

Abstract
Continuous fluorescence microphotolysis (CFM) and fluorescence correlation spectroscopy (FCS) permit measurement of molecular mobility and association reactions in single living cells. CFM and FCS complement each other ideally and can be realized using identical equipment. So far, the spatial resolution of CFM and FCS was restricted by the resolution of the light microscope to the micrometer scale. However, cellular functions generally occur on the nanometer scale. Here, we develop the theoretical and computational framework for CFM and FCS experiments using 4Pi microscopy, which features an axial resolution of ∼100 nm. The framework, taking the actual 4Pi point spread function of the instrument into account, was validated by measurements on model systems, employing 4Pi conditions or normal confocal conditions together with either single- or two-photon excitation. In all cases experimental data could be well fitted by computed curves for expected diffusion coefficients, even when the signal/noise ratio was small due to the small number of fluorophores involved.
INTRODUCTION
Thermal molecular motion has been referred to as the restless heartbeat of matter and life (1). This applies, in particular, to living cells in which many processes are driven directly and others are strongly influenced by diffusion. The experimental study of diffusion in cellular systems was fundamentally advanced in 1974 when two complementary techniques were introduced, fluorescence microphotolysis ((FM) also referred to as fluorescence recovery after photobleaching) (2) and fluorescence correlation spectroscopy (FCS) (3). In FM (2,4–10), a small area of a fluorescent specimen is illuminated by a focused laser beam at a very low, nonbleaching intensity. The fluorescence of the illuminated spot is then bleached “instantaneously” by increasing the power of the laser beam by several orders of magnitude for a short time. Subsequently, the redistribution of fluorescence by diffusion is followed at the initial nonbleaching intensity and used to derive diffusion coefficients. FCS also employs a focused laser beam to illuminate a small spot of a fluorescent specimen at a small nonbleaching intensity. However, the fluorophore concentration is kept so small that fluctuations in the number of illuminated fluorophores become apparent and can be used to derive diffusion coefficients by a correlation analysis. In the meantime, FM and FCS have been further developed into many directions (for review, see Day and Schaufele (11)). It has become clear that both techniques can provide information on molecular mobility and the association between different molecular species. However, the techniques differ, and, in fact, complement each other ideally, for instance with respect to range of accessible diffusion coefficients and fluorophore concentration. Thus, FM and FCS have become indispensable tools of cell biology and are employed at an ever-increasing pace.
In 1981, continuous fluorescence microphotolysis (CFM) was introduced (12). It employs a focused laser beam to continuously illuminate a spot of a fluorescent sample at a laser power inducing slow photobleaching. By these means the illuminated spot is, at the same time, depleted of fluorophores by photobleaching and replenished with fresh fluorophores by diffusion. Because the laser power is much higher in CFM than in the measuring phases of FM, the CFM signal quality is better. This is traded in by the necessity to extract not only the diffusion coefficient but also the photobleaching rate from the experimental data. An intriguing property of CFM is that it can be easily installed on a confocal microscope and combined with FCS without any further instrumental modifications (13). Due to the complexity of the diffusion-reaction processes studied using FM or FCS, it is often impossible to extract the diffusion coefficient using an analytical description; in such cases, numerical calculations are employed. Indeed, computational approaches proved to be reliable and are widely used nowadays for analyzing the diffusion properties observed in conventional FM (14–24), CFM (12,25,26,13,27), and correlation spectroscopy (28–30).
So far, in both CFM and FCS experiments the size of the illuminated area was limited by diffraction to ∼250 nm in the focal plane and 600 nm in direction of the optical axis. However, recently new light microscopy concepts have been developed that improve the resolution up to ≈30 nm. These techniques include 4Pi microscopy (31–36), I5M microscopy (37,38,35), and stimulated emission depletion (STED) microscopy (39–42,36). Emergence of these techniques has already provided a new level of detail observed in biological imaging (41,42,36,43), and promises to bring about a tremendous improvement for the FM technology, but to take advantage of the resolution improvement, an appropriate theoretical and computational framework has to be created. In this article, we develop the theory and computational methods for the utilization of the 4Pi microscope in CFM and FCS measurements. We also demonstrate the feasibility of CFM and FCS experiments with a 4Pi microscope, and analyze the ways of extracting the characteristics of diffusion from the experimental data using numerical calculations.
The principle of 4Pi microscopy (31,33) is illustrated in Fig. 1. Coherent light (red) from a laser is split into two beams, which are focused at the same point onto a sample. Constructive interference of the two beams enhances the focusing of the light, and the illuminated region is narrower along the optical axis in the case of the 4Pi microscope than in the case of the common confocal microscope, but accompanied by two side lobes (see Fig. 2). Two-photon illumination is usually employed to further narrow the excitation volume (31,33) by mainly reducing the height of the side lobes. In this case, the fluorescence signal (green beam in Fig. 1) is observed at the wavelengths that are shorter than the illumination wavelength. In 4Pi microscopy, various types of illumination and detection are utilized (31): type A corresponds to the illumination via two objectives with constructive interference and detection through one of the objectives in a confocal mode; in type B, illumination is performed by one beam, and detection via two objectives; for type C, both illumination and detection are performed using two objectives, with constructive interference in both cases. We use type A 4Pi microscopy, since it is the only one available commercially (Leica Microsystems, Wetzlar, Germany).
FIGURE 1.

Schematic of a 4Pi microscope. Light from a laser is split into two beams by a beam splitter (BS). Coming from opposite directions, the two beams are focused at the sample, where constructive interference of the two fields (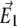 and
and 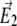 ) creates a narrow illumination spot. The fluorescence signal is deflected by the dichroic mirror (DM), and focused on a detector. Since two-photon excitation is employed (red), the fluorescence (green) is at a shorter wavelength than the illuminating light. See the literature (31,33,44) for details.
) creates a narrow illumination spot. The fluorescence signal is deflected by the dichroic mirror (DM), and focused on a detector. Since two-photon excitation is employed (red), the fluorescence (green) is at a shorter wavelength than the illuminating light. See the literature (31,33,44) for details.
FIGURE 2.

Point spread function. (a) Construction of the PSF of a 4Pi microscope is shown schematically. Light wavefronts from the top and bottom objectives coming along the z axis interfere constructively at the sample. Calculated profiles of the PSF along the x axis (b) and z axis (c) are shown for the case of the 4Pi (circles) and confocal (squares) microscopes. Experimental PSF profiles are shown as solid lines. The 4Pi PSF is substantially narrower in the z-direction compared to the PSF of a confocal microscope (c), but has a complicated form (see Eq. 5). A 3D image of the 4Pi PSF is shown in panels d and e. Isocontours of the PSF are drawn at three values: 1/20 (red), 1/2,000 (blue), and 1/20,000 (white) of the central peak height. The PSFs are symmetric in regard to rotation around the z axis; “ripples” present in panel e are due to the rectangular grid used to visualize the PSF. Objectives with half-aperture angle of 68.5° and n = 1.46 were assumed. For the confocal PSF, the illumination wavelength is 488 nm, detection wavelength is 627 nm; for the 4Pi PSF two-photon excitation is used, with light of 905 nm wavelength; the detection wavelength is 627 nm. The PSF width in the x-direction is the same for 4Pi and confocal microscopes; the different widths observed in panel b are solely due to the different wavelengths. The 3D PSF images were created with VMD (64).
The main challenge in analyzing CFM and FCS by means of 4Pi microscopy is an adequate representation of the microscope point spread function (PSF), which describes how a point gets spread by the imaging process, due to the limited resolution of the microscope (see Fig. 2 and Supplementary Material). The 4Pi PSF has a complex, nonanalytical shape in three-dimensional (3D) space (see, e.g., Hell and Stelzer (31)), which complicates the numerical solution required for simulation of the simultaneous diffusion and bleaching of fluorophores in CFM. There exist programs that allow one to solve the partial differential equations encountered in the description of CFM experiments (see, e.g., Schaff et al. (16)). However, to our knowledge, none of them is capable of handling an arbitrary form of the PSF. Therefore, we developed our own program to solve the arising differential equations numerically.
As shown below, numerical calculations reliably reproduce the experimental fluorescence signal. We find that in CFM and FCS with the 4Pi microscope, the prominent feature of the fluorescence is a poor signal/noise ratio, because of the small number of fluorophores in the irradiated area due to the narrowness of the PSF, a low detection efficiency of the optical setup, and a low fluorescence yield in two-photon excitation. This can be improved by reducing the measurement rate in an experiment, which, however, reduces the time resolution. We demonstrate that the characteristics of the diffusion can be inferred nevertheless from a single CFM or FCS measurement with a low signal/noise ratio. Overall, we show the feasibility of two independent ways of obtaining the characteristics of diffusion from the 4Pi FM measurements: analysis of the fluorescence signal and fluorescence correlation spectroscopy.
