

Abstract
Downstream processes that discriminate between DNA adducts of a third generation platinum antitumor drug oxaliplatin and conventional cisplatin are believed to be responsible for the differences in their biological effects. These different biological effects are explained by the ability of oxaliplatin to form DNA adducts more efficient in their biological effects. In this work conformation, recognition by HMG domain protein and DNA polymerization across the major 1,2-GG intrastrand cross-link formed by cisplatin and oxaliplatin in three sequence contexts were compared with the aid of biophysical and biochemical methods. The following major differences in the properties of the cross-links of oxaliplatin and cisplatin were found: i), the formation of the cross-link by oxaliplatin is more deleterious energetically in all three sequence contexts; ii), the cross-link of oxaliplatin bends DNA slightly but systematically less in all sequence contexts tested; iii), the affinity of HMG domain protein to the cross-link of oxaliplatin is considerably lower independent of the sequence context; and iv), the Klenow fragment of DNA polymerase I pauses considerably more at the cross-link of oxaliplatin in all sequence contexts tested. We have also demonstrated that the chirality at the carrier ligand of oxaliplatin can affect its biological effects.
INTRODUCTION
Since the introduction of cisplatin [cis-diamminedichloridoplatinum(II)] (Fig. 1 A), only [cis-diamminecyclobutanedicarboxylatoplatinum(II)] (carboplatin) and [(1R,2R-diamminocyclohexane)oxalatoplatinum(II)] (oxaliplatin) (Fig. 1 A) have received worldwide approval and achieved routine clinical use (1). Carboplatin is less toxic than cisplatin and can be given at a much higher dose than cisplatin. Unfortunately, carboplatin is still only active in the same range of tumors as cisplatin (2). As of yet, oxaliplatin, administered as a single agent, has not demonstrated substantial advantages over cisplatin or carboplatin although it has shown potential for use in some cisplatin-resistant tumors if administered in combination with 5-fluorouracil or folinic acid (3).
FIGURE 1.

Structures of platinum compounds and sequences of the synthetic oligodeoxyribonucleotides with their abbreviations. (A) Structures: a, cisplatin; b, oxaliplatin; c, [Pt(R,R-DACH)]2+; d, [Pt(S,S-DACH)]2+. (B) Sequences: the top and bottom strands of each pair in the figure are designated “top” and “bottom”, respectively, throughout. The boldface letters in the top strands of the duplexes indicate the platinated residues.
DNA is considered the major pharmacological target of platinum compounds (4–6). Direct analogs of cisplatin, which have already achieved routine clinical use, such as carboplatin and oxaliplatin, produce on DNA adducts similar to those produced by the parent drug, though different in their relative rates of formation (6,7). Hence, from a mechanistic DNA-binding point of view, it is not too surprising that the introduction of the new platinum antitumor drugs in the clinic does not represent a fundamental breakthrough in the treatment of cancer with platinum agents. This conclusion is in accordance with the hypothesis systematically tested by us and others that platinum agents, which bind to DNA in a fundamentally different manner, may have altered pharmacological properties (8–10). Nevertheless, the studies on DNA interactions of cisplatin, carboplatin, and oxaliplatin and their closely related analogs or models afforded a number of interesting results that broaden theoretical background needed for the design of new, more effective, platinum anticancer drugs.
For the reaction of oxaliplatin with DNA to occur, the parent compound must become aquated. The hydrolysis of oxaliplatin to form reactive diaqua species [Pt(R,R-DACH)(H2O)2]2+ (DACH = 1,2-diaminocyclohexane, Fig. 1 A) is a slower process than the hydrolysis of cisplatin. Therefore, oxaliplatin is inherently less able than cisplatin to form DNA adducts (11), but the sites in DNA of oxaliplatin adducts and their spectrum are nearly identical to the situation when DNA is modified by cisplatin (12). On the other hand, oxaliplatin adducts are removed from DNA by a nucleotide excision repair system (13) or by recombination repair (14) with similar efficiency as cisplatin adducts. In contrast, DNA adducts of oxaliplatin and cisplatin are processed by a mismatch repair system (15) and translesion DNA polymerases (16–18) differently. Despite lower DNA reactivity, oxaliplatin exhibits similar or greater cytotoxicity in several human tumor cell lines. Thus, oxaliplatin requires fewer DNA lesions than does cisplatin to achieve cell growth inhibition (11).
The overall conformational alterations induced in DNA by the major 1,2-GG intrastrand cross-link (CL) of cisplatin and the same adduct of oxaliplatin in the TGGT sequence were studied by x-ray crystallography (19). The 1,2-GG intrastrand CLs of cisplatin and oxaliplatin have been shown to be similar although they differ in several details. The bulky DACH ring of the oxaliplatin adduct fills much of the DNA major groove, making it narrower and less polar at the site of the CL. Recently, the solution structures of the 1,2-GG intrastrand CL of oxaliplatin and cisplatin formed in the AGGC sequence were solved (20,21), and again several conformational differences were observed between the cisplatin-GG adduct and the oxaliplatin-GG adduct.
The structure of an oxaliplatin-GG adduct has been so far described only for two sequence contexts, such as TGGT and AGGC, although under different conditions. Besides [Pt(R,R-DACH)]2+ (oxaliplatin), another enantiomeric form of this complex exists so that the biological activity of platinum complexes with enantiomeric amine ligands such as [PtX2(R,R-DACH)]2+ and [Pt X2(S,S-DACH)]2+ (Fig. 1 A) are also of great interest. For instance, the DACH carrier ligand has been shown to significantly affect the ability of platinum-DNA adducts to block essential processes such as replication and transcription (22). Also, importantly, the [PtCl2(S,S-DACH)]2+ complex, having an S configuration at the asymmetric carbon atoms, is markedly more mutagenic toward several strains of Salmonella typhimurium than [PtCl2(R,R-DACH)]2+ (23). Even more importantly, oxaliplatin (having λ-gauche conformation of the diamine chelate ring) exhibits higher activity toward various cancers than the S,S-enantiomer (having δ-gauche conformation) so that oxaliplatin and not its S,S-enantiomer has been approved for clinical use (24). Hence, although the asymmetry in the amine ligand in these platinum complexes does not involve the coordinated nitrogen atom but rather an adjacent carbon atom, a dependence of the biological activity on the configuration of the amine is observed.
To allow drawing a more definite conclusion relating the structural differences between the 1,2-GG intrastrand CLs of oxaliplatin and cisplatin to the biological differences between these two platinum drugs, we performed a more complex study including the effect of the sequence context on the structure of 1,2-GG intrastrand adduct of oxaliplatin using gel electrophoretic retardation (phasing) assay, chemical probes of DNA conformation, and differential scanning calorimetry (DSC). We also performed studies aimed at recognition of this adduct by HMG domain protein and the inhibitory effect of this adduct on DNA polymerization in vitro. The latter factors play an important role in the mechanism underlying antitumor effects of cisplatin (6,25). In addition, we have also given several examples of how chirality at the carbon atom of the carrier DACH ligand affects some important factors that play a significant role in the mechanism of biological effects of cisplatin and its analogs.
MATERIALS AND METHODS
Chemicals
[Pt(R,R-DACH)(H2O)2](SO4) or [Pt(S,S-DACH)(H2O)2](SO4) (Fig. 1 A) were prepared from the corresponding dichloro species (23) by treatment with Ag2SO4 (26). Cisplatin was obtained from Sigma (Prague, Czech Republic). The stock solutions of platinum compounds were prepared at a concentration of 5 × 10−4 M in 10 mM NaClO4 and stored at 4°C in the dark. The synthetic oligodeoxyribonucleotides were synthesized and purified as described previously (27). Expression and purification of domain A (residues 1–84) of the HMGB1 protein (HMGB1a) (HMG = high-mobility group) were carried out as described (28,29). T4 DNA ligase, the Klenow fragment from DNA polymerase I (exonuclease minus, mutated to remove the 3′→5′ proofreading domain) (KF−), restriction endonuclease EcoRI, and T4 polynucleotide kinase were purchased from New England Biolabs (Beverly, MA). Deoxyribonucleoside 5′-triphosphates were from Roche Diagnostics (Mannheim, Germany). Acrylamide, bis(acrylamide), urea, and NaCN were from Merck (Darmstadt, Germany). Dimethyl sulfate (DMS), KMnO4, diethyl pyrocarbonate (DEPC), KBr, and KHSO5 were from Sigma. Nonidet P-30 was from Fluka (Prague, Czech Republic). Radioactive products were from Amersham (Arlington Heights, IL). Proteinase K and ATP were from Boehringer (Mannheim, Germany).
Platinations of oligonucleotides
The duplexes containing single, central 1,2-GG intrastrand CL of [Pt(R,R-DACH)]2+, [Pt(S,S-DACH)]2+, or cisplatin in the pyrimidine-rich top strands were prepared as described (27). The platinated oligonucleotides were purified by ion-exchange fast protein liquid chromatography (FPLC). It was verified by platinum flameless atomic absorption spectrophotometry (FAAS) and by the measurements of the optical density that the modified oligonucleotides contained one platinum atom. It was also verified using DMS footprinting of platinum on DNA (30) that one DACH or cisplatin molecule was coordinated to the N7 atom of the single G in the top strands of each duplex. FPLC purification and FAAS measurements were carried out on a Pharmacia Biotech (Piscataway, NJ) FPLC System with MonoQ HR 5/5 column and a Varian (Palo Alto, CA) AA240Z Zeeman atomic absorption spectrometer equipped with a GTA 120 graphite tube atomizer, respectively. The unmodified or platinated duplexes used in the studies of recognition by HMGB1 domain proteins were still purified by electrophoresis on native 15% polyacrylamide (PAA) gel (mono/bis(acrylamide) ratio = 29:1). Other details have been described previously (27,31).
