

Abstract
Sulfation of multiply hydroxylated small organic molecules is fraught with problems of poor yield, multitude of products and long reaction times. We have developed a rapid microwave-based method for synthesis of highly sulfated small organic molecules, which affords the per-sulfated product in moderate to excellent yields and high purity. The method is expected to be of value in the discovery of per-sulfated organic molecules as mimics of glycosaminoglycans, which are being increasingly recognized as modulators of key physiological functions.
Recent work in our laboratory shows that designed highly sulfated, aromatic, small organic molecules possess interesting physico-chemical and biological properties.1-5 Biochemically, these molecules form multiple ionic as well as non-ionic interactions, which form the backbone of most protein-recognition elements. Structurally, these represent mimics of glycosaminoglycans (GAGs), which are increasingly being recognized as modulators of key physiological functions,6,7 while toxicologically, the sulfated structure represents a highly water-soluble, already-metabolized form that is expected to possess minimal toxicity. Despite these novel features, highly sulfated organic molecules remain largely unexplored.
A major limitation in exploring these novel structures is their challenging synthesis. Nearly all small organic sulfates reported in the literature are mono- or di-sulfated molecules,8-12 typically prepared using sulfur trioxide complexes with amines in a highly polar solvent (DMF or DMA). Sulfation of such organic scaffolds may require as many as 13 hrs and temperatures as high as 95 °C in the presence of a large excess of the sulfating complexes,8-12 while sugars, which contain multiple –OH groups, require reaction times in the range of 12 hrs to several days.13-19
Theoretically, this method could be extended for synthesis of highly sulfated drug-like molecules, yet practically it is a synthetic nightmare because these molecules possess significantly higher negative charge density.2 The major challenge is driving the reaction to completion in order to sulfate all available hydroxyl groups (alcoholic or phenolic) on the substrate. As the number of –OH groups increase on a small scaffold, sulfation becomes progressively difficult because of anion crowding, resulting in numerous partially sulfated side-products.
A further challenge is the isolation of the chemically pure per-sulfated product, which requires aqueous isolation techniques. Yields in the range of 11 and 100% have been reported, yet the presence of inorganic salts arising from the use of buffers and salts lead to significant inconsistencies and inaccuracies. Additionally, instability of highly anionic products introduce limitations on reaction times and temperature.13 This is likely to be especially true for highly sulfated, aromatic, small organic molecules, which are expected to be less stable than the saccharide scaffolds.
To avoid these problems with one-step sulfation, we recently synthesized some small aromatic per-sulfated structures using a two-step approach involving the 2,2,2-trichloroethyl protecting group.20 The two-step protection-deprotection protocol resolved some of the problems of the direct sulfation approach, yet required careful real-time monitoring of the reaction by RP-HPLC to prevent product degradation and was not particularly applicable to substrates that were acid and/or metal sensitive. These limitations led us to seek an alternative sulfation approach, which can be rapid, efficient, and widely applicable to a number of poly-hydroxy scaffolds.
We hypothesized that significant rate enhancements are likely to be achieved using microwaves, especially because the ionic sulfated product may couple to microwaves through ionic conduction, e.g., in CH3CN.21 CH3CN was chosen as the solvent over the commonly used DMF because a) it can be evaporated at lower temperature (thus aiding isolation) and b) it was likely to solubilize the persulfated product with an amine counter-ion. We also hypothesized that introducing free base in the reaction mixture should promote the difficult per-sulfation reaction.