METHODS
In the following, we will describe the experimental setup where fluorophores diffuse in a 3D volume Ω, and the photons emitted by fluorophores are recorded. The whole volume Ω is assumed to contain N fluorophore molecules. For each molecule i (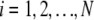 ), its position at time t is denoted by
), its position at time t is denoted by 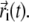 Below, fluorophores will be referred to also as “particles”.
Below, fluorophores will be referred to also as “particles”.
Modeling of the 4Pi point spread function
The illumination PSF of the microscope describes the distribution of light intensity in space and determines the strength of bleaching and fluorescence at each given point. In the 4Pi microscope, the approximately spherical light wavefronts from two opposing lenses are focused onto the sample (31), as sketched in Fig. 2 a. The constructive interference of these wavefronts sculpts the PSF (31,34,35,44,36) that generally extends over a narrower region than that illuminated in a confocal microscope (45) (see Fig. 2, b and c). The PSF specific for an instrument is expressed through the functions 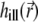 and
and 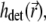 where
where 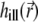 describes the illumination of the sample and
describes the illumination of the sample and 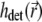 the recording of the fluorescent emission. The complete PSF
the recording of the fluorescent emission. The complete PSF 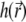 is the product of these two functions, i.e.,
is the product of these two functions, i.e., 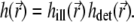 Both factors are wavelength dependent, but in experiments a range of wavelengths is detected; for the purpose of analysis, we assume a single wavelength, which corresponds to the weighted (in the detection channel) emission maximum of the fluorophore.
Both factors are wavelength dependent, but in experiments a range of wavelengths is detected; for the purpose of analysis, we assume a single wavelength, which corresponds to the weighted (in the detection channel) emission maximum of the fluorophore.
Following the seminal work of Hell and Stelzer (31), one can calculate the 4Pi PSF 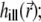 it is the result of the interference of two coherent light beams, one coming from the top and another from the bottom (Fig. 2 a), with their corresponding electric fields
it is the result of the interference of two coherent light beams, one coming from the top and another from the bottom (Fig. 2 a), with their corresponding electric fields 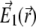 and
and 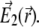 The function is given by the expression
The function is given by the expression
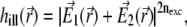 |
(1) |
where nexc is the number of photons required for the excitation of a fluorophore. We assume in the following two-photon excitation, and, thus, choose nexc = 2. The field 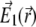 corresponds to the spherical wavefront focused around
corresponds to the spherical wavefront focused around 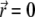 (31,46,47), expressed as follows
(31,46,47), expressed as follows
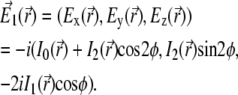 |
(2) |
The field 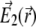 represents the coherent beam going in the opposite direction, expressed through
represents the coherent beam going in the opposite direction, expressed through
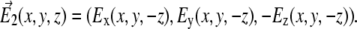 |
(3) |
Here, we used 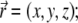 φ is the angle between the plane of oscillation of the electric field in the beam and the plane of observation; the functions I0, I1, and I2 are given by
φ is the angle between the plane of oscillation of the electric field in the beam and the plane of observation; the functions I0, I1, and I2 are given by
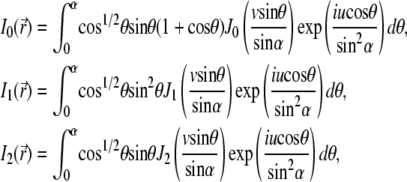 |
(4) |
with u and v being optical coordinates (u = (2πn sin2α)z/λ, 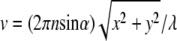 ), λ the wavelength, n the refraction index, α the aperture angle, and J0, J1, J2 Bessel functions of the first kind. The resulting 4Pi illumination PSF (31) is given by
), λ the wavelength, n the refraction index, α the aperture angle, and J0, J1, J2 Bessel functions of the first kind. The resulting 4Pi illumination PSF (31) is given by
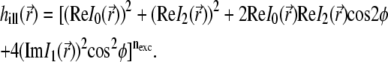 |
(5) |
The detection PSF of the type A 4Pi microscope is given by 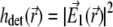 (because a confocal detection system is used), which leads to the expression
(because a confocal detection system is used), which leads to the expression
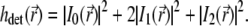 |
(6) |
It should be noted, however, that because of the Stokes shift the wavelength λ is generally different for illumination and for detection.
Profiles of the total 4Pi PSF, 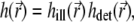 along the x and z axes are shown in Fig. 2, b and c, and isointensity surfaces are shown in Fig. 2, d and e (see also the movie in Supplementary Material). As can be seen from Eqs. 4 and 5, as well as in Fig. 2, the 4Pi PSF is a function with a complex shape in 3D that cannot be adequately reproduced by a step function or a Gaussian, as commonly used for confocal or other simple PSFs in theoretical models for continuous photobleaching. Accurate reproduction of the PSF shape in calculations is critical to take full advantage of the high resolution provided by the 4Pi microscope or other emerging microscopy techniques in photobleaching schemes.
along the x and z axes are shown in Fig. 2, b and c, and isointensity surfaces are shown in Fig. 2, d and e (see also the movie in Supplementary Material). As can be seen from Eqs. 4 and 5, as well as in Fig. 2, the 4Pi PSF is a function with a complex shape in 3D that cannot be adequately reproduced by a step function or a Gaussian, as commonly used for confocal or other simple PSFs in theoretical models for continuous photobleaching. Accurate reproduction of the PSF shape in calculations is critical to take full advantage of the high resolution provided by the 4Pi microscope or other emerging microscopy techniques in photobleaching schemes.
Model for bleaching and diffusion of fluorophores
We assume that individual fluorophores diffuse and are subject to photobleaching, in which case a particle disappears from the volume Ω. For each particle, its trajectory  is a continuous function of t. Using a unified description for all N particles, their positions at given time t are distributed in space according to the probability distribution function
is a continuous function of t. Using a unified description for all N particles, their positions at given time t are distributed in space according to the probability distribution function  In the following, we will assume that the interactions between fluorophores can be neglected and no external force is applied to the fluorophores, except the interactions between the fluorophores and the medium in which they diffuse. In this case, the distribution function,
In the following, we will assume that the interactions between fluorophores can be neglected and no external force is applied to the fluorophores, except the interactions between the fluorophores and the medium in which they diffuse. In this case, the distribution function, 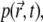 describing the probability to find the fluorophore at position
describing the probability to find the fluorophore at position 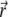 at time t, obeys the diffusion equation with an additional term accounting for the bleaching
at time t, obeys the diffusion equation with an additional term accounting for the bleaching
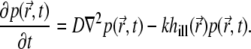 |
(7) |
Here D and k are the diffusion coefficient and bleaching constant, respectively; 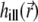 is the illumination PSF introduced in Eq. 5. Interactions between the fluorophores and the medium are accounted for by the diffusion coefficient D. In general, D can be a function of
is the illumination PSF introduced in Eq. 5. Interactions between the fluorophores and the medium are accounted for by the diffusion coefficient D. In general, D can be a function of 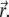 We will assume that D is constant in the whole region Ω, because usually one is interested not in spatial distribution of diffusion characteristics in a given specimen, but rather in an average characteristic of diffusion in the studied type of biological medium.
We will assume that D is constant in the whole region Ω, because usually one is interested not in spatial distribution of diffusion characteristics in a given specimen, but rather in an average characteristic of diffusion in the studied type of biological medium.
The initial distribution of particles 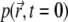 is known (below we set
is known (below we set 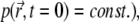 and we assume that reflective boundary conditions are enforced at the boundary ∂Ω of the volume Ω. Also,
and we assume that reflective boundary conditions are enforced at the boundary ∂Ω of the volume Ω. Also, 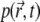 is a one-particle probability distribution function; i.e., it holds
is a one-particle probability distribution function; i.e., it holds
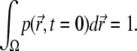 |
(8) |
For t > 0 the value of this integral decreases due to photobleaching. It is important to note that 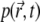 is the same for all fluorophores.
is the same for all fluorophores.
The average fluorescence signal recorded from the sample is usually written (12,25)
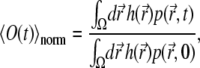 |
(9) |
where
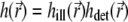 |
(10) |
is the total PSF, and 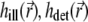 are given by Eqs. 5 and 6. Obviously, it holds
are given by Eqs. 5 and 6. Obviously, it holds 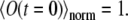 The function
The function 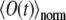 is obtained from the experiments, and also can be computed once Eq. 7 is solved numerically. Varying D and k in Eq. 7, one can fit the calculated
is obtained from the experiments, and also can be computed once Eq. 7 is solved numerically. Varying D and k in Eq. 7, one can fit the calculated 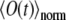 to the observed signal and, thus, find the diffusion coefficient.
to the observed signal and, thus, find the diffusion coefficient.
Numerical procedures
The numerical solution of the diffusion-bleaching equation (Eq. 7) was coded in C++. The solution is based on the method of finite differences (48,49) on a uniform 3D grid (or effectively in a two-dimensional (2D) space when symmetry allows one to reduce the 3D problem to a 2D one, as is true in this case). The volume Ω is chosen as either a rectangular parallelepiped or as a cylinder. The second-order expansion of the Laplacian is used and the time derivative is represented by the first-order finite difference with time step Δt. The diffusion-reaction Eq. 7 is propagated forward in time, assuming reflective boundary conditions at the surface ∂Ω, incorporated into the finite difference scheme with a regular lattice of step Δx. Typical values for Δt and Δx are discussed below.
