalysts in modern asymmetric transformations.1–4 The enantiomers of chiral phosphonate and phosphate esters are differentially toxic to multi-cellular organisms and these compounds are used as agricultural pesticides and chemical weapons.5–7 These applications have stimulated efforts directed at the synthesis, resolution and determination of the enantiomeric purity of chiral phosphorus compounds.8–12
Recent advances in the characterization of the catalytic properties of the bacterial phosphotriesterase (PTE) and rationally designed site-directed mutant enzymes have provided a convenient method for the kinetic resolution of racemic phosphinate, phosphonate and phosphate esters through the stereoselective hydrolysis of a single enantiomer.13–15 These enzyme libraries have facilitated the isolation of enantiomerically pure substrates of either stereochemistry, since mutant enzymes have been identified where the stereoselectivity is either enhanced or inverted relative to the wild type enzyme.16,17 The absolute configurations of the unhydrolyzed products could be reliably predicted based upon the known stereoselectivity of the wild type enzyme and the characterized mutant variants.17,18 However, experimental determination of the precise enantiomeric purity of the isolated phosphinate, phosphonate and phosphate ester products is not trivial. For example, the enantiomeric purity of the four stereoisomers of pinacolyl 4-nitrophenyl methylphosphonate and the two enantiomers of 4-acetylphenyl methyl phenylphosphonate were resolved with much effort by chiral HPLC methods and chiral electrophoresis, respectively.13,19 A more rapid and convenient method is needed, as more of such enantiomers are routinely obtained by enzymatic methods.
An attractive alternative method for the determination of the chiral purity of these compounds is the use of 31P and 1H NMR spectroscopy with a chiral solvating agent.20 The potential advantages of using chemical solvating agents for the differentiation of enantiomers, relative to other methods, such as the use of lanthanide complexes or the formation of covalent diastereomer derivatives with chiral reagents, are well recognized.21 Chiral methyl phenyl phosphinothioic acid 2 and t-butyl phenyl phosphinothioic acid 3 have been successfully used to discriminate between the enantiomers of chiral phosphorus compounds including the amide and ester derivatives of phosphonic and phosphinic acids.21–27 However, these chiral reagents are not commercially available.
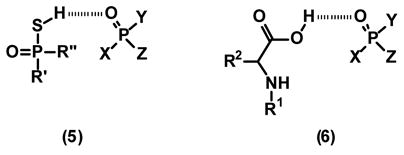
Thioic acids 2 and 3 are apparently able to form transient diastereomeric complexes in solution with chiral phosphorus esters but the specific mode of interaction is not precisely known. In an effort to mimic the molecular interactions in these complexes with commercially available chiral acids, we chose N-acyl substituted amino acids 4. Modified amino acids have been used as chemical solvating agents for the enantiodiscrimination of chiral α-arylalkylamines.28 The rationale for the choice of 4 as a potential complexing agent for the differentiation of enantiomers such as 1 is that a broad range of modified amino acids are readily available and the free carboxylic acid may function in a manner similar to the thioic acid in compounds 2 and 3. The formation of hydrogen bonded complex 5 might be the origin of the differentiation in the chemical shifts between enantiomers.21–22 In this respect, chiral carboxylic acids, such as 4, may form similar complexes in solution as presented in complex 6. Compounds of the general structure 4 are readily available as inexpensive commercial derivatives of amino acids used in solid phase peptide synthesis.29 Herein we report that 31P and 1H spectroscopy in the presence of protected L-amino acid derivatives can differentiate between the enantiomeric forms of chiral phosphorus oxide derivatives.
2. Results and discussion
To test the potential of N-acyl L-amino acid derivatives as chiral solvating agents for the differentiation of the chiral phosphorus compounds, we added N-Fmoc-L-phenylalanine to a solution of racemic 4-acetylphenyl cyclohexyl methylphosphonate 7 (see Table 1). Two signals of moderate separation without significant line broadening were observed in the 31P NMR spectrum in CDCl3 at room temperature (Figure 3C). N-Fmoc-N′-Boc-L-tryptophan (FBTrp), which has a larger side-chain group, was used next and a better resolution between the two enantiomers was obtained. To optimize the conditions for the differentiation of the two enantiomers with this compound, we titrated compound 7 with FBTrp in CDCl3 at room temperature. The observed dependence of the separation in the chemical shift (Δδ in ppb) between the two enantiomers of 7 in the presence of an increasing concentration of FBTrp is presented in Figure 1A. At saturating concentrations of FBTrp the maximum difference in chemical shift is approximately 38 ppb and the data are consistent with the formation of a 1:1 complex between compound 7 and FBTrp.
Table 1.
