

Abstract
Ca-calmodulin-dependent protein kinase II (CaMKII) was recently shown to alter Na+ channel gating and recapitulate a human Na+ channel genetic mutation that causes an unusual combined arrhythmogenic phenotype in patients: simultaneous long QT syndrome and Brugada syndrome. CaMKII is upregulated in heart failure where arrhythmias are common, and CaMKII inhibition can reduce arrhythmias. Thus, CaMKII-dependent channel modulation may contribute to acquired arrhythmic disease. We developed a Markovian Na+ channel model including CaMKII-dependent changes, and incorporated it into a comprehensive myocyte action potential (AP) model with Na+ and Ca2+ transport. CaMKII shifts Na+ current (INa) availability to more negative voltage, enhances intermediate inactivation, and slows recovery from inactivation (all loss-of-function effects), but also enhances late noninactivating INa (gain of function). At slow heart rates, with long diastolic time for INa recovery, late INa is the predominant effect, leading to AP prolongation (long QT syndrome). At fast heart rates, where recovery time is limited and APs are shorter, there is little effect on AP duration, but reduced availability decreases INa, AP upstroke velocity, and conduction (Brugada syndrome). CaMKII also increases cardiac Ca2+ and K+ currents (ICa and Ito), complicating CaMKII-dependent AP changes. Incorporating ICa and Ito effects individually prolongs and shortens AP duration. Combining INa, ICa, and Ito effects results in shortening of AP duration with CaMKII. With transmural heterogeneity of Ito and Ito downregulation in heart failure, CaMKII may accentuate dispersion of repolarization. This provides a useful initial framework to consider pathways by which CaMKII may contribute to arrhythmogenesis.
INTRODUCTION
Cardiac sodium (Na+) channels control cardiac excitability and the velocity of impulse propagation by initiating the action potential (AP). Different disorders in heart excitability have been related to derangement of the cardiac Na+ channel (1). A number of inherited diseases associated with mutations in SCN5A, the gene encoding the α-subunit of the cardiac Na+ channel, have been discovered and linked to long QT type 3 (LQT3) and Brugada (BrS) syndromes, conduction diseases, and structural defects. Notably, mutations showing overlapping phenotypes have been characterized. A single human mutation (1795InsD) in SCN5A is linked to simultaneous LQT3 and BrS features. This 1795InsD mutation in humans has been studied in expressed Na+ channels (2). The mutant Na+ current (INa) exhibits increased intermediate inactivation and slowed recovery from inactivation, and the mutant Na+ channel availability is shifted to more negative potentials, all of which would reduce INa (loss of function) and could explain Brugada-like symptoms in patients at faster heart rates. In addition, this 1795InsD mutation causes persistent INa, which fails to completely inactivate during long-duration APs. The persistent INa would produce a gain of function, and that could contribute to AP prolongation and LQT syndrome. This would be especially pronounced at slow heart rates where INa recovery from inactivation may be more complete (even with the mutation) and where the longer intrinsic AP duration (APD) would increase the impact of the late INa.
Na+ channel gating may also be altered in acquired diseases, e.g., drug-induced LQT syndrome, cardiac ischemia, and heart failure (HF). In HF, altered Na+ channel regulation could cause widespread acquired Na+ channel dysfunction and arrhythmogenesis. For example, Ca-calmodulin-dependent protein kinase II (CaMKII) is upregulated and also more active in HF (3). Wagner et al. (4) recently showed that CaMKII alters cardiac myocyte Na+ channel gating in a manner very similar to the combined LQT/Brugada genetic phenotype mentioned above (1795InsD). Adenovirus-mediated (acute) and transgenic (chronic) overexpression of the cytosolic δ isoform (CaMKIIδc) shifted voltage dependence of Na+ channel availability to more negative membrane potentials, enhanced intermediate inactivation, and slowed recovery from inactivation (and these effects were prevented by CaMKII inhibitors). In addition, CaMKIIδc markedly increased persistent (late) inward INa and intracellular [Na+] ([Na+]i). From these alterations, one might expect less Na+ channel availability at fast heart rates but more inward INa during long APs at slow heart rates.
The increased levels of CaMKII in HF may target several myocyte proteins (5). CaMKII phosphorylates Ca2+ transport proteins such as phospholamban (6), ryanodine receptors (7–9), and L-type Ca2+ channels (8,10,11). In addition, CaMKII can also modulate K+ channels (12–15). CaMKII is known to mediate Ca2+-dependent ICa facilitation (8,10,11), and Kohlhaas et al. (8) showed that CaMKII overexpression in rabbit myocytes increases peak ICa and slows inactivation (possibly related to ICa facilitation). CaMKII can also modulate transient outward K+ current (Ito). Wagner et al. (15) showed that upon CaMKII overexpression, the expression levels of Kv1.4 channels (responsible for the slow component of Ito) are enhanced, as is the current recovery from inactivation. Given the multiple CaMKII targets, it is difficult a priori to discern the relative effects of CaMKII modulation of INa, ICa, and Ito on ventricular myocyte APs. In this study, we simulate the CaMKII-induced alterations of INa, ICa, and Ito (individually and collectively) on the rabbit ventricular AP.
Mathematical models have been widely used to simulate the voltage-dependent gating of ion channels. Composite models of ion channels and excitation-contraction coupling (ECC) allow investigation of the effects of altered Na+ channel kinetic properties on myocyte electrical activity (16–18). Clancy and Rudy (16) showed that a theoretical AP model could simulate the 1795InsD mutant current behavior by including an extended formulation of their Markovian Na+ channel structure that incorporates a bursting mode. Here, we used a variation of this Markovian Na+ channel model to simulate the CaMKIIδc effects on INa reported by Wagner et al. (4) and we incorporate this into a comprehensive AP and ECC model of rabbit ventricular myocytes (19). We also incorporated CaMKII-dependent effects on ICa and Ito in rabbit ventricular myocytes to distinguish the independent and combined effects of these CaMKII targets on APD.
METHODS
Na+ current model simulation
The Markov model structure (Fig. 1) implemented in this study is based on that proposed by Clancy and Rudy (16,17). It contains two modes of gating, a background mode and a burst mode. The background mode reflects the normal sequence of activation and inactivation that most channels undergo upon depolarization and contributes predominantly to the large transient component of INa. The burst mode reflects a small population of channels that transiently fail to inactivate during the AP plateau and results in a sustained inward current. The background mode includes the nine states in the upper two rows, consisting of three closed states (C3, C2, and C1), a conducting open state (O), a fast inactivation state (IF), and two intermediate inactivation states (IM1 and IM2) that are required to reproduce the complex fast and slow recovery features of INa inactivation. The lower four states in Fig. 1 correspond to the burst or late mode (prefix L), in which inactivation does not occur. Voltage (Vm)-independent transition rates between upper and lower states represent a probability of transition between the two modes of gating.
FIGURE 1.