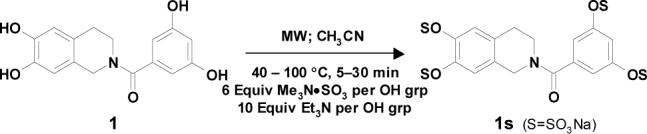
Sulfation of 122 with SO3•Me3N complex (6 equivalents per –OH group) at 100 °C in the absence of free base gave only 4.7 % of per-sulfated product 1s in 20 minutes (Table 1, entry 1).23 Inclusion of 1 equivalent of free Et3N per –OH group resulted in 13.5 % conversion (entry 2), while 79.8 % of 1s was formed with 5 equivalents of Et3N (entry 3). Further increase to 10 equivalents Et3N per –OH group had a negligible increase in the yield of 1s (entry 4). Increasing the proportion of SO3•Me3N per –OH group from 1 to 9 molar equivalents (Table 1, entries 5−8) gradually increased the yield of the per-sulfated product from 14.5 to 79.5 %, while further increase to 12 equivalent was found to be not particularly advantageous (entry 9). For further studies, 6 and 10 molar equivalents of the sulfating complex and base, respectively, were chosen.
Table 1.
| Time (min) | Temp. (°C) | Modifications to Conditions | HPLC Yield (%) | |
|---|---|---|---|---|
| 1 | 20 | 100 | No base | 4.7 |
| 2 | 20 | 100 | 1 equiv. Et3N per OH grp. | 13.5 |
| 3 | 20 | 100 | 5 equiv. Et3N per OH grp. | 79.8 |
| 4 | 20 | 100 | —a | 80.0 |
| 5 | 10 | 100 | 1 equiv. SO3•Me3N per OH grp. | 14.5 |
| 6 | 10 | 100 | 3 equiv. SO3•Me3N per OH grp. | 46.1 |
| 7 | 10 | 100 | —a | 47.4 |
| 8 | 10 | 100 | 9 equiv. SO3•Me3N per OH grp. | 79.5 |
| 9 | 10 | 100 | 12 equiv. SO3•Me3N per OH grp. | 80.7 |
| 10 | 10 | 100 | DMF as solvent | 17.2 |
| 11 | 10 | 100 | CH3NO2 as solvent | 23.2 |
| 12 | 10 | 100 | With 6 equiv SO3•py/OH grp and 10 equiv py/OH grp as base. | 82.3 |
| 13 | 10 | 40 | —a | 0 |
| 14 | 10 | 70 | —a | 18.2 |
| 15 | 10 | 120 | —a | 90.8 |
| 16 | 30 | 100 | —a | 91.2 |
Reaction conditions here are as listed above with no additional modifications.
To assess the effect of solvent, we chose to evaluate nitromethane and DMF, both of which are solvents with high dielectric constant and known to be microwave-friendly. While only 23.2 and 17.2 % of per-sulfated product 1s was formed from 1 in 10 minutes at 100 °C in CH3NO2 and DMF, respectively, 47.4% of the product was formed in CH3CN (Table 1, entries 10 and 11). Thus, our initial choice of CH3CN proved to be optimal. To assess the effect of temperature and reaction time, sulfation was performed for 10−30 minutes at 40 to 120 °C. While 30 minutes were required to yield 91.2 % of 1s at 100 °C, only 10 minutes were needed for 90.8 % conversion at 120 °C. In striking contrast, no product was detected at 40 °C within 10 minutes. Finally, SO3•py/py was found to give nearly twice as much per-sulfated product as SO3•Me3N/Et3N (entries 7 and 12) in 10 minutes at 100 °C. Since pyridine is ∼ 10,000-fold weaker base in comparison to Et3N, this result suggests general base catalysis as the predominant mechanism of sulfation rather than a process involving deprotonation of the substrate followed by nucleophilic attack.
Appropriate control reactions in the absence of microwaves using two different substrates – 1 and 3 (entries 1 and 3 in Table 2) – at 60 °C in DMF with no free base showed poor product yields. For example, it took 24 hrs in the absence of microwaves to yield 1s in 60% yield, while 3s was not detected even after 24 hrs (entry 3, Table 2). These results highlight the importance of microwaves in achieving rapid per-sulfation.
Table 2.








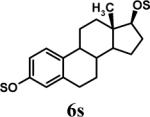


S: SO3Na.
With 9 equiv of SO3•Me3N/OH group.