Differences in the chemical shifts between enantiomers induced upon the addition of 50 mM FBTrp in CDCl3 at 25°C.
| Entry | X | Y | Z | Δδ (ppb) 31P (NMR) | Δδ (ppb)a 1H (NMR) |
|---|---|---|---|---|---|
| 7 | CH3- |
|
|
28 | 0 |
| 8 (SpSc, RpRc) | CH3- |
|
|
46 | 0 |
| 8 (RpSc, SpRc) | CH3- |
|
|
17 | 0 |
| 9 | CH3- |
|
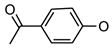
|
20 | 3 |
| 10 | CH3- |
|
|
0 | 5 |
| 11 | CH3- |
|
|
13 | 3 |
| 12 | CH3- |
|
|
54 | 0 |
| 13 | CH3CH2- |
|
|
51 | 0 |
| 14 | CH3CH2- |
|
CH3CH2O- | 0 | 7 |
| 15 | CH3O- |
|
|
8 | 2 |
| 16 | CH3O- |
|
|
5 | 0 |
| 17 | CH3O- |
|
|
0 | 3b |
| 18 | CH3- |
|
H2N— | 46 | 10 |
| 19 | CH3- |
|
H2N— | 80, 17 | 6, 4 |
| 20 | CH3- |
|
|
0 | 6 |
When the separation of the proton resonance are indicated, the specific hydrogens are highlighted in bold font.
In the presence of Fmoc-serine(trityl)-OH.
Figure 3.
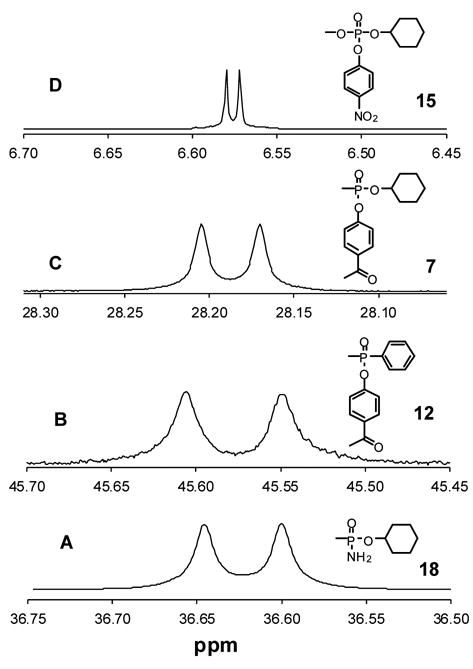
31P NMR spectra for racemic mixtures of four types of chiral phosphorus compounds utilized in this investigation in the presence of FBTrp. (A) Phosphonamidate 18. (B) phosphinate ester 12. (C) methyl phosphonate ester 7. (D) phosphate triester 15.
Figure 1.
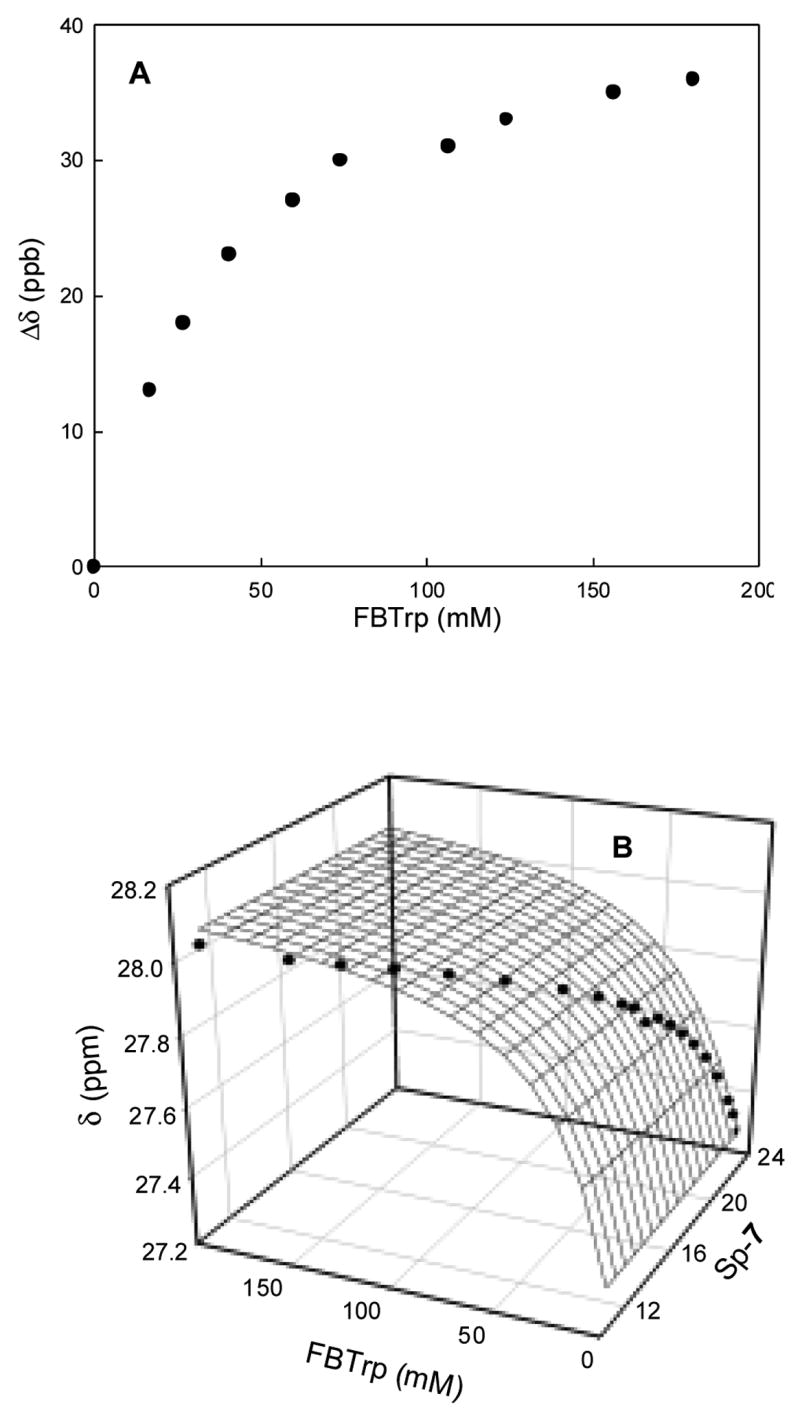
(A) Separation of the 31P NMR signals (Δδ in ppb) for racemic compound 7 (47.5 mM in 0.6 mL CDCl3) after the addition of FBTrp in CDCl3 at 25 °C. (B). Binding of variable amounts of (SP)-7 with variable amounts of FBTrp in CDCl3 at 25 °C. The data were fit to equation 1 with a Kd of 19 mM.
To determine if the solvating agent preferentially binds one of the two enantiomers over the other, enantiomerically pure (RP)-7 and (SP)-7 were titrated separately with FBTrp in CDCl3. The values of Kd for the complexes formed between (RP)-7 or (SP)-7 and FBTrp were calculated by a non-linear least squares analysis to be 21 ± 1 mM and 19 ± 1 mM, respectively, from a fit of the data to equation 1 for the formation of a 1:1 complex (Figure 1B). In equation 1, δob is the observed chemical shift for (RP)-7 or (SP)-7; δA is the chemical shift of (RP)-7 or (SP)-7 in the absence of FBTrp; δAB is the chemical shift for the complex of (RP)-7 or (SP)-7 with FBTrp; “A” is the total concentration of (RP)-7 or (SP)-7; “B” is the total concentration of FBTrp, and Kd is the corresponding dissociation constant.
| (Eq. 1) |