Markov model of the cardiac Na+ channel. The channel model contains background (upper nine states) and burst (lower four states) gating modes. The burst mode reflects a population of channels that transiently fail to inactivate and accounts for a sustained inward current (or late current).
Table 1 shows the expressions of the transition rates between the Markov model states. The corresponding parameters were identified by a fitting procedure to simulate the electrophysiological characterization of cardiac INa in βGal and in CaMKIIδc-overexpressing rabbit myocytes reported by Wagner et al. (4). The following voltage-clamp protocols were simulated for parameter identification: activation, steady-state inactivation, intermediate inactivation, recovery from inactivation, and long-depolarization voltage step to assess late INa. The model results for each protocol were compared with experimental data. The transition rate parameters that maximally influence each voltage-clamp protocol were identified with an automatic procedure. Namely, the ratio a8/b8 was modified to reproduce the experimental data accounting for late INa; a5/b5 was tuned to correctly simulate the channel availability curve; a6, b6 and a7, b7 were adjusted to represent the observed characteristics of intermediate inactivation and recovery from inactivation. The Nelder-Mead simplex direct algorithm was used to find the parameter values minimizing the sum of the least-square errors between model predictions and experimental data for each protocol. The values proposed by Clancy and Rudy (16) were chosen as initial guesses in the minimization procedure to identify the transition rate parameters that allowed the best fitting of our βGal data. The identified βGal set in Table 2 was subsequently used as an initial guess to identify the CaMKIIδc channel parameters. The parameters that influence more than one protocol were then manually tuned to optimize fitting to all the experimental data. A full list of the parameters is given in Table 2. Matlab 7 and Simulink (The MathWorks, Natick, MA) were used for all numerical computations.
TABLE 1.
Transition rate expressions (ms−1)
| Transition rates |
|---|
| a1 = (P1a1/(P2a1 exp(−Vm/17) + 0.20 exp(−Vm/150))) |
| a2 = (P1a1/(P2a1 exp(−Vm/15) + 0.23 exp(−Vm/150))) |
| a3 = (P1a1/(P2a1 exp(−Vm/12) + 0.25 exp(−Vm/150))) |
| b1 = P1b1 exp(−Vm/P2b1) |
| b2 = P1b2 exp(− (Vm − P2b2)/(P2b1)) |
| b3 = (P1b3 exp(− (Vm − P2b3)/(P2b1))) |
| a5 = P1a5 exp(−Vm/P2a5) |
| b5 = (P1b5 + P2b5Vm) |
| a4 = (P1a4 exp(Vm/P2a4)) |
| b4 = (a3a4a5)/(b3b5) |
| a6 = a4/P1a6 |
| b6 = P1b6 exp(−Vm/P2b6) |
| a7 = (P1a7 exp(Vm/P2a7)) |
| b7 = P1b7 exp(−Vm/P2b7) |
| a8 = P1a8 |
| b8 = P1b8 |
TABLE 2.
Na+ channel model parameters for βGal and CaMKII channels
| Parameters | βGal | CaMKII |
|---|---|---|
| P1a1 | 3.802 | |
| P2a1 | 0.1027 | |
| P1a4 | 9.178 | |
| P2a4 | 25 | |
| P1a5 | 3.7933e−7 | |
| P2a5 | 7.7 | |
| P1b1 | 0.1917 | |
| P2b1 | 20.3 | |
| P1b2 | 0.2 | |
| P2b2 | 5 | |
| P1b3 | 0.22 | |
| P2b3 | 10 | |
| P1b5 | 0.0042 | 0.0067 |
| P2b5 | 2e−6 | |
| P1a6 | 100 | |
| P1b6 | 8.8061e−7 | 7.0449e−7 |
| P2b6 | 11.3944 | |
| P1a7 | 0.3543e−3 | 0.6377e−3 |
| P2a7 | 23.2696 | |
| P1b7 | 0.2868e−3 | |
| P2b7 | 35.9898 | |
| P1a8 | 4.7e−7 | 11.75e−7 |
| P1b8 | 9.5e−4 | |
L-type Ca2+ current simulation
The L-type Ca2+ current formulation of Shannon et al. (19) was modified to reproduce the effects of CaMKII (8) by increasing the channel maximal conductance by 10% and mimicking Ca2+-dependent facilitation with an additional term that makes the Ca2+-dependent inactivation less complete.
In βGal-overexpressing cardiomyocytes, Ca2+-dependent inactivation (local Kd(Ca) = 7 μM) is modeled by using the variable fCa, in which dynamic changes are described as
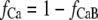 |
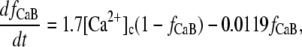 |
where [Ca2+]c is the [Ca2+] in the actual compartment (either junctional or subsarcolemmal space, in mM).
To account for CaMKII overexpression, the Ca2+-dependent inactivation variable becomes
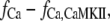 |
where
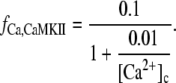 |
Transient outward K+ current simulation
The formulations of the slow and fast component of Ito were adapted from Bassani et al. (20) to account for the increased expression levels of Kv1.4 channels and for the enhanced recovery from inactivation in CaMKII-overexpressing rabbit myocytes (15). To this purpose, maximal conductance of the slow component Gto,slow was increased by 50% in the CaMKII model and the time constants of Ito,slow inactivation were modified as follows:
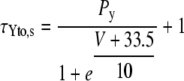 |
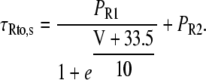 |
Values of Py, PR1, and PR2 are (in ms) 182, 8085, and 131, respectively, for βGal and 15, 3600, and 500, respectively, for CaMKII-overexpressing cardiomyocytes.
Action potential simulation
The ventricular AP was simulated using the Chicago model of rabbit ventricular myocyte (19), which includes both a cleft space (between sarcoplasmic reticulum (SR) and sarcolemmal membrane) and a separate subsarcolemmal compartment (where sarcolemmal ion channels and transporters sense higher [Ca2+]i than in the bulk cytosol). The model was implemented in Matlab 7 (The MathWorks, Natick, MA). The model was adjusted to correctly describe the ratio between slow and fast components of Ito to reproduce the experimental APD adaptation to the pacing rate observed in rabbit myocytes (21). For this purpose, the conductances of Ito,fast and Ito,slow were set to 0.02 and 0.06 mS/μF, respectively.
The INa model was replaced with the above Markov model. Maximal conductance GNa at 37°C was calculated as 23 mS/μF for both βGal and CaMKII (Q10 = 1.5 for Na+ channel conductance (22)). The kinetic rates were normalized to 37°C with a Q10 of 2.1 (22). ICa was similarly scaled up to 37°C, using our previous measurements in rabbit ventricular myocytes at 25°C and 35°C (23). Total Ito density was lower in rabbit ventricular myocytes expressing βGal after 24–36 h in culture (15), compared to freshly isolated myocytes (20). To better reflect the physiological situation for AP simulations, we used fresh myocyte Ito amplitudes (Ito,fast and Ito,slow) for control and relative changes caused by CaMKII versus βGal overexpression to simulate the CaMKII case. Pacing was simulated by a current pulse train (pulses of 5 ms in duration) of 9.5 A/F in amplitude with different frequencies (0.1, 0.25, 0.5, 1, 2, and 3 Hz). A variable order solver based on the numerical differentiation formulas was used to numerically solve the model equations (ode15s) (24). The stimulation was maintained until a steady AP was reached (typically ∼100 s).