Ligation and electrophoresis of oligonucleotides
Unplatinated 20–23-mer single strands (bottom strands of the duplexes described in the section DNA unwinding and bending) were 5′-end-labeled with [γ-32P]ATP by using T4 polynucleotide kinase. Then they were annealed with their phosphorylated complementary strands (unplatinated or containing the platinum CL). The duplexes were allowed to react with T4 DNA ligase. The resulting samples along with ligated unplatinated duplexes were subsequently examined on 8% native PAA (mono/bis(acrylamide) ratio = 29:1) electrophoresis gels. Other details of these experiments were as described in previous articles (32–34).
Chemical modifications
The modification by KMnO4, DEPC, and KBr/KHSO5 were performed as described previously (31,35–37). The strands of the duplexes were 5′-end labeled with [γ-32P]ATP. In the case of the platinated oligonucleotides, the platinum complex was removed after reaction of the DNA with the probe by incubation with 0.2 M NaCN (pH 11) at 45°C for 10 h in the dark.
Gel mobility shift assay
The 5′-end-labeled 22-bp oligonucleotide duplexes with blunt ends (their sequences were identical to those of the duplexes TGGT (23), CGGA (23), and AGGC (23) shown in Fig. 1 B except that they did not contain terminal overhanging nucleotides) either unplatinated (controls) or containing the central platinum adduct in their top strands were used, and their reaction with HMGB1a protein was performed and analyzed as described previously (38).
Inhibition of DNA polymerization
We investigated in this work DNA polymerization using the templates site specifically modified by [Pt(R,R-DACH)]2+, [Pt(S,S-DACH)]2+, or cisplatin by KF−. The DNA polymerase I class of enzymes has served as the prototype for studies on structural and biochemical mechanisms of DNA replication (39,40). In addition, as the most extensive genetic, biochemical, and structural studies have been carried out on a Klenow fragment of DNA polymerase I (including its exonuclease-deficient analog), this enzyme appears to be an ideal model system for investigating the molecular mechanisms associated with template-directed DNA synthesis (39,40).
The 23-mer templates (see Fig. 5 B, bottom) containing a single 1,2-GG intrastrand adduct of [Pt(R,R-DACH)]2+, [Pt(S,S-DACH)]2+, or cisplatin were prepared in the same way as described in the section Platinations of oligonucleotides (vide supra); 8-mer DNA primer whose sequence is also shown in Fig. 5 B was complementary to the 3′ termini of the 23-mer templates. The DNA substrates were formed by annealing templates and 5′-end-labeled primer (5 × 10−8 M) at a molar ratio of 3:1. All experiments were performed at 25°C in a volume of 50 μl in a buffer containing 50 mM Tris-HCl (pH 7.4), 10 mM MgCl2, 0.1 mM dithiothreitol, 50 μg/ml bovine serum albumin, 100 μM deoxyadenosine 5′-triphosphate, 100 μM deoxycytidine 5′-triphosphate, 100 μM deoxyguanosine 5′-triphosphate, and 100 μM thymidine 5′-triphosphate and 0.5 unit of KF−. Reactions were terminated by the addition of EDTA so that its resulting concentration was 20 mM and by heating at 100°C for 30 s. Products were resolved by denaturing 20% PAA/8 M urea gel and then visualized and quantified by using the FUJIFILM bioimaging analyzer and AIDA image analyzer software.
FIGURE 5.

Primer extension activity of KF− using the 8-mer/23-mer, primer/template duplex. (A) The experiments were conducted for the times indicated in the figure (3–60 min) using undamaged template (lanes 1–5), the template containing single, site-specific 1,2-GG intrastrand CL in the TGGT sequence of [Pt(R,R-DACH)]2+ (lanes 6–10) or [Pt(S,S-DACH)]2+ (lanes 11–15). The pause sites opposite the platinated guanines and a flanking residue on the 3′ site are marked 14, 13, and 12, respectively. (B) The nucleotide sequences of the templates containing central TGGT, CGGA, or AGGC sequences and the primer. See the text for details. (C) The time dependence of the inhibition of DNA synthesis on undamaged (control) templates (solid squares), DNA-containing 1,2-GG intrastrand CL of [Pt(R,R-DACH)]2+ (solid circles), [Pt(S,S-DACH)]2+ (open circles), or cisplatin (squares) in the TGGT (top), AGGC (middle), or CGGA (bottom) sequence. Data are means from three different experiments with two independent template preparations.
Differential scanning calorimetry
Excess heat capacity (ΔCp) versus temperature profiles for the thermally induced transitions of TGGT (15), CGGA (15), and AGGC (15) duplexes (see Fig. 1 B for their sequences) unmodified or containing a unique 1,2-GG intrastrand CL of cisplatin, [Pt(R,R-DACH)]2+, or [Pt(S,S-DACH)]2+ were measured using a VP-DSC calorimeter (Microcal, Northampton, MA). In the DSC experiments the concentrations of the duplexes were 30 μM, the heating rate was 60°C/h, and the maximum temperature was 95°C. After reaching the maximum temperature the samples were cooled at the same rate to the starting temperature of 25°C. In this study ΔCp is defined as excess heat capacity, which is baseline subtracted and concentration normalized (41). The reference scans were subtracted from the sample scans to obtain ΔCp versus temperature profiles. Enthalpies (ΔHcal) and entropies (ΔS) of duplex melting were calculated from the areas under the experimental ΔCp versus T and derived ΔCp/T versus T curves, respectively, using ORIGIN v.5.0 software (Microcal, Studio City, CA). The free energy of duplex dissociation at 25°C (ΔG25) was calculated using the standard thermodynamic relationship given in Eq. 1 and the corresponding ΔH and ΔS values:
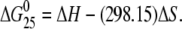 |
(1) |
The duplexes were dissolved in the buffer containing 10 mM sodium phosphate (NaH2PO4/Na2HPO4) pH 7.0 and 150 mM NaCl. It was also verified in the same way as described in previous articles (42,43) that the melting transitions of both the platinated and unmodified duplexes were fully reversible.
RESULTS
Conformational changes produced in double helical DNA by the site-specific 1,2-GG intrastrand cross-link
The goal of our work was to establish whether the steric structure of the nonleaving group of platinum DACH enantiomers could influence the distortions induced in DNA by the formation of the 1,2-GG intrastrand CL. We directed our studies on establishing distortions and other biophysical properties of oligodeoxyribonucleotide duplexes containing a single, site-specific 1,2-GG intrastrand CL of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ in three central sequences, such as TGGT, AGGC, and CGGA.
Differential scanning calorimetry
A calorimetric technique was used to characterize the influence of the 1,2-GG intrastrand CL of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ on the thermal stability and energetics of the site specifically platinated 15-mer DNA duplexes. Such thermodynamic data can reveal how the platinum adduct influences duplex stability, a property that has been shown to play a significant role in the mechanism of antitumor activity of platinum drugs (42,44,45). Recently, calorimetric and spectroscopic techniques were used to characterize the influence of the 1,2-GG intrastrand CL on the thermal stability and energetics of a 20-mer DNA duplex site specifically modified by cisplatin, antitumor dinuclear platinum complex, and [Pt(R,R-DAB)]2+ or [Pt(S,S-DAB)]2+ (DAB = 2,3-diaminobutane) (42,46,47). We expanded these studies on the oligodeoxyribonucleotide duplex containing unique 1,2-GG site-specific intrastrand adducts of the [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ complexes.
Fig. 2 shows DSC melting profiles (ΔCp versus T) for the parent, nonmodified 15-bp duplexes TGGT (15), AGGC (15), and CGGA (15) (solid curves) and the same duplexes containing single 1,2-GG intrastrand CL of cisplatin (dot and dashed curves), [Pt(R,R-DACH)]2+ (dashed curves), or [Pt(S,S-DACH)]2+ (dotted curves). Each transition shows negligible changes in the heat capacities between the initial and final states, and denaturation (heating) and renaturation (cooling) curves for the unmodified and platinated duplexes were superimposable (not shown), which is consistent with the reversibility of the melting equilibrium. The interpretation of our calorimetric data described below is also based on the assumption that all thermodynamic parameters for melting of the unmodified and platinated duplexes are ascribed to differences in the initial duplex states. This implies that the final single-stranded states should be thermodynamically equivalent at the elevated temperatures at which they are formed. This assumption has been verified similarly as in earlier reports by recording identical circular dichroic spectra for the samples of nonplatinated and platinated duplexes that were heated to high temperatures (42–44,46,48). In aggregate, meaningful thermodynamic data from our calorimetric measurements described below could be obtained.
FIGURE 2.

DSC thermograms for the (A) TGGT (15), (B) CGGA (15), and (C) AGGC duplexes unmodified (solid lines) and containing in the top strand 1,2-GG intrastrand adduct of cisplatin (dot and dashed line), [Pt(R,R-DACH)]2+ (dashed line), and [Pt(S,S-DACH)]2+ (dotted line). The concentrations of the duplexes were 30 μM, and the buffer conditions were 10 mM sodium phosphate pH 7.0 and 150 mM NaCl.
DSC melting profiles were analyzed as described in Materials and Methods to obtain the results listed in Table 1. All thermodynamic parameters discussed in this work refer to the duplex dissociation process. Differences in the dissociation thermodynamics due to the presence of the adduct are presented as “ΔΔ” parameters. These parameters are computed by subtracting the appropriate value measured for control, unmodified duplex from the value measured for the duplex containing the single, site-specific platinum adduct and are reported in Table 1 in parentheses.
TABLE 1.