These results indicate that the binding strength between FBTrp and either of the two enantiomers of 7 is approximately the same. This conclusion is consistent with the observation that the 31P NMR signals for the two diasteromers in the racemic mixture are of the same linewidth and signal intensity. Therefore, the differentiation of the two enantiomers by FBTrp is attributed to the intrinsic difference in the chemical shift of the two diasteromeric complexes formed between FBTrp and (RP)-7 or (SP)-7. The upfield signal in the 31P NMR spectra was assigned to (RP)-7 and the downfield resonance to (SP)-7 by the addition of the corresponding pure enantiomers separately into the racemic mixture in the presence of FBTrp. The NMR spectra of (RP)-7 or (SP)-7 alone in the presence of FBTrp demonstrated that the chiral purity of these compounds, prepared via a kinetic enzymatic resolution, exceeded an enantiomeric excess of 98%.
For racemic 7, the differentiation between the two enantiomers can easily be measured by 31P NMR spectroscopy, but no separation was detectable by 1H NMR spectroscopy. In addition to FBTrp and N-Fmoc-L-phenylalanine, other N-protected L-amino acids were assessed for the differentiation of racemic 7. Most of them are unable to discriminate between the two enantiomers and none gave a better separation than FBTrp when measured by 31P NMR or 1H NMR spectroscopy. A change in solvent from chloroform-D to benzene did not improve the resolution.
The ability of FBTrp to differentiate between the enantiomers of compounds which are chiral at a phosphorus center was further tested with the diasteromeric mixture of the methyl phosphonate diester 8 (Figure 2F). In the presence of FBTrp, each of the four stereoisomers of 8 exhibited nearly baseline resolved 31P NMR resonances as shown in Figure 2A. For the assignment of these resonances to specific diastereomers, each of the four isomers was added separately into the complete mixture of stereoisomers in the presence of FBTrp (Figures 2B–E). The two most downfield signals were assigned to enantiomeric pair of (SPSC)-8 and (RPRC)-8 with the (SPSC)-8 isomer as the most downfield resonance. The two upfield resonances were assigned to (SPRC)-8 and (RPSC)-8 with (RPSC)-8 as the most upfield resonance. Upon complexation with FBTrp, the chemical shifts of the two diasteromers moved about 0.65 ppm. The separation between the NMR signals of the enantiomeric pair, (SPSC)-8 and (RPRC)-8, is 46 ppb, which is significantly larger than the 17 ppb separation between the enantiomeric pair of (SPRC)-8 and (RPSC)-8. This example demonstrates that the separation of the NMR signals upon the addition of FBTrp is dependent upon the identity of the functional groups and stereochemical orientation distant from the phosphorus center.