With SO3•py (6−9 equiv/OH group) and pyridine as base.
Reaction conditions: 120°, 10 min, SO3•py (12 equiv/OH group) and pyridine as base.
S = SO3•pyH+
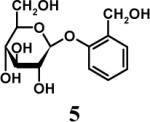
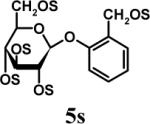
Having optimized the reaction conditions, we assessed whether the method works for a variety of different substrates. Per-sulfation of 2 proceeded smoothly in a manner identical to 1 (Table 2).24 More importantly, persulfation of 3, containing the crowded 3,4,5-trihydroxy moiety, was achieved under microwave conditions in an isolated yield of 54 %, while the conventional procedure completely failed to give 3s. Finally, microwave-assisted per-sulfation also works extremely well for substrates 5 through 8 containing one to six –OH groups. Interestingly, 5 and 6 gave a mixture of products with SO3•Me3N, but yielded the per-sulfated products with SO3•py.
Several points make the microwave-assisted synthetic protocol particularly attractive. A) The method appears to tolerate a range of functional groups including amide (Table 2, entries 1 through 4), ester (entry 4), aldehyde (entry 8) and double bond (entry 7). The relatively high isolated yields (∼70−95%) in each case make the reaction especially suitable for library construction. B) The methods works equally well for substrates containing one –OH group to those that contain six –OH groups. This is important because the small size of these molecules introduces considerable anion–anion repulsion as the number of sulfate groups increase. C) The method applies equally well to alcoholic and phenolic –OH groups, especially with SO3•py complex. D) The method provides high purity per-sulfated product that is readily isolated using an aqueous G10 filtration column. Typically, the purity of these highly water soluble, per-sulfated, small, organic molecules was found to be more than 95% using reverse polarity capillary electrophoresis (see Supplementary Material). E) The method is particularly suitable for quantitative isolation of small amounts (<10 mg) of the per-sulfated products, however could be linearly scaled up at least 20-fold without affecting the yields to a significant extent.
In summary, we have developed a rapid and high yielding microwave-based synthesis of variably functionalized, persulfated organic molecules. The protocol is expected to greatly facilitate the construction of a library of persulfated, small organic molecules for screening as glycosaminoglycan mimetics.
Acknowledgments
This work was made possible by the financial support from the National Heart, Lung and Blood Institute (RO1 HL069975 and R41 HL081972) and the American Heart Association – National Center (EIA 0640053).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Preparation and characterization (NMR, mass and capillary electrophoretic data) of the molecules prepared are available free of charge via the internet at http://______________.
References and notes
- 1.Monien BH, Henry BL, Raghuraman A, Hindle M, Desai UR. Bioorg. Med. Chem. 2006;14:7988–98. doi: 10.1016/j.bmc.2006.07.066. [DOI] [PubMed] [Google Scholar]
