

Abstract
Malic enzyme is a tetrameric protein with double dimer quaternary structure. In 3–5 M urea, the pigeon cytosolic NADP+-dependent malic enzyme unfolded and aggregated into various forms with dimers as the basic unit. Under the same denaturing conditions but in the presence of 4 mM Mn2+, the enzyme existed exclusively as a molten globule dimer in solution. Similar to pigeon enzyme (Chang, G. G., T. M. Huang, and T. C. Chang. 1988. Biochem. J. 254:123–130), the human mitochondrial NAD+-dependent malic enzyme also underwent a reversible tetramer-dimer-monomer quaternary structural change in an acidic pH environment, which resulted in a molten globule state that is also prone to aggregate. The aggregation of pigeon enzyme was attributable to Trp-572 side chain. Mutation of Trp-572 to Phe, His, Ile, Ser, or Ala abolished the protective effect of the metal ions. The cytosolic malic enzyme was completely digested within 2 h by trypsin. In the presence of Mn2+, a specific cutting site in the Lys-352-Gly-Arg-354 region was able to generate a unique polypeptide with Mr of 37 kDa, and this polypeptide was resistant to further digestion. These results indicate that, during the catalytic process of malic enzyme, binding metal ion induces a conformational change within the enzyme from the open form to an intermediate form, which upon binding of L-malate, transforms further into a catalytically competent closed form.
INTRODUCTION
Malic enzyme (ME) catalyzes a divalent metal ion (Mn2+- or Mg2+-dependent) reversible oxidative decarboxylation of L-malate to yield CO2 and pyruvate with a concomitant reduction of NAD(P)+ to NAD(P)H (1–3).
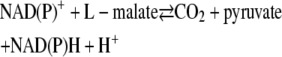 |
All ME have a similar overall double dimer quaternary structure (3) (Fig. 1). Each monomer of the enzyme comprises four structural domains. The active site is located at the interface between domains B and C. Domains A and D, on the other hand, are involved in subunit association and are essential for the overall structural integrity of the enzyme.
FIGURE 1.

Overall quaternary structure and the interfacial regions of pigeon c-NADP-ME. (A) The tetrameric pigeon cytosolic ME (pdb code: 1GQ2) is shown as a surface model that clearly illustrates the double dimer structure of the enzyme. One of the monomers located at the upper left corner is shown in ribbon diagram and illustrates the four structural domains: domain A in green, domain B in blue, domain C in red, and domain D in yellow. In the active site, the transition state analog oxalate (in green) and coenzyme NADP+ are shown in bond form, and the manganese ion is shown as a purple sphere. The unique Trp-572 is shown in bond form within domain D. The figure was generated using PyMOL (37). (B–D) Interfacial regions of c-NADP-ME. (B) A-B interface interactions. (C) A-C interface interactions. (D) A-D interface interactions. Panels B–D were generated using PDBsum (http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/). Hydrogen bonds are shown in cyan, and nonbonded interactions are shown in orange. The width of the striped line is proportional to the number of atomic contacts. The interfacial amino acid residues are colored by residue conservation as shown in the bottom of the figure. Trp-572 is a highly conserved residue among all ME.
Structural studies of human or ascarid mitochondrial NAD+-dependent malic enzyme (m-NAD-ME) have clearly indicated that conformational changes within the enzyme occur after ligand binding (4–8). There are significant structural differences between the open form (ME-NAD+ binary complex) (4) and the closed form (ME-NAD+-Mn2+-oxalate quaternary complex) (5) of m-NAD-ME. In addition, an intermediate form (ME-NAD+-Lu3+ ternary complex) has been observed, which is different from the open and closed forms (6).
The crystal structure of the cytosolic NADP+-dependent malic enzyme (c-NADP-ME) can be superimposed on m-NAD-ME (9). We have provided kinetic and spectroscopic evidence showing that Mn2+ and Lu3+ are able to induce similar slow conformational changes in c-NADP-ME and m-NAD-ME (10,11). These results imply that the enzyme's kinetic properties may be modified by metal ion-induced structural changes. We have also demonstrated the structural role of metal ions in c-NADP-ME (12). A single tryptophanyl residue (Trp-572) is found to be responsible for the stabilization role of metal ions (13). The essential role of this tryptophanyl residue is further delineated in this work. Furthermore, we provide additional evidence demonstrating the existence of a Mn2+-induced open form II and an inherently unstable closed form I, which completes the possible structural changes of ME on binding to its ligands.
MATERIALS AND METHODS
Preparation of recombinant WT and tryptophan mutants of c-NADP-ME
Expression and purification of pigeon c-NADP-ME and human m-NAD-ME were carried out as described previously (14) and are summarized in the Supplementary Material. The purified recombinant enzyme had no tag at the termini and thus had an identical amino acid sequence to that of native ME.
For the Trp-mutant construction, two complementary oligonucleotides containing the desired mutation were designed to flank the unmodified nucleotide sequence (Supplementary Table 1S). These oligonucleotide primers were annealed with the denatured plasmid using the nonstrand-displacing action of Pfu DNA polymerase, which extended and incorporated the mutagenic primers into nicked circular strands. The methylated, nonmutated parental DNA template was digested with Dpn I. The circular, nicked double-stranded DNA was transformed into Top10 F′ supercompetent Escherichia coli cells, which are able to repair the nicks in the plasmid and amplify the plasmid. These recombinant c-NADP-ME mutants were successfully expressed and purified to apparent homogeneity (Supplementary Fig. 1S).