Computation of the PSFs 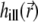 and
and 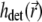 can be demanding due to their nonanalytical form and, therefore, we precompute these functions for a given set of illumination and detection angles once, saving the PSFs and reusing them when necessary. A Mathematica (Wolfram Research, Champaign, IL) script was developed for this purpose. Before passing the illumination PSF
can be demanding due to their nonanalytical form and, therefore, we precompute these functions for a given set of illumination and detection angles once, saving the PSFs and reusing them when necessary. A Mathematica (Wolfram Research, Champaign, IL) script was developed for this purpose. Before passing the illumination PSF 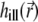 to the solver of the diffusion-bleaching Eq. 7, it is normalized so that the integral of
to the solver of the diffusion-bleaching Eq. 7, it is normalized so that the integral of 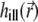 over the volume Ω is equal to one. Similarly, the total PSF
over the volume Ω is equal to one. Similarly, the total PSF 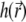 is normalized when used to calculate the fluorescence signal (Eq. 9). The constant k in Eq. 7 incorporates then the contributions from multiple factors, such as the light intensity, bleaching efficiency for a single fluorophore, etc., and we report values of k in arbitrary units (arb. u.). However, with the convention that the normalized PSF is measured in μm−3, the “arbitrary units” for k are, in fact, μm3/s.
is normalized when used to calculate the fluorescence signal (Eq. 9). The constant k in Eq. 7 incorporates then the contributions from multiple factors, such as the light intensity, bleaching efficiency for a single fluorophore, etc., and we report values of k in arbitrary units (arb. u.). However, with the convention that the normalized PSF is measured in μm−3, the “arbitrary units” for k are, in fact, μm3/s.
The discretized form of Eq. 7 reads
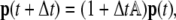 |
(11) |
where Δt is the time step, p is the vector representing the binned distribution 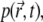 1 the identity matrix, and matrix
1 the identity matrix, and matrix 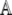 represents the operator
represents the operator 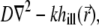 including the boundary conditions. The integral
including the boundary conditions. The integral 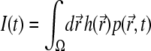 is in discretized form given by the scalar product
is in discretized form given by the scalar product
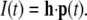 |
(12) |
Equations 11 and 12 are applied iteratively to calculate the observable 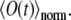
The essential part of the 4Pi microscope PSF is usually spread over a domain <0.5–1 μm wide (see Fig. 2). Accordingly, to resolve this domain we use a spatial discretization of Δx = 20–30 nm. The region Ω described in the calculations is usually of 20–30 μm size in each direction. For each calculation, we check if the increase in considered volume changes the function 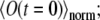 the result is considered to be the same if the average deviation in this function is below 1%. For the experiments performed (uniform diffusion of GFP in glycerin, D ∼ 1 μm2/s), a region of size
the result is considered to be the same if the average deviation in this function is below 1%. For the experiments performed (uniform diffusion of GFP in glycerin, D ∼ 1 μm2/s), a region of size 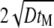 is usually large enough, where tM is the measurement time. Experiments generally monitor diffusion on a timescale of tM ≤ 100 s, i.e., in this case the computational diffusion domain should have a 20-μm width. The maximum time step one can use in the numerical solution is inversely proportional to the diffusion constant D; for the considered experiments (D ∼ 1 μm2/s), the time step we used, accordingly, was Δt = 10−4 s.
is usually large enough, where tM is the measurement time. Experiments generally monitor diffusion on a timescale of tM ≤ 100 s, i.e., in this case the computational diffusion domain should have a 20-μm width. The maximum time step one can use in the numerical solution is inversely proportional to the diffusion constant D; for the considered experiments (D ∼ 1 μm2/s), the time step we used, accordingly, was Δt = 10−4 s.
A typical calculation covering 10 s of measurement time required 8 min of computing time on a single core of an Intel Core 2 Quad CPU running at 2.66 GHz, using the Intel C/C++ compilers version 9.0. The computing time can be reduced employing parallel processors that require, however, professional support for their installation and operation. Fortunately, another less demanding and much cheaper option is available through the most recent generation of graphical processing units (GPUs) to be found soon in commodity computers. These GPUs offer about a hundred cores on which the needed computations can be carried out as fast as on a computer cluster with as many processors offering nearly teraflops of computer power; a key new feature of the new GPUs is the availability of general purpose compilers. Indeed, such GPUs were already successfully employed for scientific applications (50–52) with speed-ups of a factor 10–100 on a GPU in comparison with a CPU for various scientific computing tasks (52).
We adapted our code to work on GPUs as well as on a CPU. This was possible in a rather straightforward manner since, according to Eqs. 11 and 12, the numerical solution of the diffusion-bleaching equation involves an iterative matrix product, an operation commonly arising in the processing of graphics, and, thus, particularly suitable for parallelization on GPUs. This version of the GPU implementation of our code, on the NVIDIA GeForce 8800GTX GPU (Santa Clara, CA), performed 10 times faster than the CPU-based version, carrying out the calculation mentioned above within 45 s rather than 8 min on a CPU. The developed code is available from the authors upon request.
Experimental procedures
The GFP (S65T) was expressed in Escherichia coli and purified as described previously (53). The CFM measurements were performed in a Leica TCS 4Pi microscope (Leica Microsystems) of type A equipped with a pair of ×100/1.35 N.A. glycerol objectives at 23 ± 0.5°C. Samples were prepared by diluting GFP at concentrations from 5 to 20 μM in 87% glycerol buffered with phosphate buffered saline (PBS) and placed between two quartz coverslips that were sealed with silicone. The embedding glycerol/PBS mixture was also used as the immersion medium to ensure continuity in the refractive index between the two objectives. The lower coverslip was equipped with a mirror at the periphery, which allowed the adjustment of the objectives' correction collars for exact refractive index alignment.
Fluorescent beads of subresolution size with red-shifted emission wavelength compared to GFP (TransFluorSpheres, 0.1 μm; Molecular Probes, Eugene, OR) immobilized at the upper coverslip were used to align the focus of the two objectives and the interference phase of the microscope. The adjustment procedure was carried out for each individual sample. The beads and the GFP fluorescence were excited in two-photon mode by a Ti:Sapphire laser (MaiTai, Spectra Physics, Mountain View, CA) or in one-photon mode by the 488-nm line of an argon laser with beam expander 3 of the microscope; signal detection was performed after passing a filter cube (SP700, BS560, BP500–550, or BP607–683) by photon counting avalanche photodiodes (Perkin Elmer, Foster City, CA). The detection pinhole was set to 0.74 Airy units.
To record the fluorescence with a nonscanning parked beam the microscope was further equipped with an instrumental upgrade developed for classical FCS applications (VistaFCS, ISS, Champaign, IL). In CFM experiments the GFP was excited at 905 or 860 nm and the fluorescence was recorded for 40–60 s with a sampling rate of 1–5 kHz. In FCS experiments the wavelength was set to 860 nm and the signal was autocorrelated online for 200 s at a sampling rate of 5 kHz.
RESULTS
As shown in Fig. 2, the illuminated volume of the type A 4Pi microscope is much smaller than that of a conventional microscope. Using such a small volume leads to a possibly small number of fluorophores under the PSF, with the fluorophores being constantly bleached in CFM experiments. Indeed, for a GFP concentration of 10 μM that we normally used in the experiments with uniform GFP solution, the illuminated volume contains only ∼200 fluorophores. In this situation the continuum description of the fluorophore diffusion, bleaching, and fluorescence may not be adequate, and one may want to consider a stochastic description tracing individual particles. Also, the signal/noise ratio is rather poor for data obtained under such conditions. Therefore, we use below descriptions of individual particles in CFM experiments to derive the relation to a continuum representation. Our results suggest that although noise due to individual particles is indeed an important issue, the data from the 4Pi CFM measurements can be safely accounted for through a continuum description, and valid diffusion constants extracted.