RESULTS
Na+ current simulation
Steady-state inactivation and activation
Fig. 2 shows experimental data (7) and simulations, which confirm the adequacy of the Na+ channel Markovian model. Table 3 shows sources of experimental data used for validation of the simulations here. To assess the voltage dependence of steady-state inactivation (or availability), INa was measured during test pulses to −20 mV (20 ms) after 500-ms prepulses from −140 mV to various Vm (with 10-mV increments). As in the experimental data, CaMKIIδc overexpression shifts inactivation to more negative potentials with respect to βGal (Fig. 2 A).
FIGURE 2.

Experimental (squares) and simulated (solid lines) voltage-clamp protocols for βGal- (black) and CaMKII-modulated (gray) Na+ channels (data at 140 mM external Na+ concentration). (A) Steady-state inactivation. CaMKIIδc overexpression shifts the availability curve toward negative potentials. (B) Activation. Na+ channel activation is not affected by CaMKIIδc overexpression (traces are superimposed). (C and D) CaMKIIδc slows the recovery from inactivation (C) and enhances the intermediate inactivation (D). (E) Late INa is enhanced by CaMKIIδc overexpression. Simulated traces are shown at left, and bar graphs of simulated and experimental normalized current integrals at right. (F) Fast and slow time constants of INa decay. Acute CaMKIIδc overexpression slows the late component of fast INa inactivation. Experimental data are from Wagner et al. (15).
TABLE 3.
CaMKII overexpression data
| Figure | Measure | Source | Experiment |
|---|---|---|---|
| Fig. 2 | INa | Wagner et al. (4) | Rabbit myocyte, 22°C, ruptured patch clamp |
| Fig. 4 | [Na+]i | Wagner et al. (4) | Rabbit myocyte, 22°C, ruptured patch clamp |
| Fig. 5 | ICa | Kohlhaas et al. (8) | Rabbit myocyte, 22°C, ruptured patch clamp |
| Fig. 6 | Ito | Wagner et al. (15) | Rabbit myocyte, 22°C, ruptured patch clamp |
| Fig. 8 | AP | Wagner et al. (15) | Rabbit myocyte, 22°C, ruptured patch clamp |
Steady-state activation was not altered by CaMKIIδc in the experimental data (4). This was assessed by 40-ms depolarizations from a holding potential of −140 mV to −80/+60 mV (with 10-mV increments). Relative conductance was taken by dividing peak INa by the driving force (Vm − ENa). The resulting conductance was normalized to the maximal chord conductance. As in the experimental data, simulated CaMKIIδc overexpression does not affect INa activation with respect to βGal (Fig. 2 B; note that βGal and CaMKIIδc traces are superimposed).
Recovery from inactivation and intermediate inactivation
Recovery from inactivation was investigated using a 1000-ms depolarization to −20 mV, which initiates fast and intermediate inactivation (P1), followed by a recovery interval of variable duration and a subsequent test pulse (P2). CaMKIIδc slows recovery from inactivation in our simulations, as well as in the experimental results (Fig. 2 C). Accumulation of intermediate inactivation was assessed using depolarizations of variable duration (P1) followed by a 20-ms recovery period at −140 mV (which allows channel recovery from fast inactivation). Na+ channels that have undergone intermediate inactivation would still be largely unavailable, causing a reduction in INa at the subsequent test pulse P2 to −20 mV. In the computer simulations, as in the experimental recordings, intermediate inactivation is enhanced by CaMKIIδc (Fig. 2 D).
Late current
CaMKIIδc enhances late INa. Currents were elicited by 1000-ms depolarizations to −20 mV (from −140 mV), and normalized to peak INa. The current integral was calculated between 50 and 500 ms (Fig. 2 E) and plotted relative to the INa integral if no inactivation had occurred (i.e., peak current × 450 ms). As shown in Fig. 2 E, compared to control, simulated CaMKIIδc overexpression resulted in a larger late INa. These results fit the experimental data (4) shown for comparison in Fig. 2 E.
Fast inactivation
CaMKIIδc slows fast Na+ channel inactivation in rabbit CaMKIIδc myocytes compared to βGal (Fig. 2 F). The Na+ current decay (first 50 ms) was fitted with a double exponential function. Fits (dotted lines) to the simulated traces (solid lines) and corresponding parameters τ1 and τ2 (ms) are shown in black (βGal) and gray (CaMKIIδc). The deceleration of current decay was most prominent in the late component (at right in Fig. 2 F), so that τ2 was greater compared to control (7.8 vs. 5.6 ms), whereas τ1 appeared to be unchanged (1.3 ms), in agreement with Wagner et al. (4).
CaMKII-dependent Na+ current alterations on AP and [Na+]i
The simulations of the effect of the CaMKII-dependent Na+ channel kinetic alterations on ventricular AP are shown in Fig. 3 for three pacing rates (3, 1, and 0.25 Hz). Na+ channel modulation did not change overall AP morphology with respect to βGal. The βGal AP (black line) exhibits a characteristic AP shape and its duration (185, 288, and 354 ms) shows the typical rabbit APD dependence on pacing frequency (Fig. 3 E, black symbols). CaMKII APs (gray line) exhibit distinctive rate-dependent durations (Fig. 3 E, gray symbols). At fast rates (3 Hz), the CaMKII AP is grossly superimposable on the control AP (Fig. 3 A, APD90 = 188 ms). However, the short recovery interval limits Na+ channel recovery in the CaMKII case, resulting in reduced INa amplitude, slight decrease in AP peak, and reduced AP upstroke velocity (maximum dVm/dt; see Fig. 3 A, inset). At low frequencies, AP upstroke velocity is reduced by ∼10% by the CaMKII effect, but this decrease becomes more prominent at higher frequencies (Fig. 3 D). This Na+ channel loss of function would reduce AP rate of rise and conduction velocity, especially at fast heart rates. At lower rates (Fig. 3, B and C), APD is significantly prolonged, especially at the slowest rates (APD90 = 344 ms at 1 Hz and APD90 = 553 ms at 0.25 Hz). At these low frequencies, CaMKII-dependent impaired fast inactivation and enhanced late INa outweigh the slowed recovery from inactivation (because the long diastolic interval allows more complete Na+ channel recovery). Thus, CaMKIIδc-induced AP prolongation is especially apparent at slow heart rates, and is due to the presence of a larger inward late INa during phases 2 and 3 of the AP.
FIGURE 3.