Chemical denaturation and pH jump
For the unfolding of the enzyme, the purified pigeon c-NADP-ME was preincubated with or without ligands and then treated with urea in 30 mM Tris-acetate buffer (pH 7.4) at 25°C for 2 h. Acid-induced unfolding of the human m-NAD-ME was performed by a pH jump method. The pH jump experiments were performed by instantaneous 10-fold dilution of the enzyme solution with buffer at another pH value. The actual pH value was determined by direct measurement of the resulting mixed buffer solution.
Spectrofluorometric analysis
The fluorescence spectra of the enzyme were monitored at 30°C using a Perkin-Elmer (Foster City, CA) LS-50B luminescence spectrometer, and all spectra were corrected for buffer background. The excitation wavelength was set at 280 nm, and the fluorescence emission spectrum was scanned from 300 to 400 nm. The integrated area of the fluorescence spectrum and the average emission wavelength, which registered both a red shift and changes in fluorescence intensity (15), were used to monitor the unfolding process of the enzyme.
Circular dichroism analysis
Circular dichroism (CD) spectra were measured at 30°C with a Jasco (Tokyo, Japan) J-810 spectropolarimeter under constant N2 flush. The enzyme solution was scanned from 250 to 190 nm. When the analysis was completed, the buffer was immediately scanned to obtain the baseline. The unfolded enzyme solution was filtered through a 0.22 μm filter (Whatman, Maidstone, UK) before analysis. Mean residue ellipticity was calculated by DICHROWEB (16).
Sedimentation analysis
The sedimentation coefficient and molecular mass of the enzyme under various conditions were analyzed using a Beckman-Coulter (Palo Alto, CA) XL-A analytical ultracentrifuge (AUC) with an An50Ti rotor. Sedimentation was performed at 20°C and 40,000 rpm in standard double sectors aluminum centerpieces with the buffer contribution corrected automatically in data processing. The ultraviolet absorption of the cells was scanned every 5 min for a period of 2 h. The data were analyzed with the SEDFIT program (17). The solvent density, viscosity, and partial specific volume of the enzyme were calculated by the free software SEDNTERP (http://www.jphilo.mailway.com/). The partial specific volume used for the pigeon c-NADP-ME was 0.7403 (18). All samples were visually checked for clarity after ultracentrifugation.
ANS binding
Binding 1-anilino-8-naphthalene sulfonic acid (ANS) with the partially unfolded enzyme was used and allowed the measurement of the fluorescence enhancement due to ANS. The excitation wavelength was set at 395 nm, at which ANS has an extinction coefficient of 5620 M−1 cm−1. The emission spectrum was collected from 400 nm to 600 nm. The concentration of the enzyme and ANS was 1.5 μM and 100 μM, respectively. Before the experiment, the enzyme was incubated at 25°C for 1 h in various concentrations of urea and then incubated for a further 5 min in ANS. The area of the emission spectra was integrated from 420 to 560 nm. All measurements were corrected against the background intensity of buffer containing ANS.
Tryptic digestion
Hydrolysis of pigeon c-NADP-ME (0.75 mg/ml) by trypsin (25 μg/ml) (Qiagene, Hilden, Germany; sequencing modified) was carried out in 30 mM Tris-acetate, 2 mM 2-mercaptoethanol (pH 7.4) at 37°C. The reaction was stopped at a specific time by adding 0.1 mM tosyllysine chloromethylketone (Sigma, St. Louis, MO). The cleavage patterns were examined by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The tryptic-digested enzyme was also subjected to amino acid sequencing, mass spectrometry, and enzyme activity analyses (Supplementary Material).
RESULTS
Chemical denaturation of the pigeon c-NADP-ME
We previously discovered a characteristic biphasic denaturation of the recombinant pigeon liver c-NADP-ME in urea (12). The enzyme was incubated with different concentrations of urea for 2 h, which allowed the denaturation to reach equilibrium. The CD or average emission wavelength of wild-type (WT) c-NADP-ME showed a highly reproducible biphasic denaturation curve. Inclusion of Mn2+ significantly changed the shape of the unfolding curve (Fig. 2, A and B). A single tryptophanyl residue (Trp-572) was found to be responsible for the characteristic metal protection (13).
FIGURE 2.

Essential role of Trp-572 side chain in the metal ion protective effect. Protection of pigeon c-NADP-ME (A and B, WT; C and D, W572A; E and F, W572F; G and H, W572H; I and J, W572I; and K and L, W572S) by Mn2+ against urea-induced aggregation was monitored by average emission wavelength (panels A, C, E, G, I, and K) or CD (B, D, F, H, J, and L). (Open circles) in the absence of Mn2+; (solid circles) in the presence of 4 mM Mn2+.