Fluorescence signal calculated and recorded
The total PSF 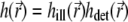 in Eqs. 9 and 10 is composed of the illumination PSF
in Eqs. 9 and 10 is composed of the illumination PSF 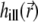 and the detection PSF
and the detection PSF 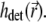 The number of photons dNphot emitted by the photoexcited particles during an infinitesimal time interval dt is
The number of photons dNphot emitted by the photoexcited particles during an infinitesimal time interval dt is
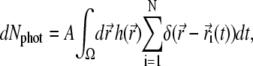 |
(13) |
where A is an unknown constant. By Nphot(t0) we denote the number of photons emitted by the whole system from time t = 0 to t = t0. The experimental observable 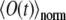 (c.f. Eq. 9) is proportional to the number of photons emitted by the system during the device operational time window Tw. Assuming that Tw is small enough so that the rate of photon emission does not change during Tw, one obtains the expression for the number of photons emitted from time t to time t + Tw, i.e., for TwdNphot(t)/dt. Therefore, the signal recorded in the experiment is directly proportional to the function O(t), defined by
(c.f. Eq. 9) is proportional to the number of photons emitted by the system during the device operational time window Tw. Assuming that Tw is small enough so that the rate of photon emission does not change during Tw, one obtains the expression for the number of photons emitted from time t to time t + Tw, i.e., for TwdNphot(t)/dt. Therefore, the signal recorded in the experiment is directly proportional to the function O(t), defined by
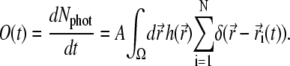 |
(14) |
The ensemble average of a signal, 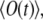 is an average over separate experiments, with all conditions being the same, denoted by
is an average over separate experiments, with all conditions being the same, denoted by 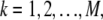 with
with 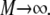 This signal is given by
This signal is given by
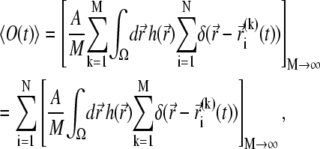 |
(15) |
where 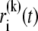 denotes the position of fluorophore i at time t during the trial k. Assuming that all fluorophores i are the same and that they do not interact, i.e., are described by the same distribution function
denotes the position of fluorophore i at time t during the trial k. Assuming that all fluorophores i are the same and that they do not interact, i.e., are described by the same distribution function 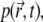 one can write
one can write
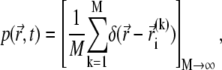 |
(16) |
and, using Eq. 15, one obtains
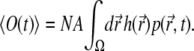 |
(17) |
Normalizing 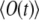 to unity at t = 0 results in an expression for
to unity at t = 0 results in an expression for 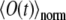 given by Eq. 9; this normalized function has been used by us to compare experimental and calculated fluorescence signals.
given by Eq. 9; this normalized function has been used by us to compare experimental and calculated fluorescence signals.
Free diffusion of GFP in an isotropic 3D solution has been assessed using CFM (10 μM of GFP in 87% glycerol solution in water). The experimental curves from single measurement runs are well reproduced by the computed 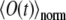 (Fig. 3 a) and an average over many independent measurements is matched well, too (Fig. 3 b). The fittings both of single measurements and of an average over multiple measurements provide values of the diffusion coefficient D for GFP in a range of 1.0–1.5 μm2/s (previous measurements using different techniques suggested a value in the range 0.5–0.9 μm2/s for 90% glycerol and 2.0–3.0 μm2/s for 80% glycerol (54,55)). The data from single measurements (Fig. 3 a) are noisy due to the small number of fluorophores within the span of the PSF, as estimated above. Combining data from multiple measurements (Fig. 3 b) helps one to reduce the noise and makes it more adequate to use expression (9) for the fluorescence signal, derived under the assumption of an infinite number of trials.
(Fig. 3 a) and an average over many independent measurements is matched well, too (Fig. 3 b). The fittings both of single measurements and of an average over multiple measurements provide values of the diffusion coefficient D for GFP in a range of 1.0–1.5 μm2/s (previous measurements using different techniques suggested a value in the range 0.5–0.9 μm2/s for 90% glycerol and 2.0–3.0 μm2/s for 80% glycerol (54,55)). The data from single measurements (Fig. 3 a) are noisy due to the small number of fluorophores within the span of the PSF, as estimated above. Combining data from multiple measurements (Fig. 3 b) helps one to reduce the noise and makes it more adequate to use expression (9) for the fluorescence signal, derived under the assumption of an infinite number of trials.
FIGURE 3.

Calculated and observed fluorescence signal as a function of time. (a) A single 4Pi CFM measurement (black) is compared with the calculated (fitted) signal (red). Best fitting is achieved with D = 1.5 μm2/s and k = 7.5 arb. u. (b) Results of 10 independent measurements are averaged (black) and fitted to a calculated curve (red, D = 1.0 μm2/s, k = 12.0 arb. u.). Curves in panels a and b are from different series of measurements, performed at different laser intensities. The calculated signal with constant k = 7.5 arb. u. and varied D are presented in panel c, from top to bottom, D = 3.0, 1.5, 1.0, 0.5, and 0.1 μm2/s. Curves for D = 0.5 μm2/s, k = 3.75 arb. u. (red) and D = 2.0 μm2/s, k = 15.0 arb. u. (green) converge to the same level as the one with D = 1.0 μm2/s, k = 7.5 arb. u. Data from calculations with constant D = 1.5 μm2/s and varied k are shown in panel d, from top to bottom, k = 3.5, 7.5, 10.0, 15.0, and 50.0 arb. u. The experimental curves were obtained with 10 μM of GFP in 87% glycerol solution in water; n = 1.46, α = 68.5°; λ = 905 nm for illumination and λ = 500–550 nm for detection.
Fig. 3, c and d, illustrates the sensitivity of the fluorescence signal in CFM measurements to variation in the diffusion constant D or bleaching constant k. Although variation between, for example, D = 1.0 μm2/s and D = 1.5 μm2/s leads to a significant difference in 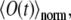 the noise in the experimental data makes it difficult to pinpoint a precise value for D. The fitting to experimental data minimizes the least square deviation between the calculated and measured curves. This procedure provides a single value of D, however, one should keep in mind that ∼±0.2 μm2/s variation in D would still reproduce the same measurement quite well, due to the noise level. Averaging over multiple measurements reduces the noise, fitting providing again a single value for D, but making it difficult in this case to estimate the error in D.
the noise in the experimental data makes it difficult to pinpoint a precise value for D. The fitting to experimental data minimizes the least square deviation between the calculated and measured curves. This procedure provides a single value of D, however, one should keep in mind that ∼±0.2 μm2/s variation in D would still reproduce the same measurement quite well, due to the noise level. Averaging over multiple measurements reduces the noise, fitting providing again a single value for D, but making it difficult in this case to estimate the error in D.
A comparison of the CFM curves obtained using 4Pi and confocal PSFs is presented in Fig. 4. To our knowledge, 4Pi microscopy has not been used for FM before, whereas confocal microscopy is commonly employed in various bioimaging applications, including FM, and has been previously demonstrated to be a reliable tool for CFM in living cells, when one-photon excitation is used (see, e.g., Wachsmuth et al. (13)). As mentioned above, the 4Pi PSF is narrower in the z-direction than the confocal PSF (see Fig. 2), which might be beneficial for many applications. As Fig. 4 demonstrates, the CFM curves for both PSF types show similar trends, suggesting that the same analysis tools can be used. However, a significant difference is that the confocal PSF can be reliably approximated by a Gaussian, whereas the 4Pi PSF has to be calculated numerically.
FIGURE 4.

Fluorescence signal recorded with one-photon and two-photon excitation using 4Pi and confocal microscopy. Measurements with a 4Pi PSF are shown in red, and with the confocal one in green; calculated curves are in black, marked with circles for the 4Pi and with squares for the confocal PSF. In panel a, results for two-photon illumination are the curves at the top, and for one-photon those at the bottom. The illumination laser power was scaled to give approximately the same count rate for measurements with 4Pi and confocal PSFs. (b) Only two-photon excitation is used, with the same laser power for 4Pi and confocal measurements, resulting in significant difference between the recorded signal decays. The experimental curves are averages over 10 measurements each. All measurements were done with the same parameters as in Fig. 3, but with λ = 860 nm for illumination in the two-photon case, λ = 488 nm for illumination in the one-photon case, and λ = 600–650 nm for detection. Calculations were done with D = 0.5 μm2/s, and with k = 1.7 arb. u. for the two-photon case in panel a, k = 9.0 arb. u. for the one-photon case, k = 2.5 arb. u. for the 4Pi PSF and k = 1.2 arb. u. for the confocal PSF in panel b.
The CFM measurements with one-photon and two-photon excitation for confocal and 4Pi illumination are shown in Fig. 4 a. For the curves shown in this panel, the laser power was scaled when changing from confocal to 4Pi PSF, so that the count rate was approximately the same. In our analysis, the illumination PSFs 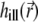 are normalized (see “Numerical Procedures”); therefore, employing the same bleaching constant k for different PSFs (see Eq. 7) corresponds to the case when the laser power is scaled for each PSF to result in the same count rate. Thus, the values of k used to fit the measurements in Fig. 4 a are the same for the two different PSFs. For the one-photon confocal PSF (13), as well as for the one-photon 4Pi and two-photon confocal and 4Pi PSFs, our calculations match experimental data well. The average signal recorded in these measurements is approximately the same for the 4Pi and confocal PSFs, despite their difference in shape, suggesting that the amount of power pumped into the sample is a primary factor determining the behavior of the CFM curves.
are normalized (see “Numerical Procedures”); therefore, employing the same bleaching constant k for different PSFs (see Eq. 7) corresponds to the case when the laser power is scaled for each PSF to result in the same count rate. Thus, the values of k used to fit the measurements in Fig. 4 a are the same for the two different PSFs. For the one-photon confocal PSF (13), as well as for the one-photon 4Pi and two-photon confocal and 4Pi PSFs, our calculations match experimental data well. The average signal recorded in these measurements is approximately the same for the 4Pi and confocal PSFs, despite their difference in shape, suggesting that the amount of power pumped into the sample is a primary factor determining the behavior of the CFM curves.