CaMKII effects on Na+ channel gating affect the AP in a rate-dependent manner. (A–C) Simulations show that at slower heart rates the enhanced late INa prolongs the AP (B and C); this effect is completely blunted at faster rates (A), where the reduced channel availability slows down the AP upstroke (A, inset). (D) The upstroke velocity (dVm/dt) is reduced upon CaMKII overexpression in a rate-dependent fashion: the faster the pacing rate, the more significant the decrease in maximum dVm/dt. (E) Quantitative summary of the APD90 dependence on the pacing rate.
Intracellular [Na+] is slightly increased at low pacing frequencies in CaMKII simulations as a result of the enhanced late INa during the AP plateau (Fig. 4 right). However, at faster rates, where APD is short and late INa is very small, [Na+]i does not change significantly between βGal and CaMKII. The experimental findings of Wagner et al. (4), on the other hand, show a markedly higher [Na+]i in CaMKII-overexpressing cardiac myocytes at all frequencies (Fig. 4 left). However, if we normalize the measured [Na+]i to the value at 2 Hz (Fig. 4 left, dotted line), we again see that the increase in [Na+]i is larger at lower frequency. The higher overall [Na+]i seen in the experimental data suggests that our simulation fails to account for an overall gain in [Na+]i in the myocytes with more active CaMKII. Possibly, CaMKII modulates background Na+ influx or efflux by another mechanism.
FIGURE 4.

Experimental (left) and simulated (right) CaMKII effects on [Na+]i induced by INa alterations. (Left) The dotted line shows the shifted experimental CaMKII data to equalize [Na+]i at 2 Hz. (Right) The dashed line represents the [Na+]i at different frequencies when INa, ICa, and Ito alterations due to CaMKII are incorporated into the model.
Ca2+ current simulation
In both acute and chronic CaMKIIδc overexpression, L-type Ca2+ current was significantly increased in amplitude and exhibited some slowing of inactivation (7,8). Data from rabbit myocytes acutely overexpressing CaMKIIδc versus LacZ rabbit myocytes are shown in Fig. 5, A and B. The simulated ICa under voltage clamp produces an increase in ICa amplitude, slowing of inactivation, and unaltered voltage dependence (Fig. 5, C and D) similar to the experimental data. The larger ICa amplitude and slower inactivation would be expected to increase APD.
FIGURE 5.

CaMKII effects on ICa. (Left) Experimental (A) and simulated (C) traces recorded upon a depolarization pulse. (Right) Experimental (B) and simulated (D) I/V relations. ICa is significantly increased in CaMKIIδc compared to control (LacZ). Inactivation is slowed by CaMKIIδc. Experimental data (upper panels) are from Kohlhaas et al. (8).
K+ current simulation
Acute CaMKIIδc overexpression in rabbit ventricular myocytes increases Ito amplitude and speeds recovery from inactivation, and these effects are primarily attributable to alterations in the slowly recovering fraction of Ito (Ito,slow versus Ito,fast (15)). Fig. 6 shows the agreement between experimental and simulated effects of CaMKIIδc overexpression on Ito. Representative traces are shown for comparison between the recordings and the simulated currents upon voltage-clamp stimulation (Fig. 6 G). The Vm dependence of Ito (Fig. 6, left and middle columns) shows an increase in total Ito in both the experiments (Fig. 6 A) and simulations (Fig. 6 D). Ito,slow amplitude was also larger in both experiments and simulations (Fig. 6, B and E), whereas no significant differences between experiments and simulations are evident for Ito,fast (Fig. 6, C and F). Ito,total recovery from inactivation was faster in CaMKIIδc-overexpressing rabbit myocytes (Fig. 6 H, symbols) as well as in silico (Fig. 6 H, lines). The combined CaMKII-dependent increase of Ito amplitude and faster Ito,slow recovery would tend to shorten APD with CaMKII activation (i.e., opposite to that caused by the INa and ICa effects discussed above).
FIGURE 6.

CaMKII effects on Ito. Experimental (left) and simulated (middle) I/V relations for total (A and D), slow (B and E), and fast (C and F) Ito. CaMKIIδc-mediated increase of the total current (solid symbols) is mainly due to CaMKIIδc effects on the slow component. (G) Representative current traces during voltage-clamp activation protocol for experimental (upper) and simulated (lower) CaMKII-mediated Ito. (H) Recovery from inactivation was investigated using a 500-ms depolarization pulse (from −80 mV holding potential to +50 mV) followed by recovery intervals of increasing duration and a subsequent test pulse. Recovery from inactivation was significantly increased by CaMKIIδc. Symbols represent experimental data, and lines indicate simulation results for βGal (black) and CaMKII (gray). Data are from Wagner et al. (15).
Impact of Na+, Ca2+, and K+ current alterations on AP and [Na+]i
In simulations (compared with experiments), it is possible to test the impact of each individual CaMKIIδc current effect on the rabbit ventricular AP, individually or collectively. Fig. 7 A shows the control (βGal) AP morphology and duration (black solid line) at 1 Hz and Fig. 7 B shows APD dependence on pacing rate (Fig. 7 B, black circles). As in Fig. 3, the effects of CaMKII on INa alone tend to prolong the AP (Fig. 7 A, dashed line) especially at slow heart rates, but less so at 2 Hz pacing (Fig. 7 B, diamonds). The CaMKII-induced gain of function of ICa also prolongs APD at 1 Hz (Fig. 7 A, dotted line) and other frequencies, though less so than the INa change (Fig. 7 B). CaMKII-dependent Ito modulation alone markedly shortens the AP, and flattens the frequency dependence of APD (Fig. 7 A and B, dash-dotted curve).
FIGURE 7.

Simulated APs at 1-Hz pacing rate (A) and APD-frequency relations (B) in βGal and CaMKIIδc-overexpressing cardiac myocytes. The separate effects of each CaMKII-induced current alteration, as well as their combined impact, have been considered. CaMKII alteration of INa prolongs the AP at slow heart rate, with no effects at a 2-Hz pacing. The APD is reduced by Ito and increased by ICa at all frequencies. The predicted overall effect of CaMKII is AP shortening.
When all three CaMKII target channels are considered (INa, ICa, and Ito), the net effect is that APD is reduced compared to βGal (Fig. 7, A and B, gray line and solid circles). The large effect of Ito enhancement to shorten APD appears to be dominant over the smaller effects on INa and ICa which prolong APD. However, the individual ion channel effects on APD modulation seem roughly additive. Fig. 8 shows experimental APD results in rabbit ventricular myocytes overexpressing CaMKIIδc: at left are the results from the study of Wagner et al. (15) and at right are those from the simulations presented in this study. CaMKIIδc overexpression shortens the rabbit ventricular APD. Over the full range of experimental measurements (0.25–2 Hz), APD was reduced by 40–50 ms in the CaMKII case compared to βGal (Fig. 8, B and D). At 0.1 Hz, the model predicts APs of similar duration for βGal and CaMKII (Fig. 8 D). It is possible that at extremely low frequencies the more prominent late INa counteracts the enhancement of Ito with almost no changes in the APD.
FIGURE 8.