To characterize the critical structural role of Trp-572, we constructed various Trp-572 mutants. The CD and fluorescence spectra of all mutants were superimposable with that of WT (not shown). The kinetic parameters were not changed by any of these Trp-572 mutants (Supplementary Table 2S). Mn2+ increased the thermal stability of all the recombinant c-NADP-ME enzymes by 1∼2°C (Supplementary Table 3S, Fig. 2S). These results indicate that mutation at Trp-572 does not change the overall structure of the enzyme. We then examined the unfolding curves of these Trp-572 mutants. All Trp-572 mutants showed a biphasic denaturation curve (Fig. 2, C–L). This characteristic unfolding feature persisted in the presence of Mn2+, confirming that Trp-572 was indeed a unique residue involved in the structural integrity of c-NADP-ME.
Changes in the quaternary structure of pigeon c-NADP-ME after ligand binding during urea denaturation
The above results indicate that c-NADP-ME was unfolded to an intermediate form at ∼3–5 M urea. AUC analysis indicated that this intermediate form was susceptible to aggregation (Supplementary Figs. 3S and 4S). Divalent metal ion-bound enzyme was resistant to aggregation, which suggests that metal ion induces the enzyme to enter a different conformation that prevents the enzyme from aggregating. To explicitly explore the enzyme's ligand-induced conformational changes, the effect of various combinations of ligands on enzyme aggregation was examined.
The native tetrameric ME can be shown to be a single species with sedimentation coefficient of 9S–10S (Fig. 3 A). The enzyme aggregated after 4 M urea treatment (Fig. 3 B). The various peaks suggest that dimers, tetramers, 6-mers, 8-mers, 10-mers etc. are formed. The enzyme remained aggregated in 4 M urea in the presence of L-malate (Fig. 3 C), NADP+ (Fig. 3 E), or both ligands (Fig. 3 H). This confirmed the existence of an unstable ME-NADP+-malate closed form I as predicted previously (3). Only the presence of metal ions was able to prevent aggregation (Fig. 3, D, F, G, and I). In the presence of Mn2+, most of the enzyme molecules existed in the dimeric form with a sedimentation coefficient of 6S. Lu3+, which has previously been used to create a metal-containing crystal structure of ME (6), gave a similar result to Mn2, albeit less effectively (Fig. 3 J). Thus the metal ion containing open form II is also confirmed.
FIGURE 3.

Effect of ligands on the quaternary structure of pigeon c-NADP-ME during urea denaturation. (A) Sedimentation velocity pattern of c-NADP-ME (0.75 mg/ml) in 30 mM Tris-acetate buffer, pH 7.4, contains 2 mM 2-mercaptoethanol. (B–J) The same concentration of c-NADP-ME was preincubated with (B) 30 mM Tris-acetate buffer/2 mM 2-mercaptoethanol (pH 7.4) only, no ligand added; (C) 5 mM malate; (D) 4 mM Mn2+; (E) 0.23 mM NADP+; (F) 5 mM malate, 4 mM Mn2+, plus 0.23 mM NADP+; (G) 5 mM malate plus 4 mM Mn2+; (H) 5 mM malate plus 0.23 mM NADP+; (I) 4 mM Mn2+ plus 0.23 mM NADP+; and (J) 60 mM Lu3+; and then treated with 4 M urea at 25°C for 2 h. The absorption signal at 280 nm of the enzyme was recorded. Some of the panels have an enlarged y axis scale to highlight the distribution of the various aggregational forms. The possible aggregational forms of the enzyme are highlighted in panels B and H. The fitting quality of these AUC data is shown in Supplementary Fig. 5S.
Exposure of the hydrophobic region of WT and W572F c-NADP-ME during urea denaturation
The above results indicated that, in the presence of Mn2+, pigeon c-NADP-ME exists exclusively as a dimer and that this has a different conformation to the open form. This metal ion involved conformation had a propensity to aggregate in the absence of metal ion. We then examined ANS binding under these conditions. ANS is a useful probe to detect the exposure of hydrophobic regions within the protein. The emission fluorescence of ANS is greatly enhanced when it interacts with exposed hydrophobic regions of a protein. Fig. 4 showed that there was a pronounced enhancement of ANS fluorescence at 3–5 M urea, which corresponds to the urea concentration at which the enzyme has a propensity to aggregate. Using WT enzyme, at a urea concentration <4 M, the ANS fluorescence area decreased noticeably in the presence of Mn2+, indicating that there was a different amount of hydrophobic area exposure after Mn2+ binding. This metal effect could not be observed with the mutant enzyme W572F (Fig. 4).
FIGURE 4.

ANS binding of pigeon c-NADP-ME during urea denaturation. The exposure of the hydrophobic regions of the enzyme in urea was examined using the integral fluorescence area in the presence of ANS; this was done either in the absence of Mn2+ (open circles, WT; open triangles, W572F) or in the presence of 4 mM Mn2+ (solid circles, WT; solid triangles, W572F).
Tryptic digestion patterns of pigeon c-NADP-ME in sodium dodecylsulfate-polyacrylamide gel electrophoresis
To further explore the enzyme's ligand-induced conformational changes, we examined the susceptibility of the enzyme to trypsin digestion under various conditions (Fig. 5). In the absence of any ligand, ME was completely digested in 4 h (Fig. 5 A). A similar digestion pattern was observed for the enzyme in the presence of NADP+ albeit with a slightly slower rate (Fig. 5 D). Malate provided ME with substantial protection against trypsin digestion (Fig. 5 B). The most prominent finding was that when the enzyme was preincubated with Mn2+ and then treated with trypsin, a unique digestion pattern was obtained (Fig. 5 C). A new polypeptide band with an Mr of ∼37 kDa appeared, and this was resistant to further digestion. The digestion pattern of ME bound with Mn2+ and malate (Fig. 5 F) was similar to that of Mn2+ only. On the other hand, inclusion of NADP+ with the Mn2+/malate form of the enzyme masked this unique cutting site (Fig. 5, E and H).