Calorimetrically derived thermodynamic parameters for dissociation (melting) of the 15-bp duplexes unmodified or containing a single, site-specific 1,2-GG intrastrand CL of cisplatin, [Pt(R,R-DACH)]2+, or [Pt(S,S-DACH)]2+
| TGGT(15) | Tm (°C) | ΔH (kcal/mol) | ΔS (cal/mol) | 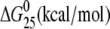 |
|---|---|---|---|---|
| no Pt (control) | 58.9 | 96.8 | 294.1 | 9.1 |
| cisplatin | 48.7 | 63.0 (−33.8) | 196.7 (−97.4) | 4.4 (−4.7) |
| [Pt(R,R-DACH)]2+ | 48.1 | 59.9 (−36.9) | 187.3 (−106.8) | 4.1 (−5.0) |
| [Pt(S,S-DACH)]2+ | 45.3 | 56.7 (−40.1) | 178.4 (−115.7) | 3.5 (−5.6) |
| CGGA(15) | Tm (°C) | ΔH (kcal/mol) | ΔS (cal/mol) | 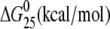 |
| no Pt (control) | 64.5 | 102.1 | 305.2 | 11.1 |
| cisplatin | 57.5 | 67.8 (−34.3) | 206.8 (−98.4) | 6.1 (−5.0) |
| [Pt(R,R-DACH)]2+ | 55.0 | 61.4 (−40.7) | 188.0 (−117.2) | 5.3 (−5.8) |
| [Pt(S,S-DACH)]2+ | 54.9 | 64.4 (−37.7) | 197.8 (−107.4) | 5.4 (−5.7) |
| AGGC(15) | Tm (°C) | ΔH (kcal/mol) | ΔS (cal/mol) | 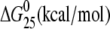 |
| no Pt (control) | 62.1 | 99.8 | 300.2 | 10.3 |
| cisplatin | 55.8 | 84.2 (−15.6) | 257.7 (−42.5) | 7.4 (−2.9) |
| [Pt(R,R-DACH)]2+ | 53.9 | 72.2 (−27.6) | 221.9 (−78.3) | 6.0 (−4.3) |
| [Pt(S,S-DACH)]2+ | 53.9 | 73.6 (−26.2) | 226.6 (−73.6) | 6.0 (−4.3) |
The ΔH and ΔS values are averages derived from three independent experiments. The experimental uncertainties of the parameters are as follows: Tm (±0.5°C), ΔHcal (±2%), ΔS (±3%), 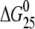 (±3%). “ΔΔ” parameters are in parentheses (these parameters are computed by subtracting the appropriate value measured for control, unmodified duplex from the value measured for the duplex containing the single, site-specific platinum adduct).
(±3%). “ΔΔ” parameters are in parentheses (these parameters are computed by subtracting the appropriate value measured for control, unmodified duplex from the value measured for the duplex containing the single, site-specific platinum adduct).
Inspection of these thermodynamic parameters reveals a number of interesting features: First, CL formation of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ reduced the duplex thermal stability. The CLs of both enatiomers reduced DNA melting temperature to the same extent that they would if they were formed in the sequences AGGC or CGGA; and this reduction was slightly greater than that induced by the CL of cisplatin formed in the same sequences. Interestingly, CL formation of cisplatin and DACH complexes reduced the duplex thermal stability more extensively if these CLs were formed in the TGGT sequence than in the other two sequences. In addition, whereas the CL of [Pt(R,R-DACH)]2+ reduced DNA melting temperature to the same extent as cisplatin, the CL of [Pt(S,S-DACH)]2+ was in this respect more efficient. Second, CL formation by cisplatin and DACH complexes resulted in a large decrease of the enthalpy of duplex dissociation (Table 1). In other words, the intrastrand CL of these platinum complexes enthalpically destabilizes the duplex relative to their nonmodified counterpart. Third, CL formation by [Pt(R,R-DACH)]2+, [Pt(S,S-DACH)]2+, or cisplatin resulted in a substantial decrease in duplex dissociation entropy (Table 1). In other words, the intrastrand CL of both DACH enantiomers and cisplatin increases the entropy of the duplex. Fourth, the net result of these enthalpic and entropic effects is that 1,2-GG intrastrand CL formation by [Pt(R,R-DACH)]2+, [Pt(S,S-DACH)]2+, or cisplatin in TGGT, CGGA, or AGGC sequences induces a decrease in the free energy for duplex dissociation at 25°C (ΔG25) (Table 1), this duplex destabilization being enthalpic in origin. In this respect, the intrastrand CLs of DACH complexes were more effective than that of cisplatin if the CLs were formed in the CGGA or AGGC sequences. On the other hand, if the CLs of [Pt(R,R-DACH)]2+ and cisplatin were formed in the TGGT sequence, the difference in the ability of these CLs to thermodynamically destabilize the duplex was notably small. Interestingly, the intrastrand CL of [Pt(S,S-DACH)]2+ formed in this latter sequence thermodynamically destabilized the duplex considerably more than its R,R counterpart.
DNA unwinding and bending
Among the alterations of secondary and tertiary structure of DNA to which it may be subjected, the role of intrinsic bending and unwinding of DNA is increasingly recognized as being potentially important in regulating replication and transcription functions through specific DNA-protein interactions. For cisplatin adducts, the structural details responsible for bending and subsequent protein recognition have recently been elucidated (49,50). Given the recent advances in our understanding of the structural basis for the bending of DNA caused by cisplatin, it is of considerable interest to examine how the character of a carrier amine in the 1,2-GG intrastrand adduct affects conformational properties of DNA, such as bending and unwinding. In this work we further performed studies on the bending and unwinding induced by single, site-specific intrastrand CLs of [Pt(DACH)]2+ enantiomers formed in oligodeoxyribonucleotide duplexes between neighboring guanine residues in three sequence contexts.
As in the previous study (47), we used electrophoretic retardation as a quantitative measure of the extent of planar curvature to analyze bending and unwinding induced by the single, site-specific 1,2-GG intrastrand CL formed by [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ in the sequences TGGT, CGGA, and AGGC. The oligodeoxyribonucleotide duplexes TGGT (20–23), CGGA (20–23), and AGGC (20–23), used for DNA bending and unwinding studies, were 20–23-bp long and had sequences identical or similar to those of the duplexes TGGT (21), CGGA (21), or AGGC (21) (Fig. 1 B), respectively; the 20-bp duplexes had one terminal C·G pair deleted, whereas one additional T·A pair was added to the 3′ end in the 22-bp duplex and T·A and C·G pairs were added to the 3′ end in the 23-bp duplex). The ligation products of these unplatinated or [Pt(DACH)]-containing duplexes were analyzed on native PAA electrophoresis gel. Experimental details of these studies are given in our recent report (47). A representative gel and its analysis showing the mobility of the ligation products of 20–23-bp duplexes containing single, site-specific 1,2-GG intrastrand CL of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ at the central sequence TGGT in a PAA gel is demonstrated in the Supplementary Material (Fig. S1). The results are summarized in Table 2. The DNA bending toward the major groove in the range of 29°–33° and unwinding in the range of 18°–22° due to the single, site-specific 1,2-GG intrastrand CL formed by [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ were similar for both enantiomers if the CLs were formed in the CGGA or AGGC sequences. The direction of the bend was determined using the 33-bp duplexes, which also contained, besides the single 1,2-GG intrastrand CL formed by either Pt-DACH enantiomer, the (A-T)5 tract located “in phase” from the CL (the cross-linked basepair and the center of the A tract were separated by 11 bp), in the same way as in our recent articles (51–53). Somewhat different results were obtained for the CLs in the TGGT sequence. The bending toward the major groove due to the single, site-specific 1,2-GG intrastrand CL formed by [Pt(R,R-DACH)]2+ was markedly greater than that induced by [Pt(S,S-DACH)]2+, and similarly the unwinding angle due to the CL of [Pt(R,R-DACH)]2+ was greater than that due to the CL of [Pt(S,S-DACH)]2+.
TABLE 2.
DNA bending and unwinding induced by the 1,2-GG intrastrand CL of cisplatin, [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ within the TGGT, CGGA, and AGGC contexts, as determined by the gel electrophoresis retardation assay
| cisplatin* | [Pt(R,R-DACH)]2+ | [Pt(S,S-DACH)]2+ | |
|---|---|---|---|
| TGGT | |||
| bending† | 34° | 31° | 23° |
| unwinding | 20° | 19° | 16° |
| CGGA | |||
| bending† | 34° | 30° | 30° |
| unwinding | 19° | 18° | 18° |
| AGGC | |||
| bending† | 33° | 28° | 34° |
| unwinding | 20° | 20° | 22° |
Data from Stehlikova et al. (64).
Toward major groove.
Chemical probing of conformational distortions
To further characterize the distortion induced in DNA by intrastrand CLs of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+, the 23-bp duplexes TGGT (23), CGGA (23), or AGGC (23) containing the 1,2-GG intrastrand CL of either enantiomer were treated with several chemical agents that are used as tools for monitoring the existence of conformations other than canonical B-DNA. These agents included KMnO4, bromine, or DEPC as probes for thymine, cytosine, and adenine/guanine residues, respectively (31,35–37,54). These chemical probes react, under the conditions used, with base residues in single-stranded DNA and distorted double-stranded DNA, but not with the base residues in intact, double-stranded DNA (31,35–37,54).
For this analysis, we used exactly the same methodology as in our recent studies dealing with DNA adducts of various antitumor platinum drugs. Thus, the details of this experiment can be found in those articles (31,55), and a representative gel showing piperidine-induced specific strand cleavage at KMnO4-modified, KBr/KHSO5-modified, and DEPC-modified bases in the 23-bp TGGT duplex unplatinated or containing single 1,2-GG intrastrand CL of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ is demonstrated in the Supplementary Material (Fig. S2). The results are schematically summarized in Fig. 3. The pattern and degree of reactivity toward the chemical probes were identical for the CLs formed by both enantiomers in the sequences CGGA or AGGC, indicating a similar character of the conformational distortion. This is in contrast to the results of the analogous experiments carried out with the duplex containing the CLs in the sequence TGGT, where the pattern and degree of reactivity toward the chemical probes was different for the CLs of the two enantiomers (Fig. 3), indicating a chirality-dependent character of the conformational distortion.