Experimental (A and B) and simulated (C and D) APs and APD-frequency relations in βGal and CaMKIIδc-overexpressing cardiac myocytes. Experimental data are from Wagner et al. (15). CaMKII shortens the AP in the entire range of pacing rates. At 0.1 Hz, where no experimental data are available, the difference between βGal and CaMKIIδc APs is negligible.
[Na+]i was also assessed during AP simulations. When ICa and Ito effects were added to the INa effect, there was slightly more CaMKII-induced rise in [Na+]i at higher frequency (Fig. 4, right, dashed line). This might be due to higher Na+ influx via Na+/Ca2+ exchange in this situation. However, the rise in [Na+]i still does not reach the CaMKII-dependent [Na+]i rise seen in the experimental data (Fig. 4, left).
APD sensitivity to the late Na+ current
We investigated the impact of different amounts of late INa on the APD. To this purpose, the value of the transition rate between background and bursting modes of INa (a8) was varied. For βGal, a range of late INa integrals (normalized to peak INa if no inactivation had occurred) from 0.02 to 0.2% was explored. Accordingly, late currents ranging from 0.17% to 0.35% were reproduced for CaMKIIδc (to keep the 0.15% differential amount of late current according to experimental data). The results of this sensitivity analysis (Fig. 9) show that the noninactivating Na+ current has a significant effect on the AP, and this effect is more pronounced at slower pacing rates (compare 0.5 and 1 Hz). Moreover, such analysis qualitatively confirms our results, since in the entire range explored, CaMKII shortens the AP with respect to βGal, independent of the actual amount of sustained INa.
FIGURE 9.

APD90 sensitivity to the late Na+ current amplitude. APs were simulated by varying the amount of late Na+ current in a range between 0.02% and 0.2% of the peak current in βGal (open symbols) and between 0.17% and 0.35% in CaMKII (solid symbols), thus keeping constant the differential current amplitude. APD changes significantly with the late current amplitude, and the sensitivity increases with the lowering of the pacing frequency (compare 0.5 and 1 Hz). Note that the results of AP shortening upon CaMKII overexpression are confirmed for any value of late Na+ current amplitudes in the explored range.
APD changes with different Ito levels
Differences in the expression pattern of the channels responsible for Ito,fast and Ito,slow in different regions of the heart produce marked differences in current density and contribute to regional variations in AP duration (25). In addition, downregulation of Ito is a central and consistent electrophysiological change in cardiac diseases (e.g., HF) (26). Thus, different levels of Ito density (both fast and slow) were simulated (Fig. 10). The simulation of CaMKII has different impacts depending on Ito density. That is, when Ito is fully expressed (Ito 100% (Fig. 8 C and Fig. 10, A and D)), CaMKII shortens the AP, whereas in cells expressing 75% or 90% Ito reduction, the effect of CaMKII-induced Ito enhancement is limited (Fig. 10, B and C). In conditions where Ito expression is <60% (Fig. 10 D), net AP prolongation occurs due to the INa and ICa effects (which then outweigh the Ito effect).
FIGURE 10.

Simulated APs at 1 Hz in βGal and CaMKIIδc-overexpressing cardiac myocytes in the presence of (A) 100%, (B) 25%, and (C) 10% Ito. (D) Ratio of CaMKII to βGal APDs for different levels of Ito. When Ito is fully expressed (A, epi), CaMKII shortens the AP, whereas the AP is prolonged due to Ito downregulation (current expression <60% (D), e.g., endo (B and C)). This could amplify transmural dispersion of repolarization (endo-epi: 66 and 78 ms for βGal versus 152 and 183 ms for CaMKII), which may predispose cells to reentry phenomena.
DISCUSSION
In this study, we used an in silico approach to investigate CaMKIIδc effects on cardiac myocyte ionic currents and APs in a comprehensive computer model of ECC (19). A motivating factor was our recent report (4) that CaMKII associates with and phosphorylates the cardiac Na+ channel and produces INa gating changes that strikingly resemble those in a human genetic mutation in Nav1.5 (1795InsD) that is linked with simultaneous arrhythmogenic LQT3 and Brugada syndrome (2). In addition, CaMKII expression is upregulated and more active in HF (3,27,28) and arrhythmias are a major cause of death in HF. There are also reports of enhanced late INa in HF (29). Thus, increased CaMKII-dependent INa modulation in HF may be an acquired arrhythmogenic Na+ channel defect that could affect millions of people with HF (as compared with the relatively small number of individuals affected by the mechanistically informative genetic Na+ channel mutations). Along these lines, overexpression of CaMKIIδc in transgenic mice leads to a lethal phenotype characterized by HF (30) and increased propensity to ventricular arrhythmias (4). Transgenic or pharmacologic CaMKII inhibition has also been shown to be protective against maladaptive remodeling from chronic β-adrenergic receptor stimulation, myocardial infarction (31), and arrhythmias (32). Thus, CaMKII may be a viable therapeutic target in the treatment of common forms of cardiac disease, and understanding how CaMKII alters ionic currents and APs is important to better understand arrhythmias that occur in conditions of CaMKII upregulation.
We focused initially on CaMKII-dependent modulation of INa (4), but we have extended the rabbit ventricular myocyte model to incorporate other channels modulated by CaMKII in heart, ICa (8) and Ito (15). This sort of computational approach has proven valuable to integrate information obtained at the molecular level to the whole cell and to the clinical phenotype in a number of inherited (16–18,33) and acquired (34–36) cardiac diseases.
Na+ channel model
A Markov model of INa was identified to simulate INa measured using whole-cell patch clamp in βGal versus CaMKIIδc-overexpressing rabbit cardiomyocytes (4). This model structure has been used successfully to simulate several INa features (16–18,33). Clancy and Rudy (16) used a nine-state model (only the background mode) for the wild-type Na+ channel. To account for long-lasting bursts experimentally observed in mutant Na+ channels they introduced an additional mode of gating (burst mode) with a second (long-lasting and noninactivating) open state (17). Thus, the burst layer, thought to be responsible for a whole-cell persistent current, was incorporated into both our βGal and CaMKIIδc Na+ channel models. The resulting model satisfactorily reproduces the characteristics of CaMKIIδc-modulated INa in rabbit ventricular myocytes (4) and seems suitable to analyze the effects of CaMKIIδc at the AP level.
Impact of Na+ current alterations on AP and [Na+]i
CaMKIIδc enhances intermediate inactivation and reduces availability, at the same time impairing fast inactivation and enhancing persistent INa. These divergent alterations of INa function cause a paradoxical phenotypic overlap of LQT3 (where INa inactivation is slowed or incomplete) and Brugada syndrome (where available INa is reduced), both thought to underlie arrhythmias. Indeed, the altered Na+ channel phenotype caused by CaMKIIδc is very similar both qualitatively and quantitatively to that caused by 1795InsD mutation in human Nav1.5, which is linked with simultaneous LQT3 and Brugada syndrome features (2).