- 2.Dantuluri M, Gunnarsson GT, Riaz M, Nguyen H, Desai UR. Anal. Biochem. 2005;336:316–22. doi: 10.1016/j.ab.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 3.Gunnarsson GT, Desai UR. Bioorg. Med. Chem. Lett. 2003;13:579–83. doi: 10.1016/s0960-894x(02)01055-7. [DOI] [PubMed] [Google Scholar]
- 4.Gunnarsson GT, Desai UR. J. Med. Chem. 2002;45:4460–70. doi: 10.1021/jm020132y. [DOI] [PubMed] [Google Scholar]
- 5.Gunnarsson GT, Desai UR. J. Med. Chem. 2002;45:1233–43. doi: 10.1021/jm020012q. [DOI] [PubMed] [Google Scholar]
- 6.Garg HG, Linhardt RJ, Hales CA. Chemistry and Biology of Heparin and Heparan Sulfate. Elsevier Science; 2005. [Google Scholar]
- 7.Raman R, Sasisekharan V, Sasisekharan R. Chem. Biol. 2005;12:267–77. doi: 10.1016/j.chembiol.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 8.Santos GA, Murray AP, Pujol CA, Damonte EB, Maier MS. Steroids. 2003;68(2):125–32. doi: 10.1016/S0039-128X(02)00166-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bogenstatter ML, A., Overman LE, Tomasi AL. J. Am. Chem. Soc. 1999;121(51):12206–7. [Google Scholar]
- 10.Kawai N, Takao K, Kobayashi S. Tetrahedron Lett. 1999;40(22):4193–6. [Google Scholar]
- 11.Liu Y, Lien IFF, Ruttgaizer S, Dove P, Taylor SD. Org. Lett. 2004;6:209–12. doi: 10.1021/ol036157o. [DOI] [PubMed] [Google Scholar]
- 12.Simpson LS, Widlanski TS. J. Am. Chem. Soc. 2006;128:1605–10. doi: 10.1021/ja056086j. [DOI] [PubMed] [Google Scholar]
- 13.Gama CI, Tully SE, Sotogaku N, Clark PM, Rawat M, Vaidehi N, Goddard WA, 3rd, Nishi A, Hsieh-Wilson LC. Nat. Chem. Biol. 2006;2:467–73. doi: 10.1038/nchembio810. [DOI] [PubMed] [Google Scholar]
- 14.Lu LD, Shie CR, Kulkarni SS, Pan GR, Lu XA, Hung SC. Org. Lett. 2006;8:5995–8. doi: 10.1021/ol062464t. [DOI] [PubMed] [Google Scholar]
- 15.Fan RH, Achkar J, Hernandez-Torres JM, Wei A. Org. Lett. 2005;7:5095–8. doi: 10.1021/ol052130o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orgueira HA, Bartolozzi A, Schell P, Litjens REJN, Palmacci ER, Seeberger PH. Chem. Eur. J. 2003;9(1):140–69. doi: 10.1002/chem.200390009. [DOI] [PubMed] [Google Scholar]
- 17.Codee JDC, van der Marel GA, van Boeckel CAA, van Boom JH. Eur. J. Org. Chem. 2002:3954–65. [Google Scholar]
- 18.Haller M, Boons GJ. J. Chem. Soc., Perkin Trans. 1. 2001;8:814–22. [Google Scholar]
- 19.Das SK, Mallet JM, Esnault J, Driguez PA, Duchaussoy P, Sizun P, Herault J-P, Herbert J-M, Petitou M, Sinaÿ P. Chem. Eur. J. 2001;7:4821–34. doi: 10.1002/1521-3765(20011119)7:22<4821::aid-chem4821>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 20.Gunnarsson GT, Riaz M, Adams J, Desai UR. Bioorg. Med. Chem. 2005;13:1783–9. doi: 10.1016/j.bmc.2004.11.060. [DOI] [PubMed] [Google Scholar]
- 21.Kappe OC, Doris Dallinger D. For a recent review on the impact of microwaves on drug discovery. Nat. Rev. Drug Discov. 2006;51:51–63. doi: 10.1038/nrd1926. [DOI] [PubMed] [Google Scholar]
- 22.See Supplementary Material for synthesis of starting materials.
- 23.RP-HPLC profile showed peaks from 4.3−6.0 min, in addition to one at 9.0 min. The peak at 4.3 was subsequently isolated after optimization of conditions and determined to be persulfated (1s). The peak at 9.0 min was identified as 1 by comparison with synthetically pure sample. Conversions (%) were determined by area normalization.