Figure 2.
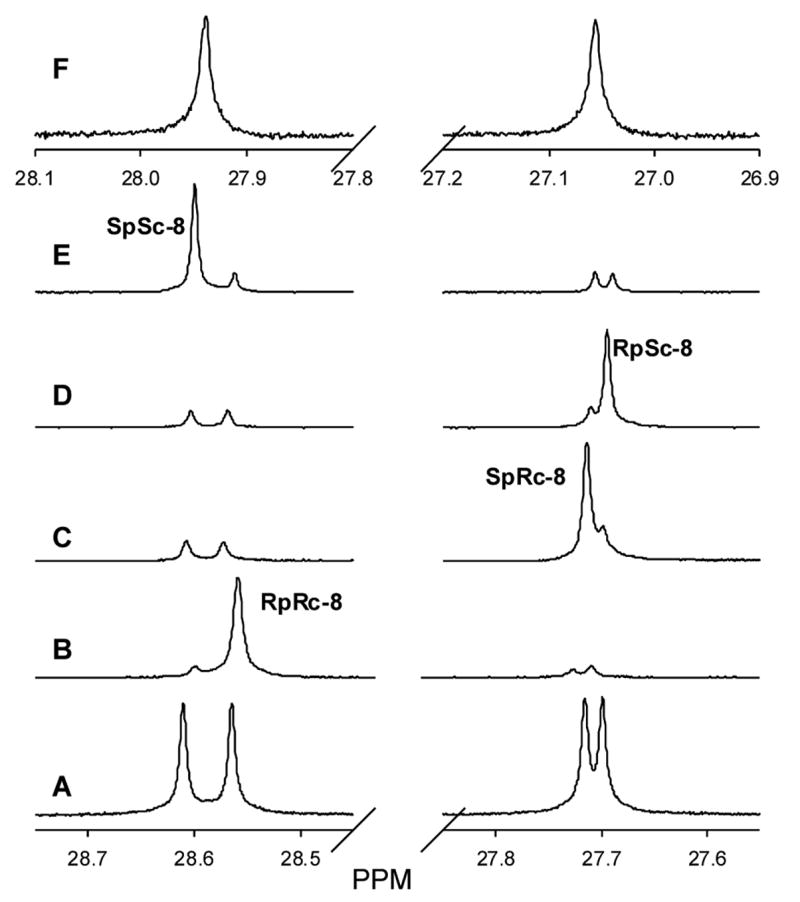
Assignment of the 31P NMR signals for the individual enantiomers within a racemic mixture of compound 8. (A) Spectrum of diastereomeric 8 in the presence of FBTrp. (B) Addition of authentic (RPRC)-8 enantiomer into diastereomeric 8. (C) Addition of (SPRC)-8 enantiomer into diastereomeric 8. (D) Addition of (RPSC)-8 enantiomer into diastereomeric 8. (E) Addition of (SPSC)-8 enantiomer into diastereomeric 8. (F) Spectrum of diastereomeric 8 in the absence of FBTrp.
The generality of using FBTrp to differentiate between chiral phosphorus compounds was tested in the resolution of phosphonates 7–11, phosphinates 12–14, phosphates 15–17, phosphonamidates 18–19 and phosphine oxides 20–21 by both 31P and 1H NMR spectroscopy. The results are summarized in Table 1. Racemic phosphonate compounds with alkoxy groups smaller than cyclohexyl and pinacolyl, 9–11, were utilized. For racemic compounds 9 and 11, the 31P resonances for the two enantiomers were distinguishable from one another with differences in chemical shift of 20 and 13 ppb, respectively. However, there was no observable difference in the chemical shifts for the individual enantiomers of 10 by 31P NMR spectroscopy. Nevertheless, for the three phosphonate esters 9–11, the 1H NMR spectra revealed distinct resonances for the methyl group directly attached to the phosphorus core. In the absence of FBTrp, the 1H NMR signals for these protons were observed as a doublet with a separation 16 Hz due to spin coupling with the adjacent phosphorus. In the presence of FBTrp, each of the resonances for the methyl phosphonate group in 9, 10, and 11 was further separated into resonances for each of the enantiomers with separations of 3, 5, and 3 ppb, respectively.