FIGURE 5.

Effect of ligands on the tryptic digestion of pigeon c-NADP-ME. The tryptic digestion pattern of c-NADP-ME (0.75 mg/ml) preincubated with (A) 30 mM Tris-acetate buffer/2 mM 2-mercaptoethanol (pH 7.4) only; (B) 5 mM malate; (C) 4 mM Mn2+; (D) 0.23 mM NADP+; (E) 5 mM malate, 4 mM Mn2+, plus 0.23 mM NADP+; (F) 5 mM malate plus 4 mM Mn2+; (G) 5 mM malate plus 0.23 mM NADP+; (H) 4 mM Mn2+ plus 0.23 mM NADP+; (I) 60 mM Lu3+; (J) 60 mM Lu3+ plus 0.23 mM NADP+; (K) 5 mM malate plus 60 mM Lu3+; (L) 5 mM malate, 60 mM Lu3+, plus 0.23 mM NADP+; and then treated with trypsin (25 μg/ml) in the same buffer at 37°C. Aliquot of the digested samples were withdrawn at 0, 1, 2, 3, and 4 h and analyzed by SDS-PAGE. The 0 h means an immediately withdrawn sample after the enzyme was mixed with trypsin, but digestion may have already taken place for 20–30 s. “M” indicates the molecular mass markers, which have sizes of 97, 66, 45, 30, 20.1, and 14.4 (in I–L) kDa.
Since the crystal structure of m-NAD-ME as a ternary complex with NAD+ and the lanthanide ion (Lu3+) is available (6), and Lu3+ is known to induce a similar structural change that creates a slow binding inhibition of both m-NAD-ME (10) and c-NADP-ME (11), we checked the trypsin digestion pattern when Lu3+ replaced Mn2+. As expected, preincubation of c-NADP-ME with Lu3+ gave a similar tryptic digestion pattern to Mn2+ (Fig. 5, I–L).
Identification of amino acid residue susceptible to tryptic digestion
Since pigeon c-NADP-ME was completely digested by trypsin (Fig. 5), there must be many exposed trypsin cutting sites. All these peptides were small and ran off the SDS-PAGE gel. The only exception was when the metal ion prevented further digestion of a polypeptide of 37 kDa as demonstrated in Fig. 5, C and F. We isolated and identified the N-terminal amino acid sequence of this polypeptide and found it was identical to the N-terminus of c-NADP-ME. Determination of molecular mass of this polypeptide by matrix-assisted laser desorption ionization-time of flight gave values of 37060.3 and 37080.2 Da (Supplementary Fig. 6S). These mass values and the known specificity of trypsin pointed to peptide cleavage at either 352 (lysine) or 354 (arginine) region. This N-terminal half of the ME was resistant to further trypsin digestion.
In accordance with the trypsin digestion results, complete digested ME lost almost all enzymatic activity in 3 h at 37°C (Supplementary Fig. 7S). NADP+ provided no protective effect. Malate or Mn2+, on the other hand, provided substantial protection upon tryptic digestion (Fig. 7S).
Structural changes of human m-NAD-ME in acidic environment
We have previously demonstrated an acidic pH-induced reversible tetramer-dimer-monomer process for pigeon c-NADP-ME (19). Acid-induced protein conformational change to a molten globule state seems to be a general phenomenon (20–22). We thus examined the structural changes of human m-NAD-ME at various pH values.
The CD spectra of m-NAD-ME at pH > 3.88 could be almost superimposed (Fig. 6 A). A significant CD spectrum change was only observed at a pH < 2.87, indicating that secondary structure of the enzyme did not change very much until a pH of 2.87 was reached. In contrast to the CD spectra, significant fluorescence spectral changes were observed over the pH range 2.87–7.2 (Fig. 6 B). This reflected significant enzyme tertiary structural changes, characteristic for a molten globule state.
FIGURE 6.

Spectral changes of human m-NAD-ME at different pH values. (A) CD spectra of the enzyme at different pH values. (Solid circles) pH 2.87; (solid squares) pH 3.88; (solid triangles) pH 4.47; (open circles) pH 5.07; (open squares) pH 6.07; (open triangles) pH 7.20. (B) Fluorescence emission spectra of the enzyme at different pH values. From top to bottom, the pH values are 2.87, 3.88, 4.47, 5.07, 6.07, and 7.20, respectively. (C), Fluorescence spectral changes of m-NAD-ME at different pH values. (Solid triangles) average emission wavelength; (solid circles) integrated fluorescence area.
The fluorescence spectral changes are presented as the integrated emission fluorescence area and average emission wavelength changes. Both changes showed a sharp change between pH 3.5 and pH 4. Furthermore, the average emission wavelength change showed a positive posttransition slope between pH 4 and pH 7, whereas the integrated fluorescence area change showed another clear transition between pH 5 and pH 7 (Fig. 6 C).