At faster heart rates, CaMKII causes Na+ channel accumulation in intermediate inactivation, with slow recovery therefrom, and reduces steady-state availability. The result is a loss of INa function, reduced rate of AP rise, and, consequently, slowed propagation velocity, consistent with a BrS phenotype at fast heart rate. At slow heart rates where the diastolic interval is longer, there is little loss of Na+ channel availability (longer recovery time), and the intrinsically longer APD is further prolonged by sustained or late INa. This results in accentuated APD prolongation at slow heart rates (Fig. 3, B, C, and E) and an LQT3 phenotype.
It is possible that increased CaMKIIδc activity in HF may alter Na+ channel gating, contributing to arrhythmogenesis. Indeed, noninactivating late INa is increased in human HF and failing canine hearts (29), and this could contribute to the prolonged APD characteristic of HF.
A higher sustained INa in HF due to CaMKIIδc could contribute to the elevated [Na+]i observed in HF (37). However, the simulations here show that the INa gating changes that prolong APD are insufficient to explain the elevated [Na+]i in either HF or CaMKII-overexpressing rabbit myocytes. Despa et al. (38) found that the elevated [Na+]i in HF was due to increased tetrodotoxin-sensitive diastolic influx. The fact that CaMKII overexpression produced a [Na+]i increase (4) similar to that seen in HF (38), where CaMKII is upregulated (3), suggests an untested hypothesis that CaMKII might enhance diastolic tetrodotoxin-sensitive Na+ influx in both situations.
The slight additional rise in [Na+]i at higher frequency when ICa and Ito effects of CaMKII were considered (versus INa alone), could be due to the close interplay between Na+ and Ca2+ regulation in myocytes. The higher ICa and SR Ca2+ release would increase Na+ influx via a Na+/Ca2+ exchanger (which is the largest normal pathway for Na+ influx in myocytes).
CaMKII has multiple targets that may alter AP
Although Na+ channel alterations can result in gain or loss of function, depending on the pacing frequency, CaMKII modulation of ICa and Ito produced only gain of function at all pacing rates. Enhanced ICa prolonged APD, whereas Ito gain of function dramatically shortened APD (Fig. 7). When considering the effects of CaMKII on INa, ICa, and Ito together, APD was shortened, indicating the stronger impact of the enhanced repolarizing Ito (compared to the combined depolarizing contributions of ICa and INa). It is noteworthy that this balance is shifted toward a net repolarizing current in a wider range of late INa, as demonstrated by the sensitivity analysis in Fig. 9.
We also included CaMKII-dependent alterations to increase the [Ca2+]i affinity of the SR Ca-ATPase (simulating phospholamban phosphorylation) and sensitization of the SR Ca2+ release channel (not shown) using parameters similar to those used by Shannon et al. (39). Although this altered Ca2+ transients and APD characteristics slightly, the APD shifts reported here were not qualitatively altered. Thus, these effects do not modify our main conclusions about INa, ICa, and Ito effects.
The CaMKII-induced enhancement of Ito would tend to prevent the LQT3 effect of late INa and ICa enhancement, and it would not ameliorate the BrS phenotype at fast heart rates. Indeed, the greater Ito might slightly further snub the latter part of the AP rise and the AP peak, accentuating the BrS phenotype. On the other hand, the concerted effect of CaMKII on Ito (with INa and ICa) may be an orchestrated response to limit APD prolongation when CaMKII is physiologically activated. In pathophysiological conditions like HF, this situation may differ. In HF there is typically a strong downregulation of Ito (26), and this would limit the ability of enhanced Ito to offset the APD prolongation resulting from INa and ICa effects (Fig. 10). The lower amplitude of SR Ca2+ release in HF would also limit the extent of Ca-dependent inactivation of ICa, which would also tend to prolong APD. These effects could make the HF patient (in whom APD is already long) more prone to this sort of acquired LQT3 and its arrhythmogenic consequences.
The AP-ECC model implemented here (19) is for a generic rabbit ventricular myocyte, without distinction for regional differences in channel density within the normal ventricular wall (40,41). However, in regions where Ito is poorly expressed (e.g., endocardium), the effects of INa and ICa might be predominant, thus potentially leading to a LQT3-like phenotype at slow heart rates (Fig. 10, B and C) and BrS phenotype at high frequencies. In regions where Ito is maximally expressed (e.g., epicardium), CaMKII activation or upregulation may lead to a markedly short AP, thus resembling an acquired form of short QT syndrome. In fact, although prolonged QT intervals are intuitively associated with the risk of life-threatening events, the notion of a short QT syndrome being arrhythmogenic is relatively new (42).
Heterogeneity of transmural ventricular repolarization in the heart has been linked to a variety of arrhythmic manifestations (43). If CaMKII prolongs APDs in the endocardium (Fig. 10, B and C) and shortens APDs in the epicardium (Fig. 10 A), this could amplify transmural dispersion of repolarization (endo-epi 66 and 78 ms for control versus 152 and 183 ms with CaMKII (Fig. 10)), thus predisposing to the development of reentrant arrhythmias. Thus, there are several potential pathways by which cardiac CaMKII activation may be arrhythmogenic, and further experimental and in silico work will be needed to clarify which pathways are most important.
Species differences and time domain of CaMKII overexpression
In striking contrast to the AP shortening shown in rabbits, we have shown that chronic CaMKII overexpression in mice significantly prolongs APD in transgenic versus wild-type mice (7,15). Mouse electrophysiology differs significantly from that of rabbit, but this difference may be simply explicable. In the mouse, with chronic overexpression of CaMKII, INa and ICa are enhanced, as in rabbit, but Ito is decreased. The result is that all three current changes considered here will tend to prolong APD in mouse. Thus, it is not surprising that CaMKII prolongs the APD in the transgenic CaMKIIδc mouse (and differs from rabbit). In rabbit myocytes, acute CaMKII overexpression increased Ito mainly by increasing Ito recovery from inactivation for both Ito,fast and Ito,slow (and this is an acute regulatory effect sensitive to CaMKII inhibitors), but there is also a minor increase in Ito,slow functional expression. In transgenic mice chronically overexpressing CaMKIIδc, the same acceleration of Ito recovery from inactivation occurs (acutely blockable by CaMKII inhibition), but down-regulation of Ito,fast (the predominant repolarizing current in mouse) and Kv4.2/3 expression causes the dominant effect to be reduced functional Ito (15). This raises the notion that CaMKII can have both acute regulatory effects on ion channels (via CaMKII-dependent phosphorylation), but can also alter the expression levels (via CaMKII effects to alter protein transcription, translation, or trafficking (44)). There may also be time-dependent differences in this regulation, since 24-h overexpression of CaMKII was sufficient to demonstrate upregulation of Ito,slow (and Kv1.4), whereas the downregulation of Ito,fast was only evident during chronic transgenic overexpression of CaMKII. Eventually, it would be valuable to incorporate these longer-term aspects of channel modulation into dynamic computational models.