Compounds 12–14 were assessed as examples of racemic phosphinate esters. For compounds 12 (Figure 3B) and 13, the differences in the 31P NMR spectra for these resonances are 54 and 51 ppb in the presence of FBTrp, about twice that observed for the phosphonate esters. However, no differentiation was observed in the 1H NMR spectra of either 12 or 13. For compound 14, no distinction between the two enantiomers was observed in the 31P NMR spectrum but a separation of 7 ppb was measured in the 1H NMR spectrum of compound 14. The protons from the methyl group of the ethoxy substituent, which is a triplet in the absence of FBTrp, are further separated from one another with a separation of 7 ppb.
Fully esterified phosphates were examined with compounds 15–17. These compounds showed the weakest separation in the 31P NMR spectra in the presence of FBTrp. For compounds 15 (Figure 3D) and 16, the separation in chemical shift values for the individual enantiomers are 8 and 5 ppb, respectively. For compound 15, the protons of the methoxy group are separated by 2 ppb in the 1H NMR spectrum but for compound 16 no separation was observed in the presence of FBTrp. For compound 17, there was no separation in either the 31P or 1H NMR spectra in the presence of FBTrp. To determine if other amino acid derivatives could differentiate between the two enantiomers of compound 17, we tested 18 other protected amino acids. We found that Fmoc-serine (trityl)-OH (FTSer) gave a moderate separation of 3 ppb for the proton resonances of the methoxy group, while no separation of the 31P NMR signal was observed.
Compounds 18 and 19 were examined as examples for phosphonamidates. In the presence of FBTrp in CDCl3, both of these chiral compounds exhibited good separation of the 31P NMR signals. The separation for compound 18 is 46 ppb for 31P (Figure 3A) and 10 ppb for the hydrogens of the methyl group. In the 31P NMR spectrum of the diastereomeric mixture of 19, two pairs of signals centered at 36.70 and 36.55 ppm were separated by 80 and 17 ppb respectively. In the 1H NMR spectrum, the differences in the chemical shifts for the protons of the methyl group are 6 and 4 ppb for each enantiomeric pair. The absolute configurations corresponding to these resonances have not been determined.
The final chiral phosphorus compounds tested were the phosphine oxides 20 and 21 (Chart 3). Neither compound showed separation in their 31P NMR spectrum in the presence of FBTrp, although complexation of the two compounds with FBTrp was indicated by a 4 ppm downfield change in the chemical shift. In the 1H NMR spectrum of compound 20, a separation of 6 ppb was observed for the protons of the methoxy group, but no separation was observed for the protons of the methyl group which is attached directly to the phosphorus center. In the 1H NMR spectrum of compound 21 (Figures 4A–D), the resonances for the proton attached to C2, centered at ~5.91 ppm and coupled to the phosphorus core with JP-H of 25 Hz (Figure 4A), were further split with a separation of 19 ppb in the presence of FBTrp (Figure 4B). The resonance for the methyl group of 21 is a singlet at 2.04 ppm (Figure 4C) in the absence of FBTrp but is a doublet with a separation of 9 ppb in the presence of FBTrp (Figure 4D). The two protons attached to C5 gave a complex set of resonances in the range of 2.26-2.13 ppm (Figure 4C) that are split into two sets of resonances at 2.40-2.29 ppm and 2.25-2.16 ppm in the presence of FBTrp (Figure 4D). No separation for the resonances of the protons attached to C4 was observed.
Chart 3.
Figure 4.
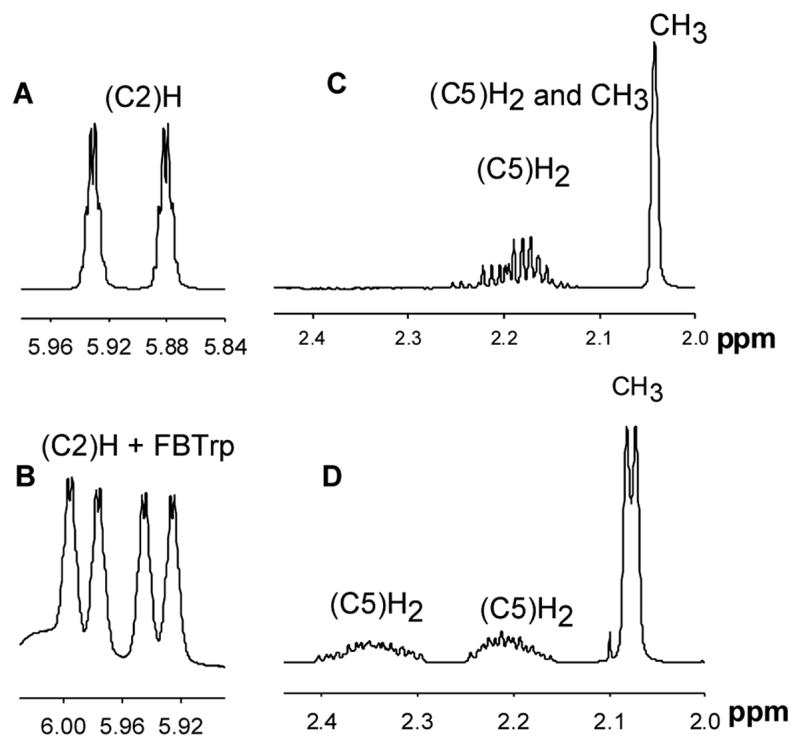
1H NMR spectra for the protons at C2, C5 and methyl group of compound 21. (A) 1H NMR signal for the proton at C2 in the absence of FBTrp. (B) 1H NMR resonance for the proton at C2 in the presence of FBTrp. (C) 1H NMR signal for the protons at C5 and the methyl group of 21 in the absence of FBTrp. (D) Same as in Figure C but in the presence of FBTrp.