FIGURE 3.

Summary of the reactivity of chemical probes with the duplexes TGGT, CGGA, and AGGC containing 1,2-GG intrastrand CL of cisplatin, [Pt(R,R-DACH)]2+, or [Pt(S,S-DACH)]2+. Solid, half-solid and open circles designate strong, medium, or weak reactivity, respectively.
Recognition by domain A of HMGB1 protein
An important feature of the mechanism of action of cisplatin is that the major adducts of this platinum drug, the 1,2-GG intrastrand CLs, are recognized by proteins containing HMG domains (6,25,56). Importantly, DNA modified by transplatin or monodentate platinum(II) compounds, such as [PtCl(dien)]+ (dien = N-(2-aminoethyl)ethane-1,2-diamine) or [PtCl(NH3)3]+, is not recognized by these cellular proteins (57). It has been shown (25,50) that the binding of these proteins to DNA treated with cisplatin may mediate the antitumor effects of this platinum drug. In addition, it has been shown (58,59) that the replacement of one or both ammine groups in cisplatin by various, mostly cyclic, nonleaving ligands altered the affinity of HMG box proteins for the 1,2-GG intrastrand CLs. Hence, it was of great interest to examine whether the replacement of the ammine ligands of cisplatin by the chiral DACH ligand could also affect the affinity of HMG box proteins for this adduct.
HMGB1 is an abundant chromosomal protein which binds preferentially without sequence specificity to bent or distorted DNA structures including CLs formed by cisplatin. HMGB1 consists of two tandem HMG box domains (A and B, HMGB1a and HMGB1b, respectively) which share a common HMG box structure. It has been shown that HMGB1a has a markedly higher binding affinity for distorted DNA including 1,2-GG intrastrand CLs. Thus, the full-length HMGB1 protein binds to the 1,2-GG intrastrand CL of cisplatin primarily through domain A (60). As it is mainly domain A that is essential for binding to the specific structure produced by the 1,2-GG intrastrand CL of cisplatin, the interactions of this domain (HMGB1a) with the 1,2-GG intrastrand CLs of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ were investigated using gel mobility shift assay. In these experiments, the 22-bp duplexes containing the central sequence TGGT, CGGA, or AGGC (these 22-bp oligonucleotide duplexes had blunt ends and their sequences were identical to those of the duplexes TGGT (23), CGGA (23), and AGGC (23) shown in Fig. 1 B except that they did not contain terminal overhanging nucleotides) were modified so that they contained a single, site-specific 1,2-GG intrastrand CL of cisplatin, [Pt(R,R-DACH)]2+, or [Pt(S,S-DACH)]2+.
The binding of the HMGB1a to these DNA probes was detected by retardation of the migration of the radiolabeled 22-bp probes through the gel (Fig. 4). HMGB1a exhibited negligible binding to the unmodified 22-bp duplexes. As indicated by the presence of a shifted band whose intensity increases with increasing protein concentration, HMGB1a recognizes the duplexes TGGT, CGGA, or AGGC containing the 1,2-GG intrastrand CL of [Pt(R,R-DACH)]2+, [Pt(S,S-DACH)]2+, or cisplatin (shown for HMGB1a and CLs formed by [Pt(R,R-DACH)]2+, [Pt(S,S-DACH)]2+, or cisplatin in the TGGT sequence in Fig. 4 A). Interestingly, HMGB1a binds the probes containing the 1,2-GG intrastrand CL in the CGGA and AGGC of either enantiomer with an affinity which is very similar for both sequences and for both enantiomers and approximately half as much as that of cisplatin (Fig. 4 B). In contrast to this observation was the affinity of HMGB1a protein to the 1,2-GG intrastrand CLs of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ formed in the 22-bp duplex containing a central TGGT sequence in the top strand. Whereas the protein binds to the CL of [Pt(R,R-DACH)]2+ and cisplatin with comparable affinities and these are, on the average, greater than those observed for the CLs formed in the CGGA or AGGC sequences, a very weak binding is observed for the 1,2-GG intrastrand CL formed by [Pt(S,S-DACH)]2+, which is even lower than those observed for the CL formed by this complex in the CGGA and AGGC sequences (Fig. 4 B).
FIGURE 4.

The differential binding of HMGB1a to the 22-bp duplexes with central TGGT, CGGA, or AGGC sequences (in the top strand) containing the single, site-specific 1,2-GG intrastrand CL of cisplatin, [Pt(R,R-DACH)]2+, or [Pt(S,S-DACH)]2+; the concentration of the duplex was 10 nM and the concentration of HMGB1a was in the range of 2–30 nM. (A) Representative gel showing binding of HMGB1a to the duplex with central TGGT sequence. (B) Bar graph illustrating Θ values (protein-bound duplex/total duplex ratio).
In aggregate, the affinity of HMGB1a protein to the 1,2-GG intrastrand CLs of either [Pt(DACH)]2+ enantiomer was considerably lower than that to the same CL of cisplatin (Fig. 4 B) except the CL formed in the TGGT sequence; the affinity of HMGB1a to the CL of cisplatin and [Pt(R,R-DACH)]2+ were very similar and relatively high, whereas that to the CL of [Pt(S,S-DACH)]2+ was particularly low.
DNA polymerization by Klenow fragment of DNA polymerase I
To learn more about the molecular basis of the replication of DNA modified by oxaliplatin, we investigated in this work DNA polymerization using the DNA templates site specifically modified with 1,2-GG intrastrand CLs of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ in the TGGT, CGGA, or AGGC sequences.
The DNA polymerization was examined by the Klenow fragment of DNA polymerase I (61). The exonuclease-deficient Klenow fragment (KF−) was selected here because translesion synthesis (TLS) proficient DNA polymerases of the X or Y families share some common properties including lack of associated 3′–5′ exonuclease proofreading activity, and, not least, the proofreading mechanism itself may introduce effects more dependent on the adduct type (62).
To assess the translesion replication capacity of KF− we investigated its capacity to elongate a 5′ 32P-labeled 8-mer primer annealed to untreated 23-mer templates or to 23-mer templates containing the 1,2-GG intrastrand CL of [Pt(R,R-DACH)]2+, [Pt(S,S-DACH)]2+, or cisplatin formed in the TGGT, CGGA, or AGGC sequences depicted in Fig. 5 B. The 3′ guanine involved in the 1,2-GG intrastrand CL on the template strand was located at its 13th position from the 3′ terminus (positioning the 3′-end of the primer five bases before the first platinated base in the template strand) (Fig. 5 B). The newly synthesized DNA products were resolved by denaturing PAA gel electrophoresis and visualized by radiography.
DNA polymerization through the single 1,2-GG intrastrand CL of [Pt(R,R-DACH)]2+, [Pt(S,S-DACH)]2+, or cisplatin formed in the TGGT, CGGA, or AGGC sequences of the templates by KF− in the presence of all four deoxyribonucleoside 5′-triphosphates was stopped at various time intervals, and the products were resolved by a sequencing gel (shown in Fig. 5 A for the CLs formed by [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ in the TGGT sequence). Polymerization proceeded rapidly up to the nucleotide opposite that preceding the 1,2-intrastrand CLs, such that the 12-, 13-, and 14-mer products accumulated to a significant extent. KF− efficiently replicated the untreated template; no intermediate products were seen with the 23-mer control templates as the full-length products were formed (Fig. 5 A). There was no accumulation of other shorter or larger DNA intermediates. Significant amounts of the full-length products were also noticed with the 23-mer templates containing the CLs of [Pt(R,R-DACH)]2+, [Pt(S,S-DACH)]2+, or cisplatin formed in the TGGT, CGGA, and AGGC sequences at longer times of incubation of the templates with KF− (60 min), although in a markedly smaller amount than with the nonmodified template; the amount of the full-length products accumulated with the template containing the CL of cisplatin was always greater than those accumulated with the templates containing the CL of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ in all sequence contexts tested (Fig. 5 C). Importantly, the amount of the full-length products noticed with the 23-mer template containing the CLs of [Pt(R,R-DACH)]2+, [Pt(S,S-DACH)]2+, or cisplatin formed in the CGGA or AGGC sequences was pronouncedly smaller than that observed for the templates containing the CLs in the TGGT sequence (Fig. 5 C).
DISCUSSION
This complex study was performed to help clarify a more definite correlation between the structural differences of 1,2-GG intrastrand CLs of oxaliplatin and cisplatin and differences in biological effects of these two platinum drugs. We focused in this work on the effects of flanking sequences on DNA conformation at the site of the adduct and in its proximity. In addition, since DNA is a chiral molecule, it may interact in a different way with the platinum complexes containing enantiomeric amine ligands, such as [Pt(R,R-DACH)]2+ (oxaliplatin) or [Pt(S,S-DACH)]2+. Hence, we also paid attention to understanding how the chirality at the carbon atoms of the carrier ligand in [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ can affect properties of the 1,2-GG intrastrand CL, which is the major DNA lesion of conventional cisplatin and most likely relevant to its antitumor effects.
DNA is considered a major pharmacological target of platinum anticancer drugs (4,25). Hence, a possible explanation for the different biological activity of oxaliplatin ([Pt(R,R-DACH)]2+) and cisplatin on one hand and [Pt(R,R-DACH)]2+ and [Pt(S,S-DACH)]2+ enantiomers on the other hand can be associated with the different conformational distortions induced in DNA by the adducts of these compounds and their different processing in the cell. To test this hypothesis the experiments described in this work were carried out.