The above results are reminiscent of the acid-induced dissociation of c-NADP-ME (19) and strongly suggested that m-NAD-ME underwent a tetramer-dimer-monomer dissociation in the acidic environment. The quaternary structural changes of m-NAD-ME over these pH ranges were therefore examined by analytical ultracentrifugation.
Excellent AUC data were obtained for m-NAD-ME at all the pH values. Some of the typical AUC results are shown in the Supplementary Material (Figs. 8S–11S). AUC provided a very reliable tool for analyzing the enzyme quaternary structure. Fig. 7 shows the continuous sedimentation velocity patterns of m-NAD-ME at different pH values and at three protein concentrations. At neutral pH, all enzyme molecules existed as a tetramer with a sedimentation coefficient of between 9 and 10 (Fig. 7 A). At pH 5.19, corresponding to the first transition as detected by the integrated fluorescence area (Fig. 6 C), the tetramer started to dissociate into dimers and monomers (Fig. 7 B). At pH 3.88, corresponding to the second transition as detected by the average emission wavelength and integrated fluorescence area (Fig. 6 C), the enzyme aggregated and this process was protein concentration dependent (Fig. 7 C). Aggregation was visible at high protein concentrations. However, when the pH was further lowered to 2.87, the aggregated protein became soluble (Fig. 7 D).
FIGURE 7.

Quaternary structural changes of human m-NAD-ME in different pH environments. Sedimentation velocity analysis of the enzyme at different pH values and at three protein concentrations is shown. The pH values are 7.20, 5.19, 3.88, and 2.87, respectively, for panels A, B, C, and D.
The quaternary structural changes of the enzyme at different pHs were analyzed in detail (Supplementary Figs. 12S–15S). A reversible tetramer-dimer-monomer process occurred in a moderate acidic environment, but the changes become irreversible at a pH below 3.58. Divalent metal ion provided substantial protection against the irreversible aggregation.
Identification of a molten globule state of human m-NAD-ME in an acidic environment
The acid-induced aggregation-prone form of human m-NAD-ME was examined in terms of its binding ability with ANS. A drastic increase in the fluorescence of the enzyme was found at pH 2.87 and 3.88 (Fig. 8 A). The detailed ANS binding to m-NAD-ME was examined over the whole pH range, and a transition was found to occur at exactly the same point as aggregation (Fig. 8 B).
FIGURE 8.

ANS binding of human m-NAD-ME. m-NAD-ME (1 μM, 0.03 mg/ml) was preincubated with 100 mM ANS at pH 2.87 (curve a), 3.88 (curve b), 5.07 (curve c), 6.07 (curve d), and 7.20 (curve e) for 10 min at 25°C. (A) The emission fluorescence spectra of ANS were monitored using an excitation wavelength at 395 nm. All spectra were corrected for the presence of ANS using a buffer blank. (B) Correlation of the integrated ANS fluorescence area with pH.
DISCUSSION
The protein unfolding-refolding process constitutes one of the most challenging areas in the life sciences (see some recent reviews (23–27)). For proteins with multiple subunits and multiple structural domains like ME, the denaturation curves are usually complex and involve multiphasic-unfolding phenomena. It is necessary to characterize each proposed unfolding intermediate in terms of quaternary structure to distinguish between a true unfolding intermediate and an aggregated species.
In this report, we have demonstrated that the unfolding of pigeon c-NADP-ME by urea and human m-NAD-ME by pH jump involves a common intermediate, the molten globule state. This intermediate state may represent a structurally inherent unstable conformation, and divalent metal ion plays an important role in stabilizing this structure. Fig. 9 summarizes the suggested quaternary structural changes of ME perturbed by urea or pH jump.
FIGURE 9.

Quaternary structural changes of ME. (A) A working model for the structural changes of ME in urea solution or in an acidic environment. The enzyme exists as a tetramer in buffer at neutral pH and starts to dissociate into dimers at 1–2 M urea or at pH 6 and then finally into monomers at 2 M urea or pH 5. A further increase of urea concentration to 3–5 M or decrease in the pH to 4.5 causes an overall structural change to a molten globule state, which has the propensity to aggregate that shunts the enzyme into a divergent misfolding pathway (off-pathway). Metal ions at this point can play a pivotal role in directing the correct refolding (on-pathway) when the structure of ME is perturbed to the folded-misfolded crossroads. (B) Hypothetical potential energy changes of the enzyme in increasing urea solution or different pH environments. Divalent metal ions are shown to bind with the molten globule state and thus prevent aggregation of the enzyme.
Numerous crystal structures for the human m-NAD-ME have been previously reported (3). Several lines of evidence suggest that MEs from different sources have similar structural properties. First, the overall structures of all MEs are similar (3), even though m-NAD-ME has extra fumarate and nucleotide binding sites at the dimer and tetramer interfaces, respectively, that endow m-NAD-ME with complex regulatory properties (28–32). Second, identical reversible tetramer-dimer-monomer dissociation in acidic environment was found for pigeon c-NADP-ME (19) and human m-NAD-ME (this work). Third, the identical inhibition pattern of lutetium ion was found for pigeon c-NADP-ME and human m-NAD-ME (6). Finally, an identical slow binding phenomenon was observed with metal ion for both pigeon c-NADP-ME (10) and human m-NAD-ME (11). We thus use the various crystal structures of human m-NAD-ME to elucidate the structural changes between different forms of ME (Fig. 10).