It should be noted that the calculated curves in Fig. 4 a were fitted to the measured ones only for the case of the 4Pi PSF. Once a good fit was obtained, the calculation was repeated with the confocal PSF, but with all other parameters kept unchanged; still, a good match with observation was obtained. An example of what happens when the laser power is not changed, once the PSF is switched, is shown in Fig. 4 b. Using the 4Pi PSF results in stronger bleaching, because the light is more concentrated as a smaller volume is illuminated. Numerically, this corresponds to using different values for k for the two PSF types, since the PSFs are normalized. The main conclusion from comparing the CFM curves obtained with the 4Pi or confocal PSFs is that it does not matter which PSF type is used in a CFM experiment designed to determine D; in either case the numerical algorithm allows for a reliable estimation of D. The 4Pi PSF should be chosen over the confocal one when the probed volume has to be smaller than the spread of the confocal PSF.
A characteristic feature of both the calculated and experimental curves in Figs. 3 and 4 is that the curves level out for 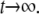 The signal's value for
The signal's value for 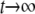 depends on D (Fig. 3 c), but also can be the same for different D-values, depending on k. Can this long-time behavior of the signal be utilized to simplify the extraction of D from experimental data? The leveling of the signal to a constant value is due to the fact that the system assumes a quasistationary state, when the rate of bleaching equals the rate of efflux of fresh fluorophores from unbleached regions. This corresponds to
depends on D (Fig. 3 c), but also can be the same for different D-values, depending on k. Can this long-time behavior of the signal be utilized to simplify the extraction of D from experimental data? The leveling of the signal to a constant value is due to the fact that the system assumes a quasistationary state, when the rate of bleaching equals the rate of efflux of fresh fluorophores from unbleached regions. This corresponds to 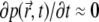 in Eq. 7, and to the convergence of the fluorophore distribution
in Eq. 7, and to the convergence of the fluorophore distribution 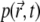 to the quasistationary solution
to the quasistationary solution 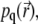 satisfying
satisfying
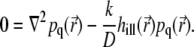 |
(18) |
Thus, the quasistationary solution 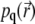 is determined by a single parameter, k/D; since the signal is defined by
is determined by a single parameter, k/D; since the signal is defined by 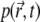 through Eq. 17, the quasistationary value of the signal at long times is also determined by k/D. As shown in Fig. 3 c, scaling D and k simultaneously by the same value (which keeps k/D constant) does not change the long-time level of the signal. Moreover, the whole signal curve shrinks or expands as if the only change was a scaling of time. The reason for this behavior is that the diffusion-bleaching equation (Eq. 7) is a first-order differential equation in time, and D and k enter the equation linearly, so that if D and k are scaled by some constant a, i.e., Dnew = aD, knew = ak, one can replace the resulting diffusion-bleaching equation by one with t replaced by tnew = at.
through Eq. 17, the quasistationary value of the signal at long times is also determined by k/D. As shown in Fig. 3 c, scaling D and k simultaneously by the same value (which keeps k/D constant) does not change the long-time level of the signal. Moreover, the whole signal curve shrinks or expands as if the only change was a scaling of time. The reason for this behavior is that the diffusion-bleaching equation (Eq. 7) is a first-order differential equation in time, and D and k enter the equation linearly, so that if D and k are scaled by some constant a, i.e., Dnew = aD, knew = ak, one can replace the resulting diffusion-bleaching equation by one with t replaced by tnew = at.
Fig. 5 compares the PSF, the fluorophore concentration, and the fluorescence signal along the z axis during the CFM experiment in Fig. 3 a. Fig. 5 b shows how the profile of the fluorophore concentration 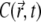 along the z axis changes with time (
along the z axis changes with time (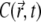 is defined through
is defined through 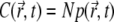 ). One recognizes that
). One recognizes that 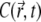 is decreasing, following to some extent the shape of the PSF, but at t = 0.2 s it reaches a quasistationary state, after which moment the shape of
is decreasing, following to some extent the shape of the PSF, but at t = 0.2 s it reaches a quasistationary state, after which moment the shape of 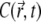 remains largely unchanged, and the overall concentration depletes slowly and uniformly. According to Eq. 17, one can define the local emission probability from the volume
remains largely unchanged, and the overall concentration depletes slowly and uniformly. According to Eq. 17, one can define the local emission probability from the volume 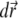 at around position
at around position 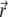 as
as 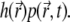 This quantity is plotted in Fig. 5 c; it also reaches a quasistationary state at the same pace as
This quantity is plotted in Fig. 5 c; it also reaches a quasistationary state at the same pace as 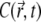 does. As a result, time evolution of the fluorescence signal follows the time evolution of
does. As a result, time evolution of the fluorescence signal follows the time evolution of 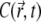 closely.
closely.
FIGURE 5.

Time evolution of fluorophore distribution and local fluorescence along the z axis. (a) The PSF profile is shown. (b) The local fluorophore concentration (normalized to unity at t = 0) is shown for successive time moments, and the local emission probability 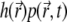 (normalized) is shown in panel c. The times for which curves are drawn in panels b and c are, from top to bottom, 0.0001, 0.0002, 0.0005, 0.001, 0.002, 0.005, 0.01, 0.02, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, and 1.0 s. All parameters are the same as in the calculation shown in Fig. 3 a.
(normalized) is shown in panel c. The times for which curves are drawn in panels b and c are, from top to bottom, 0.0001, 0.0002, 0.0005, 0.001, 0.002, 0.005, 0.01, 0.02, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, and 1.0 s. All parameters are the same as in the calculation shown in Fig. 3 a.
Calculating the long-time signal level and matching it to the experimental value can determine the value of k/D, but the value of D remains unknown. Indeed, it is impossible to obtain D from 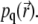 D would be known if one knew the values of k corresponding to the laser power used in an experiment, but to determine k one needs to perform separate experiments. In principle, k can be determined from the signal at t ≈ 0. Since
D would be known if one knew the values of k corresponding to the laser power used in an experiment, but to determine k one needs to perform separate experiments. In principle, k can be determined from the signal at t ≈ 0. Since 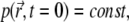 Eq. 7 reduces to
Eq. 7 reduces to
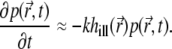 |
(19) |
This suggests an initial exponential decay. One can estimate the value of k by comparing the decay of the signal suggested by Eq. 19 with that measured in experiments. To do so, we used a least-squares fit of the numerically calculated signal (with 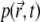 described by Eq. 19) to the measured signal over the first 0.01 s of the recording. Unfortunately, due to the high noise level in the experimental signal, the values of k obtained from fitting the signal around t = 0 are highly unreliable.
described by Eq. 19) to the measured signal over the first 0.01 s of the recording. Unfortunately, due to the high noise level in the experimental signal, the values of k obtained from fitting the signal around t = 0 are highly unreliable.
Even though the quasistationary levels of the signal can be extracted with good fidelity, providing a good estimate for k/D, using Eq. 18 to match k/D involves considerable computational effort that might be best invested into computing the numerical solution of the time-dependent Eq. 7 up to the point when 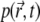 assumes the quasistationary state characterized by
assumes the quasistationary state characterized by 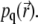 Such computation requires about the same effort as solving Eq. 18, but furnishes the initial decay of
Such computation requires about the same effort as solving Eq. 18, but furnishes the initial decay of 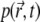 and, hence, of
and, hence, of 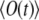 that can be matched then to the observation to yield both k and D. Therefore, the most practical route for determining D is to compute the signal as a function of time for an arbitrary starting value of D0, to vary k, calculating the signal curve for each new value of k, and to match experimental and calculated
that can be matched then to the observation to yield both k and D. Therefore, the most practical route for determining D is to compute the signal as a function of time for an arbitrary starting value of D0, to vary k, calculating the signal curve for each new value of k, and to match experimental and calculated 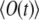 in the quasistationary regime. We judged the quality of the match by the mean square deviation between the calculation and measurement, using the t > 0.5 s part of the signal for this purpose. Then one scales the time to match the calculated signal to the experimental signal, using now the least-squares matching for all values of time t. The scaling factor a is the same for D as well as k, and one arrives at the best D- and k-values, D = aD0, k = ak0. These values can be checked by a direct calculation of
in the quasistationary regime. We judged the quality of the match by the mean square deviation between the calculation and measurement, using the t > 0.5 s part of the signal for this purpose. Then one scales the time to match the calculated signal to the experimental signal, using now the least-squares matching for all values of time t. The scaling factor a is the same for D as well as k, and one arrives at the best D- and k-values, D = aD0, k = ak0. These values can be checked by a direct calculation of 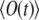 and comparison with the measurement.
and comparison with the measurement.