The presence of the 1,2-GG intrastrand adduct of cisplatin, [Pt(R,R-DACH)]2+ (oxaliplatin), and [Pt(S,S-DACH)]2+ reduces the thermal stability of the duplexes TGGT (15), CGGA (15), or AGGC (15). However, the thermal melting is not a thermodynamic parameter. DSC can also provide quantitative, model-independent characterizations of effects of the lesion on duplex thermodynamics. Introduction of the 1,2-GG intrastrand adduct of cisplatin, [Pt(R,R-DACH)]2+, and [Pt(S,S-DACH)]2+ decreases the ΔG25 for dissociation of all duplexes. Interestingly, the ΔG25 values observed for melting of the CGGA (15) and AGGC (15) duplexes containing CL of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ were identical (5.3/5.4 or 6.0 kcal/mol, respectively, Table 1), suggesting that the conformational alterations induced by the CLs formed by both enantiomers in either CGGA or AGGC sequences were similar. On the other hand, the above mentioned ΔG25 values observed for melting of the duplexes CGGA (15) and AGGC (15) containing CL of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ were considerably lower than those observed for the same duplexes containing the CL of cisplatin (6.1 or 7.4 kcal/mol, respectively). These differences in free energy represent an equilibrium preference for the CGGA (15) duplex modified by the CL of cisplatin over that modified by the CL of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ of ∼4 to 1 (∼11 to 1 for the platinated AGGC (15) duplex).
Different results were, however, obtained for melting the duplexes containing the CL in the sequence TGGT. The ΔG25 values observed for melting the TGGT (15) duplex containing the CL of [Pt(R,R-DACH)]2+ or cisplatin were similar (4.1 or 4.4 kcal/mol, Table 1). This result implies similar conformational alterations induced by the 1,2-GG intrastrand CLs of cisplatin and oxaliplatin formed in the TGGT sequence in contrast to the case when the CLs of these drugs were formed in CGGA and AGGC sequences (vide supra). Another striking difference between the 1,2-GG intrastrand CLs formed in CGGA or AGGC sequences on one hand and a TGGT sequence on the other hand consists of the different effects of the CLs of [Pt(R,R-DACH)]2+ and [Pt(S,S-DACH)]2+ enantiomers, the latter being more effective in destabilizing double-helical DNA, but only if it was formed in the TGGT sequence (ΔG25 of 3.5 kcal/mol, Table 1).
Inspection of Table 1 shows that the melting of each duplex is accompanied by unfavorable free energy terms which result from characteristic compensation of unfavorable enthalpy and favorable entropy terms. In general, the unfavorable (endothermic) enthalpy results primarily from the endothermic heats for disrupting basepairs and base-base stacks of the duplex, whereas the favorable entropy terms arise from contributions of the favorable dissociation into two separate strands. Relative to the duplexes containing the 1,2-GG intrastrand CL of cisplatin, those containing the CL of oxaliplatin {[Pt(R,R-DACH)]2+} are higher in enthalpy for all sequences tested, mostly if the CL was formed in the AGGC sequence, whereas the least perturbation was observed for the CL formed in the TGGT sequence. Hence, the formation of the 1,2-GG intrastrand adduct by oxaliplatin is more deleterious energetically than the formation of the same adduct by cisplatin. This observation may be explained in terms of a higher decrease in stacking interactions due to the CL of oxaliplatin than due to that of cisplatin although the extent of this decrease appears to be dependent on the sequence context.
The changes in free energy of the duplexes (TGGT, CGGA, and AGGC), ΔG25, as a consequence of the formation of the single, site-specific intrastrand CL of cisplatin, [Pt(R,R-DACH)]2+, and [Pt(S,S-DACH)]2+ between guanine residues in the top strands (ΔΔG25), reflect a combination of enthalpic (ΔΔH) and entropic (ΔΔS) effects (see the values in parentheses in Table 1). The magnitudes of these effects vary with the platinum complex and sequence context. The observed ΔΔG25 values are significantly smaller than the observed ΔΔH values (the values of ΔΔH range from −40.7 to −15.6 kcal/mol, whereas the values of ΔΔG25 range from only −5.8 to −2.9 kcal/mol). This result is the consequence of a considerable, but not complete, compensation of the change in ΔΔS. Interestingly, the higher ΔΔH is accompanied by the higher ΔΔS.
Structural features of DNA, such as local bending and unwinding at the site of the 1,2-GG intrastrand CLs of cisplatin, play a very important role in the mechanism of antitumor effects of this metallodrug (63). Interestingly, local bending and unwinding due to the 1,2-GG intrastrand CL of cisplatin are very little affected by the bases flanking the CL (64). On the other hand, the bending and unwinding angles due to the 1,2-GG intrastrand CLs of the chiral analogs ([Pt(DACH)]2+) as well as the closely related [Pt(DAB)]2+ (DAB = 2,3-diaminobutane) are dependent on the sequence context (65). For instance, the character of distortions determined by chemical probes and phasing assay due to the single, site-specific 1,2-GG intrastrand CL formed by the chiral analog of cisplatin [Pt(DAB)]2+ in the CGGA sequence were identical for both enantiomers; in contrast, these properties of the 1,2-GG CLs formed in the sequence context of TGGT by [Pt(DAB)]2+ depend pronouncedly on the chirality of the diamine (47). It has been shown (65) that the latter structural and chiral differentiation of DNA adducts of [Pt(DAB)]2+ originates from clashes of the 5′-oriented methyl group of the DAB ligand with the methyl group of the 5′-thymine of the TGGT sequence. This explanation was fully consistent with the observation that this chiral differentiation disappeared if the 5′ thymine was replaced by cytosine such as in the sequence CGGA (65). The strict analogy between DACH and DAB complexes suggests that the dependence of distortion, including DNA bending and unwinding angles due to the 1,2-GG intrastrand CLs of [Pt(DACH)]2+ on sequence context and on [Pt(DACH)]2+ chirality, can be explained in a similar way. Hence, the structural differentiation also observed for [Pt(DACH)]2+ enantiomers is likely to originate from clashes of the 5′-oriented methylene group of [Pt(S,S-DACH)]2+ with the methyl group of the 5′-thymine in the TGGT sequence. On the other hand, owing to the absence of such clashes in the CGGA sequence no effect of chirality of DACH compounds on the bending and unwinding angles is observed (Table 2).
We also examined conformational alterations induced in DNA by the 1,2-GG intrastrand CL of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ formed in a third sequence, AGGC. The overall character of these structural distortions induced in DNA by the CL of [Pt(R,R-DACH)]2+ is very similar to that observed for this enantiomer or cisplatin in both TGGT and CGGA sequences. The only difference between the 1,2-GG intrastrand CLs of [Pt(R,R-DACH)]2+ and cisplatin consists of a smaller bending angle (by 3°–4°) found for the CL of oxaliplatin in all sequence contexts tested in this work. It is possible that the bulky DACH group located in the major groove restricts to some degree the DNA bending toward the major groove required for the formation of the 1,2-intrastrand CL. In aggregate, we have found no distinctly pronounced dependence of the structural properties of 1,2-GG intrastrand CLs of [Pt(R,R-DACH)]2+ (oxaliplatin), such as local DNA bending and unwinding on the sequence context, as it was also observed earlier in our laboratory for the same CLs of cisplatin (64).
On the other hand, these structural features of 1,2-GG intrastrand CLs of [Pt(DACH)]2+ formed at an AGGC sequence are chirality dependent, but apparently differently than in the TGGT sequence. Thus, DNA bending and unwinding due to the 1,2-GG intrastrand CL of [Pt(S,S-DACH)]2+ are considerably more dependent on sequence context than the same adducts of cisplatin or oxaliplatin. Further structural studies are needed to explain this observation.
An important feature of the mechanism of action of cisplatin is that the 1,2-intrastrand CLs formed by this drug are specifically recognized by various proteins, including those containing HMG domains (6,25,56). We have found (47) that the recognition by domains of HMGB1 protein of the 1,2-GG intrastrand CLs formed in the sequence context of TGGT by the chiral [Pt(DAB)]2+ (which is closely related to [Pt(DACH)]2+) depends on the chirality of the asymmetric centers (66). We have also found (65) that this structural differentiation originates from clashes of the 5′-oriented methyl group of the DAB ligand with the methyl group of the 5′-thymine and that this chiral differentiation disappears in sequences XGG, with X being different from T. On the other hand, the domains of HMGB1 proteins have been shown to bind to the GG CL of [Pt(R,R-DAB)]2+ and [Pt(S,S-DAB)]2+within the CGGA sequence with the same affinity (65). We have demonstrated in this and earlier (65) work that the recognition by HMGB1a, which dominates the interaction between HMGB1 and platinum-damaged DNA (58), of the 1,2-GG intrastrand CLs formed in the sequence context of TGGT and CGGA by the chiral [Pt(DACH)]2+ depends on the chirality of the asymmetric centers in a way very similar to that of [Pt(DAB)]2+, i.e., only if the CL is formed in the TGGT sequence different binding of HMGB1a protein to the CLs of the two enantiomers is observed. In addition, no marked differences in the recognition by HMGB1a of the 1,2-GG intrastrand CLs formed in the sequence context of AGGC was observed (Fig. 4).
As shown for the 1,2-GG intrastrand CL of cisplatin (49), HMGB1a binds to DNA around the site of this adduct in the minor groove whereas the ammine ligands coordinated to platinum reside in the major groove. It is therefore unlikely that the affinity of HMGB1a to the 1,2-GG intrastrand CLs of [Pt(R,R-DACH)]2+ and [Pt(S,S-DACH)]2+ is affected in a dominant way by direct interactions of the enantiomeric nonleaving ligands with the protein. The very important structural elements that contribute to recognition of cisplatin adducts by cellular proteins are bending toward major groove and unwinding of the DNA helix (56). However, no apparent correlation exists between the binding affinity of HMGB1a to the 1,2-GG intrastrand CL formed by [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ in various sequence contexts and the bend or unwinding angles induced by these adducts (Table 2). This result may be interpreted to mean that DNA prebending toward the major groove due to the 1,2-GG intrastrand CL of [Pt(R,R-DACH)]2+ or [Pt(S,S-DACH)]2+ plays an important role in its recognition by HMG domain proteins, but other factors contribute to the stability of the complex formed between platinated DNA and these proteins.