FIGURE 10.

Ligand-induced structural changes of ME. Interconversion of the open form I (pdb code: 1QR6), open form II (pdb code: 1PJL), closed form II (pdb code: 1DO8), and the putative closed form I. (A) Superimposition of the open form I (in blue) and open form II (in green) showing the tetramer reorganization and the specific trypsin-cutting site exposure upon binding of metal ion (in purple sphere). (B) Superimposition of the open form II (in gray) and closed form II (in red) showing the active site closure after substrate binding (here in the figure is the transition state analog oxalate in the green sphere). The superimposition was generated by Swiss-PDBViewer v. 3.7 (38). (C) Zoom-in area showing the tetramer reorganization. (D) Zoom-in area showing the exposure of the specific cutting site in the open form II. (E) Zoom-in area showing the closure of active site. (F) Proposed model for the structural interconversion of ME. The enzyme binds the nucleotide first forming open form I. Mn2+ and L-malate can then be bound in any order forming open form II by tetramer reorganization or closed form I by the active-site closure. Without Mn2+, close form I (the putative molten globule state) is inherently unstable and is prone to divergence into aggregation (off-pathway). Mn2+ plays a protective role by helping the enzyme enter the correct pathway (on-pathway) leading to its catalytically competent closed form II.
The metal binding site constitutes the central part of the active center of ME. This metal site is located at the interface of the two central β-sheets of domains B and C. Binding the metal ion to the enzyme can induce rearrangement at the interfacial regions and thus affects subunit association affinity (6). The structural basis for the metal-induced open form II of ME is obvious from the crystal structure of the ME-NAD+-Lu3+ ternary complex. This involves rigid-body movements of residues in domain A and domain D, which are located at the dimer and tetramer interface, respectively (Figs. 1 A and 10 A). Interestingly, the tetramer organization observed for the ternary complex is essentially the same as that of the quaternary complex with NAD+, Mn2+, and oxalate (Fig. 10 B). The amino acid residues in domain C relative to domain B of the ternary complex (the open form II) are similar to those in the binary complex (open form I), and the active site region is open to the solvent, which is different to the quaternary complex (closed form II) (Fig. 10). All available structural information indicates that the major effect of L-malate and NAD(P)+ binding to the enzyme is a change in the subdomain structure but not the subunit interface (3,5). It is conceivable that only metal ions are able to induce specific alterations in the quaternary structure.
Analysis of the Protein Data Bank (pdb) file of the pigeon c-NADP-ME (1GQ2.pdb) reveals that the major subunit interactions involve hydrophobic contacts and a few hydrogen bonds. No salt bridge or disulfide bond is found in any subunit interface (Fig. 1, B–D). There are a total of 215 nonbonded contacts and 14 hydrogen bonds in the A-B interface, 100 nonbonded contacts and 4 hydrogen bonds in the A-C interface, and 131 nonbonded contacts and 7 hydrogen bonds in the A-D interface. Trp-572 at domain D is not involved in the A-B interface but contributes 8 nonbonded contacts with Leu-42, His-51, Phe-58, and Pro-56 in the A-C interface and 13 nonbonded contacts with Thr-136, His-138, and Asp-139 in the A-D interface. Trp-572 is ∼50 Å away from any of the catalytic Mn2+ in subunit A, B, or C. It is unclear at the present time whether there is a second metal binding site that plays the structural role in ME. It would be useful to examine the metal per subunit stoichiometry to determine if there might exist a previously unrecognized metal site to explain the impact of mutation to Trp-572.
This work shows that a unique trypsin digestion pattern of ME with Mn2+ or Lu3+ (Fig. 5) can be identified. Lu3+ and Mn2+ gave a similar protective effect on urea-induced unfolding (Fig. 3, D and J). We have demonstrated that AUC is an essential tool in the characterization of the folding-unfolding process. Our results confirmed that the binding of L-malate or Mn2+ is able to induce different structural changes within the enzyme. The metal ion transformed the putative closed form I to closed form II. Without Mn2+, closed form I is inherently unstable and possesses all the criteria that define the generally observed protein unfolding intermediate, a molten globule state (20–22). These are as follows: 1), that the secondary structure is maintained, whereas there are significant tertiary structural changes; 2), that the exposed hydrophobic regions can be accessed and show ANS binding (Fig. 4); and 3), that due to exposure of the hydrophobic regions, this form is prone to aggregate as detected by AUC (Fig. 3).
The ability of metal ions to stabilize the urea-induced partially unfolded form supports the hypothesis that metal ions have an impact on the quaternary structure of ME. In the absence of metal ion, the closed form I is prone to aggregation. The asymmetric unit that forms the closed form ME (1DO8.pdb) contains two copies of the macromolecules. Each independent copy consists of a dimeric assembly. The dimeric form is likely to be the biological unit of the quaternary structure that makes up the catalytically competent ME. The crystal structure of the ME-NAD+-Lu3+ complex is packed as an octamer (1PJL.pdb), and the cytosolic ME-NADP+-Mn2+-oxalate complex has been solved as a 16-mer with a molecular weight of 1 million (1GQ2.pdb). Our previous cross-linking results also indicate the presence of larger aggregates of the enzyme (33), and these results generally agree with the previously observed aggregational states of ME (34).