For compound 21, the 13C NMR spectra were also recorded in the presence and absence of FBTrp (Figures 5A–D). The 13C NMR resonance for C3 of compound 21, centered at 164.96 ppm with JP-C = 25.3 Hz as a doublet (Figure 5A), divided into two pairs of doublets with a separation of 42 ppb in the presence of FBTrp (Figure 5B). The 13C resonances for the methyl group, centered at 21.22 ppm with JP-C = 17.2 Hz as a doublet (Figure 5C), split into two pairs of doublets with a separation of 25 ppb in the presence of FBTrp (Figure 5D). However, no separation was observed for the 13C resonances of C2, C4 or C5.
Figure 5.
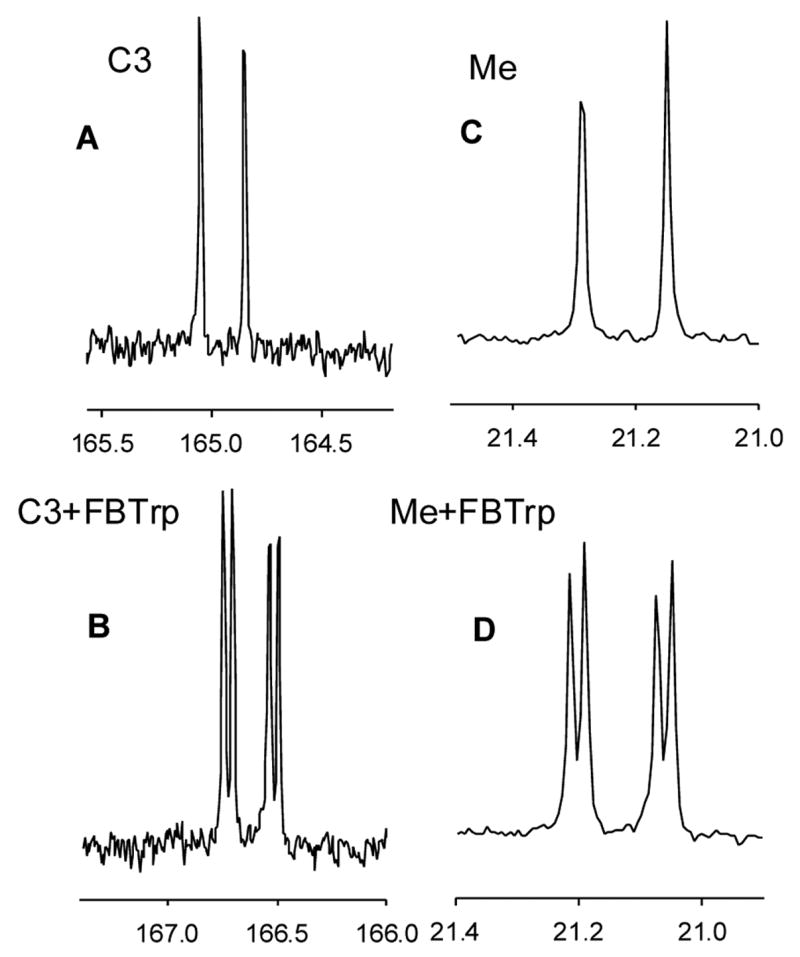
13C NMR spectra for C3 and the methyl group of compound 21. (A) 13C NMR signal for C2 in the absence of FBTrp. (B) Same as in Figure A but in the presence of FBTrp. (C) 13C NMR signal of the methyl group in the absence of FBTrp. (D) Same as in Figure C but in the presence of FBTrp.
A comparison of atypical separation in the 31P NMR spectra induced by FBTrp for the five types of chiral phosphorus compounds tested in this investigation is presented in Table 1. The magnitude of the chemical shift differences between the pairs of enantiomers are: phosphinates ~ phosphonamidates > phosphonates > phosphates>phosphine oxides. The downfield chemical shift changes in the 31P NMR spectra suggest the complexation of amino acid derivatives with the phosphorus compounds. The discrimination between enantiomers by the modified amino acids is attributed to the chemical shift differences of the diasteromeric complexes because the dissociation constants for the binding of the (RP)- and (SP)-enantiomers of compound 7 to FBTrp are essentially identical. However, we do not understand why some enantiomers can be differentiated by 31P NMR and others by 1H or 13C NMR spectroscopy. A direct correlation of the absolute configuration at phosphorus relative to the chemical shift of the diastereomeric complex formed between each enantiomer and FBTrp has not been determined. The direct interaction between these phosphorus compounds and FBTrp is expected to be facilitated by hydrogen bonding. In this respect, FBTrp might function as a versatile chemical solvating agent in the 1H NMR spectroscopy for the enantio-differentiation of chiral compounds, such as alcohols and amines.