DNA adducts of antitumor platinum drugs, such as cisplatin, are believed to exert their cytotoxic effects by inhibiting both replication and/or transcription and inducing programmed cell death (apoptosis) or necrosis (67). On the other hand, in vitro bypass by replicative DNA polymerases of helix-distorting DNA lesions, including DNA adducts of some platinum compounds, has been observed as well (68–71). Several DNA polymerases have been shown to bypass 1,2-GG intrastrand CLs of oxaliplatin with different efficiency than cisplatin-CLs (17,72), which has been proposed to contribute to the different biological effects of these drugs.
We have demonstrated in this work that DNA polymerase KF− paused at the site of the 1,2-GG intrastrand CL formed by [Pt(DACH)]2+ enantiomers in all sequence contexts tested in this work considerably more than at the CL of cisplatin (Fig. 5). This result is consistent with a higher TLS observed past cisplatin adducts compared to that past oxaliplatin adducts observed in several tumor cell lines (73,74). It is possible that bulkier DACH lesions can interfere with the conformational change of the polymerase to the closed catalytically active ternary complex (DNA-polymerase-dNTP) more than cisplatin lesions. Presumably, these bulky adducts may alter the structure and stability of the ternary complex, thus affecting an important step that is crucial in determining the rate of the nucleotide incorporation step.
The TLS past 1,2-GG intrastrand CL of [Pt(DACH)]2+, which was considerably smaller than that past the CL of cisplatin, was also chirality dependent (Fig. 5). More pronounced differences were observed mainly if the CL was formed in the TGGT sequence (Fig. 5 C) where TLS past the CL of [Pt(R,R-DACH)]2+ was markedly more extensive than that past the CL of [Pt(S,S-DACH)]2+. Much information is available regarding the TLS efficiencies of various lesions and polymerases, but a number of important details of the structural features of the DNA polymerase-DNA substrate interaction that are required to accomplish TLS have yet to be revealed. The inability of a polymerase to carry out DNA synthesis past DNA damage may reflect the polymerase's inability to position a lesion-containing basepair in a geometric conformation that allows active site assembly and catalysis. Thus, inhibition of synthesis may also be due to the inability of the polymerase to extend the 3′ lesion-containing terminus. Interestingly, the pattern and degree of reactivity toward the chemical probes were different, but only for the CLs of the two enantiomers formed in the TGGT sequence (Fig. 3). This observation indicates a chirality-dependent character of the conformational distortion in this sequence, which correlates with a chirality-dependent TLS past the [Pt(DACH)]2+-CL (Fig. 5) only observed in this sequence context. The striking feature of the structure of the 1,2-GG intrastrand CL of [Pt(DACH)]2+ enantiomers revealed by chemical probes is that distortions induced by these CLs are different mainly on the 3′ side of the lesion, the distortion being more extensive on this side of the CL for the [Pt(R,R-DACH)]2+ enantiomer. Thus, our data support the idea that an important factor facilitating TLS by KF− past 1,2-GG intrastrand CL of cisplatin and its direct analogs is an extensive distortion on the 3′ side of this lesion.
CONCLUSIONS
DNA is considered a major pharmacological target of antitumor effects of platinum drugs. Downstream processes that discriminate between DNA adducts of oxaliplatin and cisplatin are believed to be responsible for the differences in biological effects of these two metallodrugs used clinically, such as antitumor activity and mutagenicity. In addition, different biological effects of oxaliplatin in comparison with cisplatin are often explained by the ability of oxaliplatin to form fewer DNA adducts, but due to their different structural properties these adducts are more effective for biological effects. The major DNA adduct of cisplatin is an intrastrand CL formed between neighboring purine bases so that considerable attention has been paid to differences between properties of this adduct formed by cisplatin and oxaliplatin. Some structural differences have already been demonstrated (19,20), but a definite conclusion about factors involved in the mechanism underlying different cytotoxic and mutagenic response to these metallodrugs cannot yet be drawn. In this work we focused on comparison of conformation, recognition by HMG domain protein, and DNA polymerization across 1,2-GG intrastrand CL formed by cisplatin and oxaliplatin in three sequence contexts with the aid of biophysical and biochemical methods. We found the following:
The formation of the 1,2-GG intrastrand adduct by oxaliplatin is more deleterious energetically than the formation of the same adduct by cisplatin in all three sequence contexts. The calorimetric data suggest that oxaliplatin CLs exhibit a higher efficiency to reduce stacking interactions in DNA than the CLs of cisplatin, although the extent of this reduction appears to be dependent on the sequence context.
The 1,2-GG intrastrand CLs of oxaliplatin {[Pt(R,R-DACH)]2+ enantiomer} bends DNA slightly, but systematically less than the CL of cisplatin (by 3°–4°) in all sequence contexts tested in this work. It is suggested that the bulky DACH group located in the major groove restricts in some degree the DNA bending toward the major groove required for the formation of the 1,2-intrastrand CL. On the other hand, the structural properties of 1,2-GG intrastrand CLs of (oxaliplatin), such as local DNA bending and unwinding, are almost independent of the sequence context as observed earlier in our laboratory for the same CLs of cisplatin (64).
The affinity of HMG domain protein (HMGB1a) to the 1,2-GG intrastrand CLs of oxaliplatin is considerably lower than that to the same CL of cisplatin independent of the sequence context. It is suggested that the bulky DACH group in the 1,2-GG intrastrand CL restricts to some extent the additional DNA bending required for the protein binding.
DNA polymerase KF−, which serves as the prototype for studies on structural and biochemical mechanisms of DNA replication, pauses at the site of the 1,2-GG intrastrand CL of oxaliplatin in all sequence contexts tested in this work considerably more than at the CL of cisplatin. It is suggested that bulkier DACH lesions can interfere with the conformational change of the polymerase to the closed catalytically active ternary complex (DNA-polymerase-dNTP) more than cisplatin lesions.
We hypothesize that if not all, at least some of the differences between properties of the 1,2-GG intrastrand CL of cisplatin and oxaliplatin and its processing observed in this work could be involved in the different biological effects of these two metallodrugs. However, cisplatin and its antitumor analogs also form other types of adducts on DNA. The relative efficacy of DNA adducts in antitumor effects of these metallodrugs has not been univocally determined. Thus, it cannot be excluded that there are still distinct differences in the properties of other adducts of cisplatin and oxaliplatin and in their cellular processing. Revelation of these differences will complement theoretical background needed to explain differences in biological effects of these clinically used metallodrugs and could also help in the design of new platinum drugs.
In addition, we have also demonstrated examples of how the chirality at the carrier DACH ligand can affect some important factors that play a significant role in the mechanism of biological effects of cisplatin and its analogs. Interestingly, we notice specific properties of the 1,2-GG intrastrand CLs formed by [Pt(DACH)]2+ complexes in the TGGT sequence, which may give rise to specific, hitherto undisclosed biological effects of these lesions. The information contained in this work can be useful for better understanding how the stereochemistry of the carrier amine ligands of cisplatin analogs can modulate their anticancer and mutagenic properties.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit http://www.biophysj.org.
Supplementary Material
Acknowledgments
The authors acknowledge that their participation in the European Cooperation in the field of Scientific and Technical Research Actions D39 enabled them to regularly exchange the most recent ideas in the field of platinum anticancer drugs with several European colleagues.
This research was supported by the Grant Agency of the Czech Republic (Grants 305/05/2030 and 203/06/1239), the Grant Agency of the Academy of Sciences of the Czech Republic (Grant B400040601), Ministry of Education of the Czech Republic (MSMT LC06030), and the Academy of Sciences of the Czech Republic (Grants 1QS500040581 and KAN200200651) and the University of Bari.
Editor: Jonathan B. Chaires.
References
- 1.Lokich, J. 2001. What is the “best” platinum: cisplatin, carboplatin, or oxaliplatin? Cancer Invest. 19:756–760. [DOI] [PubMed] [Google Scholar]
- 2.Boulikas, T., and M. Vougiouka. 2003. Cisplatin and platinum drugs at the molecular level. Oncol. Rep. 10:1663–1682 [review]. [PubMed] [Google Scholar]
- 3.Di Francesco, A. M., A. Ruggiero, and R. Riccardi. 2002. Cellular and molecular aspects of drugs of the future: oxaliplatin. Cell. Mol. Life Sci. 59:1914–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson, N. P., J.-L. Butour, G. Villani, F. L. Wimmer, M. Defais, V. Pierson, and V. Brabec. 1989. Metal antitumor compounds: the mechanism of action of platinum complexes. Prog. Clin. Biochem. Med. 10:1–24. [Google Scholar]
- 5.Brabec, V., and J. Kasparkova. 2005. Modifications of DNA by platinum complexes: relation to resistance of tumors to platinum antitumor drugs. Drug Resist. Updat. 8:131–146. [DOI] [PubMed] [Google Scholar]
- 6.Brabec, V. 2002. DNA modifications by antitumor platinum and ruthenium compounds: their recognition and repair. Prog. Nucleic Acid Res. Mol. Biol. 71:1–68. [DOI] [PubMed] [Google Scholar]
- 7.Brabec, V., and J. Kasparkova. 2005. DNA interactions of platinum anticancer drugs. Recent advances and mechanisms of action. In Metal Compounds in Cancer Chemotherapy. J.-M. Perez-Martin, M. A. Fuertes, and C. Alonso, editors. Research Signpost, Trivandrum, Kerala, India. 187–218.