The signal for CFM consists of an initial (t ≈ 0) regime dominated by k, an intermediate regime determined by both parameters, D and k, and a long-time regime dependent on k/D. The most interesting regime for determining D is the intermediate one, which bears the signature of both bleaching and diffusion. The CFM technique resolves the intermediate regime and, hence, permits measurement of D.
Correlation function
Fluorescent correlation spectroscopy (56–59) can be employed in the 4Pi microscope by working in the nonbleaching limit. Without bleaching (k = 0 in Eq. 7), the expression for the correlation function (Eq. 26) is simplified, as the operator 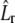 describes now free diffusion. The conditional probability (Green's function) for free diffusion is a Gaussian, namely,
describes now free diffusion. The conditional probability (Green's function) for free diffusion is a Gaussian, namely,
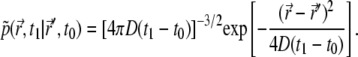 |
(20) |
Using this expression one can rewrite Eq. 26, normalizing the correlation function by 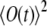 (58),
(58),
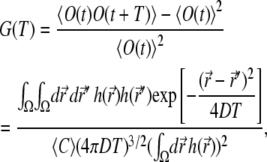 |
(21) |
where 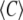 is the average concentration of the fluorophores, and we chose t0 = t, t1 = t + T.
is the average concentration of the fluorophores, and we chose t0 = t, t1 = t + T.
We measured the correlation function G(T) in the nonbleaching limit, and, because of the simplification brought about by setting k to zero, we were able to also calculate G(T) numerically. The experimental and calculated correlation functions are compared in Fig. 6. Two features of the correlation function require attention when one fits the calculated curve to the experimental one, the value at T = 0 and the decay time. One can express 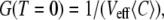 where Veff is the effective focal volume (58) defined through
where Veff is the effective focal volume (58) defined through 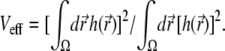 Thus, G(T = 0) is an inverse number of particles in the effective focal volume. According to Fig. 6 (for both the experiment and calculation), the number of particles in the effective focal volume is ∼100 (which would become 200 for the 10-μM concentration used in the measurements of the CFM signal above). Fig. 6 demonstrates a good agreement between the calculated curves and the experimental one near T = 0. The decay of the correlation function is also reproduced with acceptable diffusion constants in the range 0.5–2.0 μm2/s. This range is wider than the one that matched the CFM signal (1.0–1.5 μm2/s; see Fig. 3), but includes the latter range.
Thus, G(T = 0) is an inverse number of particles in the effective focal volume. According to Fig. 6 (for both the experiment and calculation), the number of particles in the effective focal volume is ∼100 (which would become 200 for the 10-μM concentration used in the measurements of the CFM signal above). Fig. 6 demonstrates a good agreement between the calculated curves and the experimental one near T = 0. The decay of the correlation function is also reproduced with acceptable diffusion constants in the range 0.5–2.0 μm2/s. This range is wider than the one that matched the CFM signal (1.0–1.5 μm2/s; see Fig. 3), but includes the latter range.
FIGURE 6.

Correlation function for the case k = 0 (no bleaching). Experimental data (circles with error bars) are the average of correlation functions from five separate measurements. Error bars represent the averaging root mean-square deviation. Setup parameters for these measurements were the same as for those presented in Fig. 3, besides the GFP concentration, which was 5 μM for these measurements, and the excitation wavelength, which was switched to 860 nm. Calculated correlation functions are shown as continuous lines (D = 0.5 μm2/s, 1.0 μm2/s, and 2.0 μm2/s, from top to bottom).
The FCS measurements can be compared also for the 4Pi and confocal PSFs, as shown in Fig. 7. Numerically calculated correlation functions reproduce the experimental ones well. As was the case for the CFM signal (Fig. 4), parameters for the numerical solution were varied only to match the curve corresponding to the 4Pi case; once a good match was obtained, switching the numerical solver to employ the confocal PSF instead of the 4Pi one resulted in a good match for the confocal case, too. The laser power was scaled in these experiments to have the same count rate for the 4Pi and confocal PSFs (see also Fig. 4).
FIGURE 7.

Correlation function for the 4Pi (•) and confocal (▪) PSF (two-photon in both cases), measured on the same sample. Numerical fits for both cases are shown as solid black lines, corresponding to D = 0.5 μm2/s (k = 0). Experimental data are the average of correlation functions from 10 separate measurements. The GFP concentration is 15 μM; all other parameters are the same as in Fig. 6.
The effective focal volume differs substantially for 4Pi and confocal PSFs, being only half the size for the 4Pi PSF (see also Fig. 2), as demonstrated in Fig. 7 by the values of G(T = 0). Indeed, according to these values, the number of particles in the effective focal volume for the confocal PSF is ≈550, and for the 4Pi PSF is ≈300, with the same fluorophore concentration of 15 μM (in case of Fig. 6, the number of particles in the effective focal volume for the 4Pi PSF was ∼100 at a 5-μM concentration). When the two curves in Fig. 7 are scaled to start at the same value at T = 0, it appears that the 4Pi correlation function decays faster. This difference is prominent for intermediate times 0.001–0.1 s. The faster decay in the 4Pi case is due to the smaller effective focal volume. The smaller the volume, the shorter is the time spent by a fluorophore in that volume, and, thus, the correlations in the signal are limited to shorter average times that the fluorophores spend in the focal spot.
Although the 4Pi correlation function decays faster on a timescale of 0.001–0.1 s, for longer, and especially for shorter times, the confocal and 4Pi correlation functions are quite close (when scaled to coincide at T = 0). Due to the fact that the diffusion studied here is rather slow, and, accordingly, the acquisition rate is slow too, fast photophysical phenomena such as transition into a nonradiating triplet state (which can happen even though the laser power is very low) are not resolved, and the correlation functions do not exhibit a difference in their short-time behavior. In principle, such a difference could be used to distinguish between photophysics and diffusion because the ratio of the correlation functions for the patterned 4Pi PSF and the confocal PSF should be independent of the spectroscopic properties of the particles (34). In the case studied here, the short-time behavior of the two correlation functions is similar; however, the numerical calculations are able to distinguish between the two cases, in regard to both decay rate and absolute values of the correlation function.
CONCLUSIONS AND PERSPECTIVES
The biological cell features a complex and sophisticated nanostructure that is dynamic and subject to frequent and rapid reorganization. An overwhelming body of data supports the contention that it is this dynamics on the nanometer scale that plays a crucial role in cell function. For these reasons one needs techniques that capture dynamic processes in living cells at the highest possible spatial resolution.
Both FM and FCS have much contributed to the unraveling of dynamic processes in single living cells. However, spatial resolution remains a central issue. One exemplary case is the lateral mobility of proteins in the plasma membrane. Already some time ago (60) it has been observed that the lateral mobility of membrane proteins as measured by FM depends on the size of the illuminated area, pointing to an intricate organization of the plasma membrane on the nanometer level. It has been suggested (61) that the plasma membrane features nanoscopic lipid aggregates (“rafts”) that serve as a basis for organizing membrane proteins in clusters and, thus, have far reaching functional implications, particularly in signal transduction. However, so far the spatial resolution of FM and FCS has been insufficient to unambiguously verify the existence of lipid rafts and their existence remains debated (62). Similar arguments for the need of improved resolution of FM and FCS methods hold for most other cellular processes.
In this study the spatial resolution of CFM and FCS were increased by combining the techniques with 4Pi microscopy. The 4Pi microscope features a point spread function (Fig. 2) with a central peak of ∼≥100 nm width in the axial direction and ≥220 nm width in the focal plane. In addition, the point spread function has smaller secondary maxima spaced on the optical axis at a distance of about half a wavelength from the main maximum. The essential point here is (c.f. Fig. 5 c) that >90% of the signal measured in both CFM and FCS experiments derives from the main peak. Thus, the observation volume is virtually coincident with that of the main maximum of the point spread function.
This does not imply that 4Pi CFM and FCS measurements have an axial resolution of ≥100 nm. In fact, the environment of the observation volume affects diffusion in the observation volume and is an integral part of the system. In CFM measurements the depletion of fluorophores reaches further and further into the environment with time (Fig. 5 b). Nevertheless, the spatial resolution in CFM and FCS measurements scales with the size of the observation volume and, therefore, can be said to be improved by 4Pi microscopy by a factor of 5–7 as compared to normal confocal microscopy.
A major problem in the adaptation of CFM and FCS to 4Pi microscopy was to account for the complicated point spread function of the 4Pi microscope. For that purpose a theoretical and computational framework was developed for data analysis. The numerical approach developed and described here is characterized by flexibility; i.e., the computation scheme can be adopted to point spread functions of arbitrary shape. This is particularly relevant with respect to the fast development of high-resolution light microscopy culminating recently in a resolution of ≈30 nm (63). The numerical approach suggested here permits one also to take other conditions into account that are relevant in cell biological studies. For example, in addition to the pure and unrestricted diffusion of one molecular species one can take the association of the diffusing species with immobile binding sites into account. Furthermore, one can consider situations in which the diffusion space is limited. The approach can also be extended to two-color microscopy, a powerful means to quantify reversible bimolecular association reactions.