On the basis of the above discussion, the structural interconversion of ME that has been proposed previously (3) can be expanded to include a divergence into aggregation of closed form I (off-pathway). The ME structural changes are summarized in Fig. 10 F. ME follows a sequential bi-ter kinetic mechanism with malate binding last (35). Binding of ME with nucleotide forms the open form I. The metal ion transforms the open form I enzyme into open form II, which, in the presence of bound L-malate, changes into the catalytic competent closed form II. In the absence of metal ion, the enzyme is induced by malate to form an inherently unstable closed form I. Thus, metal ions play an important protective role with this enzyme and direct the correct pathway (on-pathway) that the structure of ME should follow when it is perturbed; this occurs when the enzyme stands at the folded-misfolded-unfolded crossroads.
Significantly, aggregation of ME does not occur in vivo under normal conditions because of the protective role of divalent metal ions, which stabilize the molten globule state of the enzyme. The concentration of Mg2+ in vivo is usually ∼5–8 mM (36). It is striking that divalent metal ions, which are essential for catalysis, also have a tremendous effect on structure stability. The dual functional roles of metal ions in ME demonstrate the intricate nature of the role of metal ions in biological systems.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Prof. Ralph Kirby for critically reading the manuscript.
This work was supported by the National Science Council, Republic of China (NSC 95-2320-B-010-0523-MY3).
The amino acid sequence numbering system of human m-NAD-ME was used.
Editor: Heinrich Roder.
References
- 1.Frenkel, R. 1975. Regulation and physiological functions of malic enzymes. Curr. Top. Cell. Regul. 9:157–181. [DOI] [PubMed] [Google Scholar]
- 2.Hsu, R. Y. 1982. Pigeon liver malic enzyme. Mol. Cell. Biochem. 43:3–26. [DOI] [PubMed] [Google Scholar]
- 3.Chang, G. G., and L. Tong. 2003. Structure and function of malic enzymes, a new class of oxidative decarboxylases. Biochemistry. 42:12721–12733. [DOI] [PubMed] [Google Scholar]
- 4.Xu, Y., G. Bhargava, H. Wu, G. Loeber, and L. Tong. 1999. Crystal structure of human mitochondrial NAD(P)+-dependent malic enzyme: a new class of oxidative decarboxylases. Structure. 7:877–889. [PubMed] [Google Scholar]
- 5.Yang, Z., D. L. Floyd, G. Loeber, and L. Tong. 2000. Structure of a closed form of human malic enzyme and implications for catalytic mechanism. Nat. Struct. Biol. 7:251–257. [DOI] [PubMed] [Google Scholar]
- 6.Yang, Z., R. Batra, D. L. Floyd, H. C. Hung, G. G. Chang, and L. Tong. 2000. Potent and competitive inhibition of malic enzymes by lanthanide ions. Biochem. Biophys. Res. Commun. 274:440–444. [DOI] [PubMed] [Google Scholar]
- 7.Coleman, D. E., G. S. Rao, E. J. Goldsmith, P. F. Cook, and B. G. Harris. 2002. Crystal structure of the malic enzyme from Ascaris suum complexed with nicotinamide adenine dinucleotide at 2.3 Å resolution. Biochemistry. 41:6928–6938. [DOI] [PubMed] [Google Scholar]
- 8.Rao, G. S., D. E. Coleman, W. E. Karsten, P. F. Cook, and B. G. Harris. 2003. Crystallographic studies on Ascaris suum NAD-malic enzyme bound to reduced cofactor and identification of an effector site. J. Biol. Chem. 278:38051–38058. [DOI] [PubMed] [Google Scholar]
- 9.Yang, Z., H. Zhang, H. C. Hung, C. C. Kuo, L. C. Tsai, H. S. Yuan, W. Y. Chou, G. G. Chang, and L. Tong. 2002. Structural studies of the pigeon cytosolic NADP+-dependent malic enzyme. Protein Sci. 11:332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hung, H. C., G. G. Chang, Z. Yang, and L. Tong. 2000. Slow binding of metal ions to pigeon liver malic enzyme: a general case. Biochemistry. 39:14095–14102. [DOI] [PubMed] [Google Scholar]
- 11.Kuo, C. W., H. C. Hung, L. Tong, and G. G. Chang. 2004. Metal-induced reversible structural interconversion of human mitochondrial NAD(P)+-dependent malic enzyme. Proteins. 54:404–411. [DOI] [PubMed] [Google Scholar]
- 12.Chang, H. C., W. Y. Chou, and G. G. Chang. 2002. Effect of metal binding on the structural stability of pigeon liver malic enzyme. J. Biol. Chem. 277:4663–4671. [DOI] [PubMed] [Google Scholar]
- 13.Chang, H. C., and G. G. Chang. 2003. Involvement of single residue tryptophan 548 in the quaternary structural stability of pigeon cytosolic malic enzyme. J. Biol. Chem. 278:23996–24002. [DOI] [PubMed] [Google Scholar]