3. Conclusion
In conclusion, we have found that FBTrp is a versatile chemical solvating agent that can be used for the differentiation of chiral phosphorus centers in compounds that include phosphine oxides, phosphinates, phosphonates, phosphates and phosphonamidates by 31P, 13C and 1H NMR spectroscopy. The separations of the resonances that are induced by FBTrp in the 31P NMR spectra are substantial except for the chiral phosphates and phosphine oxides, for which the separation is relatively small. This observation has allowed the determination of the enantiomeric purity for chirally enriched phosphorus compounds. Since FBTrp is commercially available and relatively inexpensive, it might be used as a convenient new chemical solvating agent for other chiral compounds that may include chiral sulfoxides.30 FBTrp or other amino acid derivatives may also be applied in development of new methods for the rapid and convenient determination of enantiomeric excess.31
4. Experimental
4.1. General
All of the 31P and 1H NMR experiments were carried out with a Varian Inova-400 Broad Band Spectrometer unless mentioned otherwise. For the 31P NMR spectra, the acquisition time was set to 5–8 seconds with 2 second delay. Aqueous phosphoric acid (85%) was used as an external reference. For titration of the single enantiomers of (RP)-7 or (SP)-7, aqueous phosphoric acid (85%) in a sealed capillary was inserted into the NMR tube as an internal reference. For collection of 1H and 13C NMR spectra, standard parameters were used. The amino acid derivatives Fmoc-Trp(Boc)-OH, Fmoc-Ser(Trityl)-OH were used as purchased from EMD Biosciences. (4-Methoxyphenyl)methylphenylphosphine oxide 20 was purchased from ASDI Inc. 3-Methyl-1-phenyl-2-phospholene 1-oxide 21 was purchased from TCI America. The racemic phosphinates, phosphonates and phosphates ester were prepared following known procedures.13 Phosphonamidates 18 and 19 were made and characterized as described below. Enantiomeric phosphonates were obtained by enzymatic resolution of their racemic mixtures.19
4.2. Cyclohexyl methylphosphonamidate 18
To a solution of cyclohexanol (10 mmol) in ethyl ether (20 mL) in a dry ice/acetone bath was added butyl lithium (10 mmol, 2.5 M in hexanes). To the suspension was added a solution of methylphosphonic dichloride (10 mmol) in ethyl ether (40 mL). The mixture was stirred for 20 minutes in a dry ice/acetone bath and then at room temperature for 1 hour. After removal of the solvent, concentrated aqueous ammonia (20 equivalents) was added to the residue and the mixture was stirred at room temperature for 30 minutes. After the ammonia and water were removed under reduced pressure, the residue was resuspended in ethyl ether, filtered and washed with ethyl ether. A solution of ethyl ether was collected and condensed to dryness. Recrystalization from ethyl ether yielded the desired compound 18 as colorless crystals in 80% yield. 1H NMR (CDCl3, δ in ppm): 4.50-4.35 (2H, m); 2.83 (2H, broad); 2.05–2.14 (14H, m). 31P (CDCl3, δ in ppm, 85% aqueous H3PO4 as external reference): 32.73.
4.3. Pinacolyl methylphosphonamidate 19
The diasteromeric mixture was prepared in an 85% yield following the procedure described for 18. 1H NMR (CDCl3, δ in ppm): 4.25-4.17 (1H, CH, m); 2.96, 2.91 (2H, NH2, s, broad), 1.49, 1.51 (3H, CH3-P, two doublets, JP-H = 16.7 Hz); 1.27, 1.24 (3H CH3-C, two doublets, JH-H = 6.70 Hz); 0.87, 0.88 (9H, t-butyl, two singlets). 31P (CDCl3, δ in ppm, 85% aqueous H3PO4 as external reference): 33.11; 32.77.
Supplementary Material
Chart 1.
Chart 2.