- 8.Vrana, O., V. Brabec, and V. Kleinwächter. 1986. Polarographic studies on the conformation of some platinum complexes: relations to anti-tumour activity. Anticancer Drug Des. 1:95–109. [PubMed] [Google Scholar]
- 9.Farrell, N., Y. Qu, and M. P. Hacker. 1990. Cytotoxicity and antitumor activity of bis(platinum) complexes: a novel class of platinum complexes active in cell lines resistant to both cisplatin and 1,2- diaminocyclohexane complexes. J. Med. Chem. 33:2179–2184. [DOI] [PubMed] [Google Scholar]
- 10.Farrell, N. 1993. Nonclassical platinum antitumor agents: perspectives for design and development of new drugs complementary to cisplatin. Cancer Invest. 11:578–589. [DOI] [PubMed] [Google Scholar]
- 11.Woynarowski, J. M., S. Faivre, M. C. S. Herzig, B. Arnett, W. G. Chapman, A. V. Trevino, E. Raymond, S. G. Chaney, A. Vaisman, M. Varchenko, and P. E. Juniewicz. 2000. Oxaliplatin-induced damage of cellular DNA. Mol. Pharmacol. 58:920–927. [DOI] [PubMed] [Google Scholar]
- 12.Woynarowski, J. M., W. G. Chapman, C. Napier, M. C. S. Herzig, and P. Juniewicz. 1998. Sequence- and region-specificity of oxaliplatin adducts in naked and cellular DNA. Mol. Pharmacol. 54:770–777. [DOI] [PubMed] [Google Scholar]
- 13.Reardon, J. T., A. Vaisman, S. G. Chaney, and A. Sancar. 1999. Efficient nucleotide excision repair of cisplatin, oxaliplatin, and bis-aceto-ammine-dichloro-cyclohexylamine-platinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res. 59:3968–3971. [PubMed] [Google Scholar]
- 14.Wu, H. I., J. A. Brown, M. J. Dorie, L. Lazzeroni, and J. M. Brown. 2004. Genome-wide identification of genes conferring resistance to the anticancer agents cisplatin, oxaliplatin, and mitomycin C. Cancer Res. 64:3940–3948. [DOI] [PubMed] [Google Scholar]
- 15.Vaisman, A., M. Varchenko, A. Umar, T. A. Kunkel, J. I. Risinger, J. C. Barrett, T. C. Hamilton, and S. G. Chaney. 1998. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 58:3579–3585. [PubMed] [Google Scholar]
- 16.Vaisman, A., C. Masutani, F. Hanaoka, and S. G. Chaney. 2000. Efficient translesion replication past oxaliplatin and cisplatin GpG adducts by human DNA polymerase η. Biochemistry. 39:4575–4580. [DOI] [PubMed] [Google Scholar]
- 17.Vaisman, A., and S. G. Chaney. 2000. The efficiency and fidelity of translesion synthesis past cisplatin and oxaliplatin GpG adducts by human DNA polymerase β. J. Biol. Chem. 275:13017–13025. [DOI] [PubMed] [Google Scholar]
- 18.Bassett, E., A. Vaisman, J. M. Havener, C. Masutani, F. Hanaoka, and S. G. Chaney. 2003. Efficiency of extension of mismatched primer termini across from cisplatin and oxaliplatin adducts by human DNA polymerases beta and eta in vitro. Biochemistry. 42:14197–14206. [DOI] [PubMed] [Google Scholar]
- 19.Spingler, B., D. A. Whittington, and S. J. Lippard. 2001. 2.4 Å crystal structure of an oxaliplatin 1,2-d(GpG) intrastrand cross-link in a DNA dodecamer duplex. Inorg. Chem. 40:5596–5602. [DOI] [PubMed] [Google Scholar]
- 20.Wu, Y., P. Pradhan, J. Havener, G. Boysen, J. A. Swenberg, S. L. Campbell, and S. G. Chaney. 2004. NMR solution structure of an oxaliplatin 1,2-d(GG) intrastrand cross-link in a DNA dodecamer duplex. J. Mol. Biol. 341:1251–1269. [DOI] [PubMed] [Google Scholar]
- 21.Wu, Y., D. Bhattacharyya, C. L. King, I. Baskerville-Abraham, S.-H. Huh, G. Boysen, J. A. Swenberg, B. Temple, S. L. Campbell, and S. G. Chaney. 2007. Solution structures of a DNA dodecamer duplex with and without a cisplatin 1,2-d(GG) intrastrand cross-link: comparison with the same DNA duplex containing an oxaliplatin 1,2-d(GG) intrastrand cross-link. Biochemistry. 46:6477–6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Page, J. D., I. Husain, A. Sancar, and S. G. Chaney. 1990. Effect of the diaminocyclohexane carrier ligand on platinum adduct formation, repair, and lethality. Biochemistry. 29:1016–1024. [DOI] [PubMed] [Google Scholar]
- 23.Fanizzi, F. P., F. P. Intini, L. Maresca, G. Natile, R. Quaranata, M. Coluccia, L. Di Bari, D. Giordano, and M. A. Mariggio. 1987. Biological activity of platinum complexes containing chiral centers on the nitrogen or carbon atoms of a chelate diamine ring. Inorg. Chim. Acta. 137:45–51. [Google Scholar]
- 24.Misset, J. L. 1998. Oxaliplatin in practice. Br. J. Cancer. 77(S4):4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jamieson, E. R., and S. J. Lippard. 1999. Structure, recognition, and processing of cisplatin-DNA adducts. Chem. Rev. 99:2467–2498. [DOI] [PubMed] [Google Scholar]
- 26.Benedetti, M., L. G. Marzilli, and G. Natile. 2005. Rotamer stability in cis-[Pt(diA)G2] complexes (diA=diamine derivative and G=guanine derivative) mediated by carrier-ligand amine stereochemistry as revealed by circular dichroism spectroscopy. Chem. Eur. J. 11:5302–5310. [DOI] [PubMed] [Google Scholar]
- 27.Brabec, V., J. Reedijk, and M. Leng. 1992. Sequence-dependent distortions induced in DNA by monofunctional platinum(II) binding. Biochemistry. 31:12397–12402. [DOI] [PubMed] [Google Scholar]
- 28.Stros, M. 1998. DNA bending by the chromosomal protein HMG1 and its high mobility group box domains. Effect of flanking sequences. J. Biol. Chem. 273:10355–10361. [PubMed] [Google Scholar]
- 29.Stros, M. 2001. Two mutations of basic residues within the N-terminus of HMG-1 B domain with different effects on DNA supercoiling and binding to bent DNA. Biochemistry. 40:4769–4779. [DOI] [PubMed] [Google Scholar]
- 30.Kasparkova, J., K. J. Mellish, Y. Qu, V. Brabec, and N. Farrell. 1996. Site-specific d(GpG) intrastrand cross-links formed by dinuclear platinum complexes. Bending and NMR studies. Biochemistry. 35:16705–16713. [DOI] [PubMed] [Google Scholar]
- 31.Brabec, V., M. Sip, and M. Leng. 1993. DNA conformational distortion produced by site-specific interstrand cross-link of trans-diamminedichloroplatinum(II). Biochemistry. 32:11676–11681. [DOI] [PubMed] [Google Scholar]
- 32.Koo, H. S., H. M. Wu, and D. M. Crothers. 1986. DNA bending at adenine · thymine tracts. Nature. 320:501–506. [DOI] [PubMed] [Google Scholar]
- 33.Bellon, S. F., and S. J. Lippard. 1990. Bending studies of DNA site-specifically modified by cisplatin, trans-diamminedichloroplatinum(II) and cis-Pt(NH3)2(N3-cytosine)Cl+. Biophys. Chem. 35:179–188. [DOI] [PubMed] [Google Scholar]
- 34.Kasparkova, J., N. Farrell, and V. Brabec. 2000. Sequence specificity, conformation, and recognition by HMG1 protein of major DNA interstrand cross-links of antitumor dinuclear platinum complexes. J. Biol. Chem. 275:15789–15798. [DOI] [PubMed] [Google Scholar]
- 35.Bailly, C., D. Gentle, F. Hamy, M. Purcell, and M. J. Waring. 1994. Localized chemical reactivity in DNA associated with the sequence-specific bisintercalation of echinomycin. Biochem. J. 300:165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross, S. A., and C. J. Burrows. 1996. Cytosine-specific chemical probing of DNA using bromide and monoperoxysulfate. Nucleic Acids Res. 24:5062–5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bailly, C., and M. J. Waring. 1997. Diethylpyrocarbonate and osmium tetroxide as probes for drug-induced changes in DNA conformation in vitro. In Drug-DNA Interaction Protocols. K. R. Fox, editor. Humana Press, Totowa, NJ. 51–79. [DOI] [PubMed]
- 38.He, Q., U.-A. Ohndorf, and S. J. Lippard. 2000. Intercalating residues determine the mode of HMG1 domains A and B binding to cisplatin-modified DNA. Biochemistry. 39:14426–14435. [DOI] [PubMed] [Google Scholar]
- 39.Lam, W. C., E. J. C. Van der Schans, L. C. Sowers, and D. P. Millar. 1999. Interaction of DNA polymerase I (Klenow fragment) with DNA substrates containing extrahelical bases: implications for proofreading of frameshift errors during DNA synthesis. Biochemistry. 38:2661–2668. [DOI] [PubMed] [Google Scholar]
- 40.Patel, P. H., M. Suzuki, E. Adman, A. Shinkai, and L. A. Loeb. 2001. Prokaryotic DNA polymerase I: evolution, structure, and “base flipping” mechanism for nucleotide selection. J. Mol. Biol. 308:823–837. [DOI] [PubMed] [Google Scholar]
- 41.Leharne, S. A., and B. Z. Chowdhry. 1998. Thermodynamic background to differential scanning calorimetry. In Biocalorimetry: Applications of Calorimetry in the Biological Sciences. J. E. Ladbury and B. Z. Chowdhry, editors. J. Wiley & Sons, Chichester, UK. 157–182.