CaMKII modeling in cardiac myocytes
In our model, we simply turned on the CaMKII-modified version of the modulated currents, without considering either dynamic or local changes of its activity or association with target ion channels. Recently, Hund and Rudy (45) proposed a novel theoretical model of the canine ventricular epicardial AP and Ca2+ cycling, including a CaMKII regulatory pathway. They investigated aspects of Ca2+ transient and APD rate dependence, and concluded that CaMKII is an important determinant of the rate dependence of Ca2+ transient but not of APD (although neither INa or Ito were considered as CaMKII targets). They also showed that elevated CaMKII activity increased the cell propensity to Ca2+ transient and APD alternans (36).
Saucerman and Bers (2007) integrated mathematical descriptions of calmodulin-dependent regulation of CaMKII and calcineurin with the Chicago ECC model in the rabbit ventricular myocyte (46). They showed how CaMKII is especially well activated in regions where local [Ca2+]i is transiently high, especially in the junctional cleft near ICa and SR Ca release sites. The subsarcolemmal region, where most other ion channels (e.g., INa and Ito) are localized, also exhibits higher [Ca2+]i than bulk cytosolic [Ca2+]i (47) and would thus also be expected to exhibit more dynamic CaMKII activation.
In conclusion, we present a computational modeling approach to study how CaMKII-dependent changes in ionic currents (INa, ICa, and Ito) influence APD at different heart rates. This may provide novel mechanistic insights into cellular bases of acquired arrhythmogenesis in HF and other pathophysiological settings. It may also help to identify or tailor novel potential therapeutic targets.
Acknowledgments
We thank Drs. Fei Wang and Jeffrey J. Saucerman for their help in the implementation of the ECC model.
This work was supported by grants from the National Institutes of Health (HL30077 and HL80101 to D.B.) and Deutsche Forschungsgemeinschaft (MA 1982/1-5 and MA 1982/2-1 to L.S.M.).
Editor: David A. Eisner.
References
- 1.Tan, H. 2006. Sodium channel variants in heart disease: expanding horizons. J. Cardiovasc. Electrophysiol. 17(Suppl 1):S151–S157. [DOI] [PubMed] [Google Scholar]
- 2.Veldkamp, M. W., P. C. Viswanathan, C. Bezzina, A. Baartscheer, A. A. M. Wilde, and J. R. Balser. 2000. Two distinct congenital arrhythmias evoked by a multidysfunctional Na+ channel. Circ. Res. 86:e91–e97. [DOI] [PubMed] [Google Scholar]
- 3.Ai, X., J. W. Curran, T. R. Shannon, D. M. Bers, and S. M. Pogwizd. 2005. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 97:1314–1322. [DOI] [PubMed] [Google Scholar]
- 4.Wagner, S., N. Dybkova, E. C. L. Rasenack, C. Jacobshagen, L. Fabritz, P. Kirchhof, S. K. G. Maier, T. Zhang, G. Hasenfuss, J. H. Brown, D. M. Bers, and L. S. Maier. 2006. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116:3127–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maier, L. S., and D. M. Bers. 2007. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 73:631–640. [DOI] [PubMed] [Google Scholar]
- 6.DeSantiago, J., L. S. Maier, and D. M. Bers. 2004. Phospholamban is required for CaMKII-dependent recovery of Ca transients and SR Ca reuptake during acidosis in cardiac myocytes. J. Mol. Cell. Cardiol. 36:67–74. [DOI] [PubMed] [Google Scholar]
- 7.Maier, L. S., T. Zhang, L. Chen, J. DeSantiago, J. H. Brown, and D. M. Bers. 2003. Transgenic CaMKIIδC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 92:904–911. [DOI] [PubMed] [Google Scholar]
- 8.Kohlhaas, M., T. Zhang, T. Seidler, D. Zibrova, N. Dybkova, A. Steen, S. Wagner, L. Chen, J. Heller Brown, D. M. Bers, and L. S. Maier. 2006. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ. Res. 98:235–244. [DOI] [PubMed] [Google Scholar]
- 9.Guo, T., T. Zhang, R. Mestril, and D. M. Bers. 2006. Ca2+/calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ. Res. 99:398–406. [DOI] [PubMed] [Google Scholar]
- 10.Yuan, W., and D. M. Bers. 1994. Ca-dependent facilitation of cardiac Ca current is due to Ca-calmodulin-dependent protein kinase. Am. J. Physiol. Heart Circ. Physiol. 267:H982–H993. [DOI] [PubMed] [Google Scholar]
- 11.Anderson, M. E., A. P. Braun, H. Schulman, and B. A. Premack. 1994. Multifunctional Ca2+/calmodulin-dependent protein kinase mediates Ca2+-induced enhancement of the L-type Ca2+ current in rabbit ventricular myocytes. Circ. Res. 75:854–861. [DOI] [PubMed] [Google Scholar]
- 12.Tessier, S., P. Karczewski, E.-G. Krause, Y. Pansard, C. Acar, M. Lang-Lazdunski, J.-J. Mercadier, and S. N. Hatem. 1999. Regulation of the transient outward K+ current by Ca2+/calmodulin-dependent protein kinases II in human atrial myocytes. Circ. Res. 85:810–819. [DOI] [PubMed] [Google Scholar]
- 13.Li, J., C. Marionneau, R. Zhang, V. Shah, J. W. Hell, J. M. Nerbonne, and M. E. Anderson. 2006. Calmodulin kinase II inhibition shortens action potential duration by upregulation of K+ currents. Circ. Res. 99:1092–1099. [DOI] [PubMed] [Google Scholar]
- 14.Colinas, O., M. Gallego, R. Setien, J. R. Lopez-Lopez, M. T. Perez-Garcia, and O. Casis. 2006. Differential modulation of Kv4.2 and Kv4.3 channels by calmodulin-dependent protein kinase II in rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 291:H1978–H1987. [DOI] [PubMed] [Google Scholar]
- 15.Wagner, S., E. Hacker, N. Dybkova, L. Fabritz, P. Kirchhof, D. M. Bers, and L. S. Maier. 2006. Ca/calmodulin kinase II differentially modulates transient outward potassium current in heart failure. Circulation. 114(Suppl. II):60. [Google Scholar]
- 16.Clancy, C. E., and Y. Rudy. 2002. Na+ channel mutation that causes both Brugada and long-QT syndrome phenotypes: a simulation study of mechanism. Circulation. 105:1208–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clancy, C. E., M. Tateyama, and R. S. Kass. 2002. Insights into the molecular mechanisms of bradycardia-triggered arrhythmias in long QT-3 syndrome. J. Clin. Invest. 110:1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vecchietti, S., E. Grandi, S. Severi, I. Rivolta, C. Napolitano, S. G. Priori, and S. Cavalcanti. 2007. In silico assessment of Y1795C and Y1795H SCN5A mutations: implication for inherited arrhythmogenic syndromes. Am. J. Physiol. Heart Circ. Physiol. 292:H56–H65. [DOI] [PubMed] [Google Scholar]