Altogether this study provides the tools for installing FM and FCS on light microscopes that are able to extend or even completely overcome classical resolution limits. Accordingly, the theoretical and computational framework will contribute to the further unraveling of the cellular nanomachinery that lies at the heart of life and thus holds the key for future progress in biomedicine.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
The authors are grateful to Nathan A. Baker, Petros Koumoutsakos, Ivo F. Sbalzarini, and Ingo Lepper for useful suggestions and discussions. We also thank John Stone and Kirby Vandivort for help with numerical algorithms, and Peter Freddolino for suggestions on visualizing the PSF.
This work was supported by a grant from the National Institutes of Health (PHS-5-P41-RR05969). K.S. was supported by the Humboldt Foundation. The work was further supported by the National Institutes of Health (grant GMO 71329), the Deutsche Forschungsgemeinschaft (grants PE138/19 and PE138/21), and the Volkswagenstiftung (grant I/79 248).
APPENDIX A: CORRELATION FUNCTION IN A GENERAL CASE
We seek to derive the expression for the correlation function 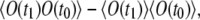 in the general case of t1 ≠ t0 and nonnegligible bleaching. The average signal
in the general case of t1 ≠ t0 and nonnegligible bleaching. The average signal 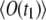 or
or 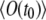 is given by Eq. 17. The correlation term,
is given by Eq. 17. The correlation term, 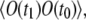 is expressed as follows
is expressed as follows
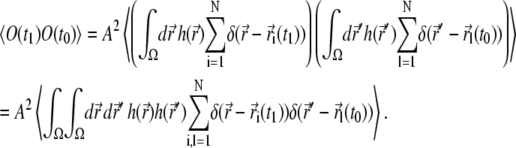 |
(22) |
This can be rewritten as
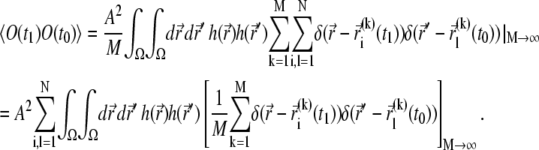 |
(23) |
The product of delta-functions arising here corresponds to a probability that at time t1 particle i is found at 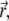 and at time t0 particle l is found at
and at time t0 particle l is found at 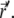 In the case of i ≠ l, these two events are independent (particles do not interact), and the sum over k with
In the case of i ≠ l, these two events are independent (particles do not interact), and the sum over k with 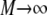 becomes the product of two probability distributions,
becomes the product of two probability distributions, 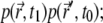 the same probability distribution p is used because the particles behave identically. In the case of i = j, the probability of the two events is conditional, so that in the limit
the same probability distribution p is used because the particles behave identically. In the case of i = j, the probability of the two events is conditional, so that in the limit 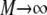 the sum becomes
the sum becomes 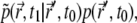 where
where 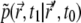 is the probability for a single particle to move from
is the probability for a single particle to move from 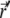 at time t0 to
at time t0 to 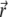 at time t1 (Green's function). Inserting the relation
at time t1 (Green's function). Inserting the relation 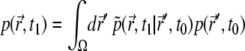 into Eq. 7, and using the initial condition
into Eq. 7, and using the initial condition 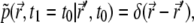 one finds that
one finds that 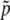 can be expressed
can be expressed
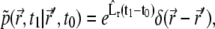 |
(24) |
where 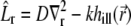 is the diffusion-bleaching operator in Eq. 7, in this case acting on the vector
is the diffusion-bleaching operator in Eq. 7, in this case acting on the vector 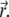 Using these results in Eq. 23, one obtains
Using these results in Eq. 23, one obtains
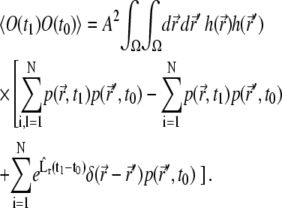 |
(25) |
The elements under the sums over i and l depend neither on i nor on l, so the sums reduce to the multiplication of corresponding terms by N or N2. Combining this with the expression for 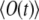 (Eq. 17), one obtains the final result for the correlation function
(Eq. 17), one obtains the final result for the correlation function
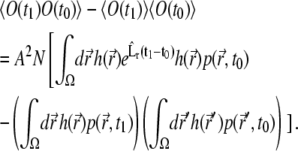 |
(26) |
The correlation function in Eq. 26 could be used for the analysis of diffusion, but we found that in its general form, t1 ≠ t0, the utilization of the correlation function is not practical, due to the difficulties with its computational implementation and high noise level in the measurements.
Editor: Petra Schwille.
References
- 1.Haw, M. 2006. Middle World: The Restless Heart of Matter and Life. McMillan Publishers, Houndsmills, UK.
- 2.Peters, R., J. Peters, K. H. Tews, and W. Bähr. 1974. A microfluorimetric study of translational diffusion in erythrocyte membranes. Biochim. Biophys. Acta. 367:282–294. [DOI] [PubMed] [Google Scholar]
- 3.Magde, D., E. L. Elson, and W. W. Webb. 1974. Fluorescence correlation spectroscopy. 2. Experimental realization. Biopolymers. 13:29–61. [DOI] [PubMed] [Google Scholar]
- 4.Edidin, M., Y. Zagyansky, and T. J. Lardner. 1976. Measurement of membrane protein lateral diffusion in single cells. Science. 191:466–468. [DOI] [PubMed] [Google Scholar]
- 5.Jacobson, K., Z. Derzko, E. S. Wu, Y. Hou, and G. Poste. 1976. Measurement of the lateral mobility of cell surface components in single, living cells by fluorescence recovery after photobleaching. J. Supramol. Struct. 5:565–576. [DOI] [PubMed] [Google Scholar]
- 6.Axelrod, D., D. E. Koppel, J. Schlessinger, E. Elson, and W. W. Webb. 1976. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 16:1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters, R., and U. Kubitscheck. 1999. Scanning microphotolysis: three-dimensional diffusion measurement and optical single-transporter recording. Methods. 18:508–517. [DOI] [PubMed] [Google Scholar]
- 8.Reits, E. A. J., and J. J. Neefjes. 2001. From fixed to FRAP: measuring protein mobility and activity in living cells. Nat. Cell Biol. 3:E145–E147. [DOI] [PubMed] [Google Scholar]
- 9.Lippincott-Schwartz, J., E. Snapp, and A. Kenworthy. 2001. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2:444–456. [DOI] [PubMed] [Google Scholar]
- 10.Chen, Y., B. C. Lagerholm, B. Yang, and K. Jacobson. 2006. Methods to measure the lateral diffusion of membrane lipids and proteins. Methods. 39:147–153. [DOI] [PubMed] [Google Scholar]
- 11.Day, R. N., and F. Schaufele. 2005. Imaging molecular interactions in living cells. Mol. Endocrinol. 19:1675–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters, R., A. Brünger, and K. Schulten. 1981. Continuous fluorescence microphotolysis: a sensitive method for study of diffusion processes in single cells. Proc. Natl. Acad. Sci. USA. 78:962–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wachsmuth, M., T. Weidemann, G. Müller, U. W. Hoffmann-Rohrer, T. A. Knoch, W. Waldeck, and J. Langowski. 2003. Analyzing intracellular binding and diffusion with continuous fluorescence photobleaching. Biophys. J. 84:3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saxton, M. J. 1996. Anomalous diffusion due to binding: a Monte Carlo study. Biophys. J. 70:1250–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siggia, E. D., J. Lippincott-Schwartz, and S. Bekiranov. 2000. Diffusion in inhomogeneous media: theory and simulations applied to whole cell photobleach recovery. Biophys. J. 79:1761–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaff, J. C., B. M. Slepchenko, and L. M. Loew. 2000. Physiological modeling with virtual cell framework. Meth. Enzym. 321:1–23. [DOI] [PubMed] [Google Scholar]
- 17.Saxton, M. J. 2001. Anomalous subdiffusion in fluorescence photobleaching recovery: a Monte Carlo study. Biophys. J. 81:2226–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith, A. E., B. M. Slepchenko, J. C. Schaff, L. M. Loew, and I. G. Macara. 2002. Systems analysis of Ran transport. Science. 295:488–491. [DOI] [PubMed] [Google Scholar]
- 19.Carrero, G., D. McDonald, E. Crawford, G. de Vries, and M. J. Hendzel. 2003. Using FRAP and mathematical modeling to determine the in vivo kinetics of nuclear proteins. Methods. 29:14–28. [DOI] [PubMed] [Google Scholar]
- 20.Weiss, M. 2004. Challenges and artifacts in quantitative photobleaching experiments. Traffic. 5:662–671. [DOI] [PubMed] [Google Scholar]