Acknowledgments
This work was supported in part by the NIH (GM 68550).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kolodiazhnyi OI, Grishkun EV. Tetrahedron: Asymmetry. 1996;7:967–970. [Google Scholar]
- 2.Moriyama M, Bentrude WG. J Am Chem Soc. 1983;105:4727–4733. [Google Scholar]
- 3.Lewis RA, Mislow K. J Am Chem Soc. 1969;91:7009–7012. [Google Scholar]
- 4.Korpiun O, Lewis RA, Chickos J, Mislow K. J Am Chem Soc. 1968;90:4842–4846. [Google Scholar]
- 5.Eto M. Organophosphorus Pesticides: Organic and Biological Chemistry. CRC Press; Cleveland, OH: 1974. pp. 123–368. [Google Scholar]
- 6.Chambers JE, Oppenheimer SF. Toxicological Science. 2004;77:185–187. doi: 10.1093/toxsci/kfh060. [DOI] [PubMed] [Google Scholar]
- 7.Abou-Donia MB. Ann Rev Pharmacol Toxicol. 1981;21:511–548. doi: 10.1146/annurev.pa.21.040181.002455. [DOI] [PubMed] [Google Scholar]
- 8.Hayakawa Y, Hyodo M, Kimura K, Kataoka M. Chem Commun. 2003:1704–1705. [Google Scholar]
- 9.Kurihara N, Miyamoto J, Paulson GD, Zeeh B, Skidmore MW, Hollingworth RM, Kuiper HA. Pure & Appl Chem. 1997;69:1335–1348. [Google Scholar]
- 10.Battershill JM, Edwards PM, Johnson MK. Food and Chemical Toxicology. 2004;42:1279–1285. doi: 10.1016/j.fct.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Lum KT, Huebner HJ, Li Y, Phillips TD, Raushel FM. Chem Res Toxicol. 2003;16:953–957. doi: 10.1021/tx034047k. [DOI] [PubMed] [Google Scholar]
- 12.Kolodiazhnyi OI. Tetrahedron: Asymmetry. 1998;9:1279–1332. [Google Scholar]
- 13.Nowlan C, Li Y, Hermann JC, Evens T, Carpenter J, Ghanem E, Shoichet BK, Raushel FM. J Am Chem Soc. 2006;128:15892–15920. doi: 10.1021/ja0658618. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Aubert SD, Raushel FM. J Am Chem Soc. 2003;125:7526–7527. doi: 10.1021/ja035625m. [DOI] [PubMed] [Google Scholar]
- 15.Li WS, Li Y, Hill CM, Lum KT, Raushel FM. J Am Chem Soc. 2002;124:3498–3499. doi: 10.1021/ja017840d. [DOI] [PubMed] [Google Scholar]
- 16.Hong SB, Raushel FM. Biochemistry. 1999;38:1159–1165. doi: 10.1021/bi982204m. [DOI] [PubMed] [Google Scholar]
- 17.Chen-Goodspeed M, Sogorb MA, Wu F, Raushel FM. Biochemistry. 2001;40:1332–1339. doi: 10.1021/bi001549d. [DOI] [PubMed] [Google Scholar]
- 18.Hermann JC, Ghanem E, Li Y, Raushel FM, Irwin JJ, Shoichet BK. J Am Chem Soc. 2006;128:15882–15891. doi: 10.1021/ja065860f. [DOI] [PubMed] [Google Scholar]
- 19.Li W-S, Lum KT, Chen-Goodspeed M, Sogorb MA, Raushel FM. Bioorg Med Chem. 2001;9:2083–2091. doi: 10.1016/s0968-0896(01)00113-4. [DOI] [PubMed] [Google Scholar]
- 20.Parker D. Chem Rev. 1991;91:1441–1457. [Google Scholar]
- 21.Omelanczuk JO, Mikolajczyk M. Tetrahedron: Asymmetry. 1996;7:2687–2694. [Google Scholar]
- 22.Harger MJP. Tetrahedron Lett. 1978;19:2927–2928. [Google Scholar]
- 23.Harger MJP. J Chem Soc Perkin II. 1980:1505–1511. [Google Scholar]
- 24.Bentrude WG, Moriyama M, Mueller MD, Sopchik AE. J Am Chem Soc. 1983;105:6053–6061. [Google Scholar]
- 25.Omelanczuk J, Spochik AE, Lee SG, Akutagawa K, Cairns SM, Bentrude WG. J Am Chem Soc. 1988;110:6908–6909. [Google Scholar]
- 26.Drabowicz J, Duzinski B, Mikolajczyk M. Tetrahedron: Asymmetry. 1992;3:1231–1234. [Google Scholar]
- 27.Drabowicz J, Dudzinski B, Mikolajczyk M, Colonna S, Gaggero N. Tetrahedron: Asymmetry. 1997;8:2267–2270. [Google Scholar]
- 28.Luo Z, Li B, Fang X, Hu K, Wu X, Fu E. Tetrahedron Lett. 2007;48:1753–1756. [Google Scholar]
- 29.Chan WC, White PD. Fmoc solid phase peptide synthesis: a practical approach. Oxford University Press; Oxford: 2000. pp. 9–40. [Google Scholar]
- 30.Deshmukh M, Dunach E, Juge S, Kagan HB. Tetrahedron Lett. 1984;25:3467–3470. [Google Scholar]
- 31.Finn MG. CHIRALITY. 2002;14:534–540. doi: 10.1002/chir.10101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.