- 42.Hofr, C., N. Farrell, and V. Brabec. 2001. Thermodynamic properties of duplex DNA containing a site-specific d(GpG) intrastrand crosslink formed by an antitumor dinuclear platinum complex. Nucleic Acids Res. 29:2034–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hofr, C., and V. Brabec. 2001. Thermal and thermodynamic properties of duplex DNA containing site-specific interstrand cross-link of antitumor cisplatin or its clinically ineffective trans isomer. J. Biol. Chem. 276:9655–9661. [DOI] [PubMed] [Google Scholar]
- 44.Pilch, D. S., S. U. Dunham, E. R. Jamieson, S. J. Lippard, and K. J. Breslauer. 2000. DNA sequence context modulates the impact of a cisplatin 1,2-d(GpG) intrastrand cross-link and the conformational and thermodynamic properties of duplex DNA. J. Mol. Biol. 296:803–812. [DOI] [PubMed] [Google Scholar]
- 45.Brabec, V., K. Stehlikova, J. Malina, M. Vojtiskova, and J. Kasparkova. 2006. Thermodynamic properties of damaged DNA and its recognition by xeroderma pigmentosum group A protein and replication protein A. Arch. Biochem. Biophys. 446:1–10. [DOI] [PubMed] [Google Scholar]
- 46.Poklar, N., D. S. Pilch, S. J. Lippard, E. A. Redding, S. U. Dunham, and K. J. Breslauer. 1996. Influence of cisplatin intrastrand crosslinking on the conformation, thermal stability, and energetics of a 20-mer DNA duplex. Proc. Natl. Acad. Sci. USA. 93:7606–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malina, J., C. Hofr, L. Maresca, G. Natile, and V. Brabec. 2000. DNA interactions of antitumor cisplatin analogs containing enantiomeric amine ligands. Biophys. J. 78:2008–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hofr, C., and V. Brabec. 2005. Thermal stability and energetics of 15-mer DNA duplex interstrand cross-linked by trans-diamminedichloroplatinum(II). Biopolymers. 77:222–229. [DOI] [PubMed] [Google Scholar]
- 49.Ohndorf, U. M., M. A. Rould, Q. He, C. O. Pabo, and S. J. Lippard. 1999. Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature. 399:708–712. [DOI] [PubMed] [Google Scholar]
- 50.Zamble, D. B., and S. J. Lippard. 1999. The response of cellular proteins to cisplatin-damaged DNA. In Cisplatin. Chemistry and Biochemistry of a Leading Anticancer Drug. B. Lippert, editor. VHCA, Wiley-VCH, Zürich, Weinheim, Germany. 73–110.
- 51.Kasparkova, J., J. Zehnulova, N. Farrell, and V. Brabec. 2002. DNA interstrand cross-links of the novel antitumor trinuclear platinum complex BBR3464. Conformation, recognition by high mobility group domain proteins, and nucleotide excision repair. J. Biol. Chem. 277:48076–48086. [DOI] [PubMed] [Google Scholar]
- 52.Kasparkova, J., O. Novakova, N. Farrell, and V. Brabec. 2003. DNA binding by antitumor trans-[PtCl2(NH3)(thiazole)]. Protein recognition and nucleotide excision repair of monofunctional adducts. Biochemistry. 42:792–800. [DOI] [PubMed] [Google Scholar]
- 53.Loskotova, H., and V. Brabec. 1999. DNA interactions of cisplatin tethered to the DNA minor groove binder distamycin. Eur. J. Biochem. 266:392–402. [DOI] [PubMed] [Google Scholar]
- 54.Nielsen, P. E. 1990. Chemical and photochemical probing of DNA complexes. J. Mol. Recognit. 3:1–24. [DOI] [PubMed] [Google Scholar]
- 55.Zehnulova, J., J. Kasparkova, N. Farrell, and V. Brabec. 2001. Conformation, recognition by high mobility group domain proteins, and nucleotide excision repair of DNA intrastrand cross-links of novel antitumor trinuclear platinum complex BBR3464. J. Biol. Chem. 276:22191–22199. [DOI] [PubMed] [Google Scholar]
- 56.Kartalou, M., and J. M. Essigmann. 2001. Recognition of cisplatin adducts by cellular proteins. Mutat. Res. 478:1–21. [DOI] [PubMed] [Google Scholar]
- 57.Pil, P. M., and S. J. Lippard. 1992. Specific binding of chromosomal protein-HMG1 to DNA damaged by the anticancer drug cisplatin. Science. 256:234–237. [DOI] [PubMed] [Google Scholar]
- 58.Wei, M., S. M. Cohen, A. P. Silverman, and S. J. Lippard. 2001. Effects of spectator ligands on the specific recognition of intrastrand platinum-DNA cross-links by high mobility group box and TATA-binding proteins. J. Biol. Chem. 276:38774–38780. [DOI] [PubMed] [Google Scholar]
- 59.Kasparkova, J., O. Novakova, Y. Najajreh, D. Gibson, J.-M. Perez, and V. Brabec. 2003. Effects of a piperidine ligand on the mechanism of action of antitumor cisplatin. Chem. Res. Toxicol. 16:1424–1432. [DOI] [PubMed] [Google Scholar]
- 60.Jung, Y. W., and S. J. Lippard. 2003. Nature of full-length HMGB1 binding to cisplatin-modified DNA. Biochemistry. 42:2664–2671. [DOI] [PubMed] [Google Scholar]
- 61.Turner, R. M., N. D. F. Grindley, and C. M. Joyce. 2003. Interaction of DNA polymerase I (Klenow fragment) with the single-stranded template beyond the site of synthesis. Biochemistry. 42:2373–2385. [DOI] [PubMed] [Google Scholar]
- 62.Villani, G., N. T. Le Gac, L. Wasungu, D. Burnouf, R. P. Fuchs, and P. E. Boehmer. 2002. Effect of manganese on in vitro replication of damaged DNA catalyzed by the herpes simplex virus type-1 DNA polymerase. Nucleic Acids Res. 30:3323–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cohen, S. M., and S. J. Lippard. 2001. Cisplatin: from DNA damage to cancer chemotherapy. Prog. Nucleic Acid Res. Mol. Biol. 67:93–130. [DOI] [PubMed] [Google Scholar]
- 64.Stehlikova, K., H. Kostrhunova, J. Kasparkova, and V. Brabec. 2002. DNA bending and unwinding due to the major 1,2-GG intrastrand cross-link formed by antitumor cis-diamminedichloroplatinum(II) are flanking-base independent. Nucleic Acids Res. 30:2894–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Delalande, O., J. Malina, V. Brabec, and J. Kozelka. 2005. Chiral differentiation of DNA adducts formed by enantiomeric analogues of antitumor cisplatin is sequence-dependent. Biophys. J. 88:4159–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malina, J., J. Kasparkova, G. Natile, and V. Brabec. 2002. Recognition of major DNA adducts of enantiomeric cisplatin analogs by HMG box proteins and nucleotide excision repair of these adducts. Chem. Biol. 9:629–638. [DOI] [PubMed] [Google Scholar]
- 67.Fuertes, M. A., J. Castilla, C. Alonso, and J. M. Perez. 2003. Cisplatin biochemical mechanism of action: from cytotoxicity to induction of cell death through interconnections between apoptotic and necrotic pathways. Curr. Med. Chem. 10:257–266. [DOI] [PubMed] [Google Scholar]
- 68.Hoffmann, J.-S., M.-J. Pillaire, D. Garcia-Estefania, S. Lapalu, and G. Villani. 1996. In vitro bypass replication of the cisplatin-d(GpG) lesion by calf thymus DNA polymerase beta and human immunodeficiency virus type I reverse transcriptase is highly mutagenic. J. Biol. Chem. 271:15386–15392. [DOI] [PubMed] [Google Scholar]
- 69.Suo, Z., S. Lippard, and K. Johnson. 1999. Single d(GpG)/cis-diammineplatinum(II) adduct-induced inhibition of DNA polymerization. Biochemistry. 38:715–726. [DOI] [PubMed] [Google Scholar]
- 70.Novakova, O., J. Kasparkova, J. Malina, G. Natile, and V. Brabec. 2003. DNA-protein cross-linking by trans-[PtCl2(E-iminoether)2]. A concept for activation of the trans geometry in platinum antitumor complexes. Nucleic Acids Res. 31:6450–6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marini, V., P. Christofis, O. Novakova, J. Kasparkova, N. Farrell, and V. Brabec. 2005. Conformation, protein recognition and repair of DNA interstrand and intrastrand cross-links of antitumor trans- [PtCl2(NH3)(thiazole)]. Nucleic Acids Res. 33:5819–5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vaisman, A., S. E. Lim, S. M. Patrick, W. C. Copeland, D. C. Hinkle, J. J. Turchi, and S. G. Chaney. 1999. Effect of DNA polymerases and high mobility group protein 1 on the carrier ligand specificity for translesion synthesis past platinum-DNA adducts. Biochemistry. 38:11026–11039. [DOI] [PubMed] [Google Scholar]
- 73.Gibbons, G. R., W. K. Kaufmann, and S. G. Chaney. 1991. Role of DNA replication in carrier-ligand-specific resistance to platinum compounds in L1210 cells. Carcinogenesis. 12:2253–2257. [DOI] [PubMed] [Google Scholar]
- 74.Mamenta, E. L., E. E. Poma, W. K. Kaufmenn, D. A. Delmastro, H. L. Grady, and S. G. Chaney. 1994. Enhanced replicative bypass of platinum-DNA adducts in cisplatin-resistant human ovarian carcinoma cell lines. Cancer Res. 54:3500–3505. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.