- 21.Schwartz, P., D. Adalsteinsson, P. Colella, A. P. Arkin, and M. Onsum. 2005. Numerical computation of diffusion on a surface. Proc. Natl. Acad. Sci. USA. 102:11151–11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sbalzarini, I. F., A. Mezzacasa, A. Helenius, and P. Koumoutsakos. 2005. Effects of organelle shape on fluorescence recovery after photobleaching. Biophys. J. 89:1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sbalzarini, I. F., A. Hayer, A. Helenius, and P. Koumoutsakos. 2006. Simulations of (an)isotropic diffusion on curved biological surfaces. Biophys. J. 90:878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saxton, M. J. 2007. A biological interpretation of transient anomalous subdiffusion. I. Qualitative model. Biophys. J. 92:1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brünger, A., R. Peters, and K. Schulten. 1985. Continuous fluorescence microphotolysis to observe lateral diffusion in membranes: theoretical methods and applications. J. Chem. Phys. 82:2147–2160. [Google Scholar]
- 26.Ferrieres, X., A. Lopez, A. Altibelli, L. Dupou-Cezanne, J. L. Lagouanelle, and J. F. Tocanne. 1989. Continuous fluorescence microphotolysis of anthracene-labeled phospholipids in membranes. Theoretical approach of the simultaneous determination of their photodimerization and lateral diffusion rates. Biophys. J. 55:1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delon, A., Y. Usson, J. Derouard, T. Biben, and C. Souchier. 2006. Continuous photobleaching in vesicles and living cells: a measure of diffusion and compartmentation. Biophys. J. 90:2548–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wawrezinieck, L., H. Rigneault, D. Marguet, and P.-F. Lenne. 2005. Fluorescence correlation spectroscopy diffusion laws to probe the submicron cell membrane organization. Biophys. J. 89:4029–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiss, M., H. Hashimoto, and T. Nilsson. 2003. Anomalous protein diffusion in living cells as seen by fluorescence correlation spectroscopy. Biophys. J. 84:4043–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kolin, D. L., D. Ronis, and P. W. Wiseman. 2006. K-space image correlation spectroscopy: a method for accurate transport measurements independent of fluorophore photophysics. Biophys. J. 91:3061–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hell, S., and E. H. K. Stelzer. 1992. Fundamental improvement of resolution with a 4Pi-confocal fluorescence microscope using two-photon excitation. Opt. Commun. 93:277–282. [Google Scholar]
- 32.Egner, A., S. Jakobs, and S. W. Hell. 2002. Fast 100-nm resolution three-dimensional microscope reveals structural plasticity of mitochondria in live yeast. Proc. Natl. Acad. Sci. USA. 99:3370–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hell, S. W. 2003. Toward fluorescence nanoscopy. Nat. Biotechnol. 21:1347–1355. [DOI] [PubMed] [Google Scholar]
- 34.Schönle, A. 2003. PSF engineering in fluorescence spectroscopy. PhD thesis. Ruprecht-Karls-Universität Heidelberg, Heidelberg, Germany.
- 35.Bewersdorf, J., R. Schmidt, and S. W. Hell. 2006. Comparison of I5M and 4Pi microscopy. J. Microsc. 222:105–117. [DOI] [PubMed] [Google Scholar]
- 36.Hell, S. W. 2007. Far-field optical nanoscopy. Science. 316:1153–1158. [DOI] [PubMed] [Google Scholar]
- 37.Gustafsson, M. G. L., D. A. Agard, and J. W. Sedat. 1995. Sevenfold improvement of axial resolution in 3d wide-field microscopy using two objective lenses. Proc. SPIE. 2412:147–156. [Google Scholar]
- 38.Gustafsson, M. G. L., D. A. Agard, and J. W. Sedat. 1999. I5M: 3D widefield light microscopy with better than 100 nm axial resolution. J. Microsc. 195:10–16. [DOI] [PubMed] [Google Scholar]
- 39.Hell, S. W., and J. Wichmann. 1994. Breaking the diffraction resolution limit by stimulated emission. Opt. Lett. 19:780–782. [DOI] [PubMed] [Google Scholar]
- 40.Westphal, V., and S. W. Hell. 2005. Nanoscale resolution in the focal plane of an optical microscope. Phys. Rev. Lett. 94:143903. [DOI] [PubMed] [Google Scholar]
- 41.Willig, K. I., S. O. Rizzoli, V. Westphal, R. Jahn, and S. W. Hell. 2006. STED-microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature. 440:935–939. [DOI] [PubMed] [Google Scholar]
- 42.Kittel, R. J., C. Wichmann, T. M. Rasse, W. Fouquet, M. Schmidt, A. Schmid, D. A. Wagh, C. Pawlu, R. R. Kellner, K. I. Willig, S. W. Hell, E. Buchner, et al. 2006. Bruchpilot promotes active zone assembly, Ca2+-channel clustering, and vesicle release. Science. 312:1051–1054. [DOI] [PubMed] [Google Scholar]
- 43.Ivanchenko, S., S. Glaschick, C. Röcker, F. Oswald, J. Wiedenmann, and G. U. Nienhaus. 2007. Two-photon excitation and photoconversion of EosFP in dual-color 4Pi confocal microscopy. Biophys. J. 92:4451–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Egner, A. 2002. Multifocale hochauflösende 3D-Fluoreszenzmikroskopie. PhD thesis, Heidelberg Universität, Heidelberg, Germany.
- 45.Wilson, T. 1990. Confocal Microscopy. Academic Press, New York.
- 46.Wolf, E. 1959. Electromagnetic diffraction in optical systems. I. An integral representation of the image field. Proc. R. Soc. Lond. A. (Math. Phys. Sci.). 253:349–357. [Google Scholar]
- 47.Richards, B., and E. Wolf. 1959. Electromagnetic diffraction in optical systems. II. Structure of the image field in an aplanatic system. Proc. R. Soc. Lond. A. (Math. Phys. Sci.). 253:358–379. [Google Scholar]
- 48.Heath, M. T. 2002. Scientific Computing: An Introductory Survey, 2nd Ed. McGraw-Hill, New York.
- 49.Braess, D. 2001. Finite Elements. Theory, Fast Solvers, and Applications in Solid Mechanics, 2nd Ed. Cambridge University Press, Cambridge, UK.
- 50.Horn, D. R., M. Houston, and P. Hanrahan. 2005. ClawHMMER: a streaming HMMer-search implementation. In SC '05: Proceedings of the 2005 ACM/IEEE Conference on Supercomputing. IEEE Computer Society, Washington, DC. 11.
- 51.Elsen, E., M. Houston, V. Vishal, E. Darve, P. Hanrahan, and V. Pande. 2006. N-body simulation on GPUs. In SC '06 Proceedings. IEEE Computer Society, Washington, DC.
- 52.Stone, J. E., J. C. Phillips, P. L. Freddolino, D. J. Hardy, L. G. Trabuco, and K. Schulten. 2007. Accelerating molecular modeling applications with graphics processors. J. Comput. Chem. 28:2618–2640. [DOI] [PubMed] [Google Scholar]
- 53.Siebrasse, J. P., and R. Peters. 2002. Rapid translocation of NTF2 through the nuclear pore of isolated nuclei and nuclear envelopes. EMBO Rep. 3:887–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kubitscheck, U., O. Kückmann, T. Kues, and R. Peters. 2000. Imaging and tracking of single GFP molecules in solution. Biophys. J. 78:2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pucadyil, T. J., and A. Chattopadhyay. 2006. Confocal fluorescence recovery after photobleaching of green fluorescent protein in solution. J. Fluoresc. 16:87–94. [DOI] [PubMed] [Google Scholar]
- 56.Magde, D., E. Elson, and W. W. Webb. 1972. Thermodynamic fluctuations in a reacting system: measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett. 29:705–708. [Google Scholar]
- 57.Webb, W. W. 2001. Fluorescence correlation spectroscopy: inception, biophysical experimentations, and prospectus. Appl. Opt. 40:3969–3983. [DOI] [PubMed] [Google Scholar]
- 58.Schwille, P., and E. Haustein. 2002. Fluorescence correlation spectroscopy: a tutorial for the biophysics textbook online (BTOL). http://www.biophysics.org/education/techniques.htm. Accessed October 10, 2007.
- 59.Ries, J., and P. Schwille. 2006. Studying slow membrane dynamics with continuous wave scanning fluorescence correlation spectroscopy. Biophys. J. 91:1915–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yechiel, E., and M. Edidin. 1987. Micrometer-scale domains in fibroblast plasma membranes. J. Cell Biol. 105:755–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simons, K., and W. L. Vaz. 2004. Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct. 33:269–295. [DOI] [PubMed] [Google Scholar]
- 62.Jacobson, K., O. G. Mouritsen, and R. G. Anderson. 2007. Lipid rafts: at a crossroad between cell biology and physics. Nat. Cell Biol. 9:7–14. [DOI] [PubMed] [Google Scholar]
- 63.Donnert, G., C. Eggeling, and S. W. Hell. 2007. Major signal increase in fluorescence microscopy through dark-state relaxation. Nat. Methods. 4:81–86. [DOI] [PubMed] [Google Scholar]
- 64.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.