- 14.Chou, W. Y., S. M. Huang, and G. G. Chang. 1997. Functional roles of the N-terminal amino acid residues in the Mn(II)-L-malate binding and subunit interactions of pigeon liver malic enzyme. Protein Eng. 10:1205–1211. [DOI] [PubMed] [Google Scholar]
- 15.Royer, C. A., C. J. Mann, and C. R. Matthews. 1993. Resolution of the fluorescence equilibrium unfolding profile of trp aporepressor using single tryptophan mutants. Protein Sci. 2:1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitmore, L., and B. A. Wallace. 2004. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32:W668–W673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuck, P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78:1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nevaldine, B. H., A. R. Bassel, and R. Y. Hsu. 1974. Mechanism of pigeon liver malic enzyme. Biochim. Biophys. Acta. 336:283–293. [DOI] [PubMed] [Google Scholar]
- 19.Chang, G. G., T. M. Huang, and T. C. Chang. 1988. Reversible dissociation of the catalytically active subunits of pigeon liver malic enzyme. Biochem. J. 254:123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ptitsyn, O. B. 1995. Molten globule and protein folding. Adv. Protein Chem. 47:83–229. [DOI] [PubMed] [Google Scholar]
- 21.Kuwajima, K. 1996. The molten globule state of alpha-lactalbumin. FASEB J. 10:102–109. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura, H. 1996. Roles of electrostatic interaction in proteins. Q. Rev. Biophys. 29:1–90. [DOI] [PubMed] [Google Scholar]
- 23.Foguel, D., and J. L. Silva. 2004. New insights into the mechanisms of protein misfolding and aggregation in amyloidogenic diseases derived from pressure studies. Biochemistry. 43:11361–11370. [DOI] [PubMed] [Google Scholar]
- 24.Daggett, V., and A. Fersht. 2003. Is there a unifying mechanism for protein folding? Trends Biochem. Sci. 28:18–25. [DOI] [PubMed] [Google Scholar]
- 25.Jahn, T. R., and S. E. Radford. 2005. The Yin and Yang of protein folding. FEBS J. 272:5962–5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindorff-Larsen, K., P. Rogen, E. Paci, M. Vendruscolo, and C. M. Dobson. 2005. Protein folding and the organization of the protein topology universe. Trends Biochem. Sci. 30:13–19. [DOI] [PubMed] [Google Scholar]
- 27.Oliveberg, M., and E. I. Shakhnovich. 2006. Folding and binding: the conformational repertoire of proteins: folding, aggregation and structural recognition. Curr. Opin. Struct. Biol. 16:68–70. [DOI] [PubMed] [Google Scholar]
- 28.Yang, Z., C. W. Lanks, and L. Tong. 2002. Molecular mechanism for the regulation of human mitochondrial NAD(P)+-dependent malic enzyme by ATP and fumarate. Structure. 10:951–960. [DOI] [PubMed] [Google Scholar]
- 29.Hsu, W. C., H. C. Hung, L. Tong, and G. G. Chang. 2004. Dual functional roles of ATP in the human mitochondrial malic enzyme. Biochemistry. 43:7382–7390. [DOI] [PubMed] [Google Scholar]
- 30.Hung, H. C., M. W. Kuo, G. G. Chang, and G. Y. Liu. 2005. Characterization of the functional role of allosteric site residue Asp102 in the regulatory mechanism of human mitochondrial NAD(P)+-dependent malate dehydrogenase (malic enzyme). Biochem. J. 392:39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hung, H. C., Y. C. Chien, J. Y. Hsieh, G. G. Chang, and G. Y. Liu. 2005. Functional roles of ATP-binding residues in the catalytic site of human mitochondrial NAD(P)+-dependent malic enzyme. Biochemistry. 44:12737–12745. [DOI] [PubMed] [Google Scholar]
- 32.Karsten, W. E., J. E. Pais, G. S. Rao, B. G. Harris, and P. F. Cook. 2003. Ascaris suum NAD-malic enzyme is activated by L-malate and fumarate binding to separate allosteric sites. Biochemistry. 42:9712–9721. [DOI] [PubMed] [Google Scholar]
- 33.Chang, G. G., T. M. Huang, S. M. Huang, and W. Y. Chou. 1994. Dissociation of pigeon-liver malic enzyme in reverse micelles. Eur. J. Biochem. 225:1021–1027. [DOI] [PubMed] [Google Scholar]
- 34.Chang, G. G., T. M. Huang, H. J. Lee, W. Y. Chou, and T. C. Chang. 1994. Pigeon liver malic enzyme. Quaternary structure and reversible dissociation of the subunits. Biotechnol. Appl. Biochem. 19:17–29. [Google Scholar]
- 35.Hsu, R. Y., H. A. Lardy, and W. W. Cleland. 1967. Pigeon liver malic enzyme. V. Kinetic studies. J. Biol. Chem. 242:5315–5322. [PubMed] [Google Scholar]
- 36.Alberts, B., D. Bray, J. Lewis, M. Raff, K. Roberts, and J. D. Watson. 1983. Molecular Biology of the Cell. Garland, New York. 286.
- 37.DeLano, W. L. 2002. The PyMOL Molecular Graphic System. DeLano Scientific, San Carlos, CA.
- 38.Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 18:2714–2723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.