- 19.Shannon, T. R., F. Wang, J. Puglisi, C. Weber, and D. M. Bers. 2004. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys. J. 87:3351–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bassani, R. A., J. Altamirano, J. L. Puglisi, and D. M. Bers. 2004. Action potential duration determines sarcoplasmic reticulum Ca2+ reloading in mammalian ventricular myocytes. J. Physiol. 559:593–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pogwizd, S. M., K. Schlotthauer, L. Li, W. Yuan, and D. M. Bers. 2001. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual β-adrenergic responsiveness. Circ. Res. 88:1159–1167. [DOI] [PubMed] [Google Scholar]
- 22.Maltsev, V. A., and A. I. Undrovinas. 2006. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc. Res. 69:116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puglisi, J. L., R. A. Bassani, J. W. Bassani, J. N. Amin, and D. M. Bers. 1996. Temperature and relative contributions of Ca transport systems in cardiac myocyte relaxation. Am. J. Physiol. Heart Circ. Physiol. 270:H1772–H1778. [DOI] [PubMed] [Google Scholar]
- 24.Shampine, L. F., and M. W. Reichelt. 1997. The MATLAB ODE suite. SIAM J. Sci. Comput. 18:1–22. [Google Scholar]
- 25.Oudit, G. Y., Z. Kassiri, R. Sah, R. J. Ramirez, C. Zobel, and P. H. Backx. 2001. The molecular physiology of the cardiac transient outward potassium current (Ito) in normal and diseased myocardium. J. Mol. Cell. Cardiol. 33:851–872. [DOI] [PubMed] [Google Scholar]
- 26.Rose, J., A. A. Armoundas, Y. Tian, D. DiSilvestre, M. Burysek, V. Halperin, B. O'Rourke, D. A. Kass, E. Marban, and G. F. Tomaselli. 2005. Molecular correlates of altered expression of potassium currents in failing rabbit myocardium. Am. J. Physiol. Heart Circ. Physiol. 288:H2077–H2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirchhefer, U., W. Schmitz, H. Scholz, and J. Neumann. 1999. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc. Res. 42:254–261. [DOI] [PubMed] [Google Scholar]
- 28.Currie, S., C. M. Loughrey, M. A. Craig, and G. L. Smith. 2004. Calcium/calmodulin-dependent protein kinase IIδ associates with the ryanodine receptor complex and regulates channel function in rabbit heart. Biochem. J. 377:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valdivia, C. R., W. W. Chu, J. Pu, J. D. Foell, R. A. Haworth, M. R. Wolff, T. J. Kamp, and J. C. Makielski. 2005. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell. Cardiol. 38:475–483. [DOI] [PubMed] [Google Scholar]
- 30.Zhang, T., L. S. Maier, N. D. Dalton, S. Miyamoto, J. Ross, Jr., D. M. Bers, and J. H. Brown. 2003. The δC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 92:912–919. [DOI] [PubMed] [Google Scholar]
- 31.Zhang, R., M. S. C. Khoo, Y. Wu, Y. Yang, C. E. Grueter, G. Ni, E. E. Price, W. Thiel, S. Guatimosim, L.-S. Song, E. C. Madu, A. N. Shah, T. A. Vishnivetskaya, J. B. Atkinson, V. V. Gurevich, G. Salama, W. J. Lederer, R. J. Colbran, and M. E. Anderson. 2005. Calmodulin kinase II inhibition protects against structural heart disease. Nat. Med. 11:409–417. [DOI] [PubMed] [Google Scholar]
- 32.Wu, Y., J. Temple, R. Zhang, I. Dzhura, W. Zhang, R. Trimble, D. M. Roden, R. Passier, E. N. Olson, R. J. Colbran, and M. E. Anderson. 2002. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 106:1288–1293. [DOI] [PubMed] [Google Scholar]
- 33.Flaim, S. N., W. R. Giles, and A. D. McCulloch. 2006. Contributions of sustained INa and IKv43 to transmural heterogeneity of early repolarization and arrhythmogenesis in canine left ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 291:H2617–H2629. [DOI] [PubMed] [Google Scholar]
- 34.Gima, K., and Y. Rudy. 2002. Ionic current basis of electrocardiographic waveforms: a model study. Circ. Res. 90:889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez, B., J. M. Ferrero, Jr., and B. Trenor. 2002. Mechanistic investigation of extracellular K+ accumulation during acute myocardial ischemia: a simulation study. Am. J. Physiol. Heart Circ. Physiol. 283:H490–H500. [DOI] [PubMed] [Google Scholar]
- 36.Livshitz, L. M., and Y. Rudy. 2007. Regulation of Ca2+ and electrical alternans in cardiac myocytes: role of CaMKII and repolarizing currents. Am. J. Physiol. Heart Circ. Physiol. 292:H2854–H2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pieske, B., L. S. Maier, V. Piacentino III, J. Weisser, G. Hasenfuss, and S. Houser. 2002. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 106:447–453. [DOI] [PubMed] [Google Scholar]
- 38.Despa, S., M. A. Islam, C. R. Weber, S. M. Pogwizd, and D. M. Bers. 2002. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 105:2543–2548. [DOI] [PubMed] [Google Scholar]
- 39.Shannon, T. R., F. Wang, and D. M. Bers. 2005. Regulation of cardiac sarcoplasmic reticulum Ca release by luminal [Ca] and altered gating assessed with a mathematical model. Biophys. J. 89:4096–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McIntosh, M. A., S. M. Cobbe, and G. L. Smith. 2000. Heterogeneous changes in action potential and intracellular Ca2+ in left ventricular myocyte sub-types from rabbits with heart failure. Cardiovasc. Res. 45:397–409. [DOI] [PubMed] [Google Scholar]
- 41.Antzelevitch, C., and J. Fish. 2001. Electrical heterogeneity within the ventricular wall. Basic Res. Cardiol. 96:517–527. [DOI] [PubMed] [Google Scholar]
- 42.Gaita, F., C. Giustetto, F. Bianchi, C. Wolpert, R. Schimpf, R. Riccardi, S. Grossi, E. Richiardi, and M. Borggrefe. 2003. Short QT syndrome: a familial cause of sudden death. Circulation. 108:965–970. [DOI] [PubMed] [Google Scholar]
- 43.Antzelevitch, C. 2005. Cardiac repolarization. The long and short of it. Europace. 7(s2):S3–S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang, T., and J. H. Brown. 2004. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc. Res. 63:476–486. [DOI] [PubMed] [Google Scholar]
- 45.Hund, T. J., and Y. Rudy. 2004. Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation. 110:3168–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saucerman, J. J., and D. M. Bers. 2007. CaM mediates differential sensitivity of CaM kinase and calcineurin to local Ca2+. Biophys. J. 92:365a. (Abstr.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weber, C. R., V. Piacentino III, K. S. Ginsburg, S. R. Houser, and D. M. Bers. 2002. Na+-Ca2+ exchange current and submembrane [Ca2+] during the cardiac action potential. Circ. Res. 90:182–189. [DOI] [PubMed] [Google Scholar]