- 24.Representative procedure for per-sulfation: To a stirred solution of the poly-alcohol (20 mg, 0.066 mmol) in MeCN (1 mL) at rt, Et3N (0.4 mL, 2.9 mmol) and Me3N•SO3 (220 mg, 1.6 mmol) was added. The reaction vessel was sealed and micro-waved (CEM-Discover microwave synthesizer) for 20 minutes at 100 °C. The reaction was repeated for 4 times and the reaction mixture was pooled for isolation of the product. The MeCN layer was decanted and pooled, while the residue was washed with MeCN (5 mL) and centrifuged. The combined MeCN layers were concentrated in vacuo. Water (5 mL) was added to the residue and stirred for 10 min. The water layer was concentrated to approximately 2 mL, loaded onto a Sephadex G10 column (∼ 160 cm) and chromatographed using water as eluent. Fractions were combined based on RP-HPLC profiles, concentrated and reloaded onto a SP Sephadex C25 column for sodium exchange. Appropriate fractions were pooled, concentrated in vacuo and lyophilized to obtain a white powder. Spectral characteristics of the final sulfated compounds are as follows: 1s: 1H NMR (DMSO, 400 MHz) δ: 7.29−7.30 (m, 2H), 6.94−6.97 (m, 3H), 4.58 (s, 2H, isomer I), 4.48 (s, 2H, isomer II), 3.58 (s, 2H, isomer II), 3.50 (s, 2H, isomer I), 2.66 (br, 2H, isomer I & II); ESI (-ve) m/z calcd for C16H11NNa4O17S4 [(MNa)−] 685.86, found 686.1; 2s: 1H NMR (DMSO, 400 MHz) δ: 7.65 (d, J = 2.4 Hz, 1H), 7.57 (d, J = 8.4 Hz, 1H), 7.29 (s, 2H), 6.99 (dd, J = 8.4, 1.6 Hz, 1H), 4.54 (s, 2H), 3.70 (br, 2H), 2.69 (t, J = 4.8 Hz, 2H); ESI (-ve) m/z calcd for C16H11NNa4O17S4 [(M-Na)−] 685.86, found 686.0; 3s: 1H NMR (DMSO, 400 MHz) δ: 7.37 (s, 2H), 7.29 (s, 2H), 4.54 (s, 2H), 3.53 (s, 2H), 2.68 (s, 2H); ESI (-ve) m/z calcd for C16H10NNa5O21S5 [(M-Na)−] 803.79, found 804.1; 4s: 1H NMR (DMSO, 400 MHz) δ:6.96−7.70 (m, 5H), 4.83−5.16 (m, 1H isomers I - III), 4.18−4.44 (m, 2H, isomers I–III), 3.55−3.61 (m, 2H, isomers I–III), 2.99−3.13 (m, 2H, isomers I–III), 1.08−1.16 (m, 3H, isomers I–III); ESI (-ve) m/z calcd for C19H15NNa4O19S4 [(M-Na)−] 757.88, found 757.5; 5s: 1H NMR (DMSO, 400 MHz) δ: 7.34 (d, 1H, J=8.0 Hz), 7.16 (t, 1H, J=8.0 Hz), 6.95 (t, 1H, J=8.0 Hz), 6.89 (d, 1H, J=8.0 Hz), 5.24 (d, 1H, J = 8.0 Hz), 4.86 (s, 2H), 4.62 (d, 2H, J=4.0 Hz), 4.35 (m, 1H), 4.21−4.27 (m, 1H), 3.93−3.99 (m, 1H), 3.75 (m, 1H); ESI (-ve) m/z calcd for C16H10NNa5O21S5 [(M-Na)−] 772.81, found 772.7; 6s: 1H NMR (DMSO, 400 MHz) δ: 7.1 (d, J = 8.4 Hz, 1H), 6.81−6.83 (m, 2H), 4.02 (t, J = 8.0 Hz, 1H), 2.71−2.74 (m, 2H), 2.21−2.25 (m, 1H), 2.08−2.12 (m, 1H), 1.87−2.00 (m, 2H), 1.75−1.78 (m, 1H), 1.48−1.60 (m, 2H), 1.09−1.34 (m, 6H), 0.66 (s, 3H); ESI (-ve) m/z calcd for C18H22Na2O8S2 [(M-Na)−] 453.07, found 453.1; 7s: 1H NMR (DMSO, 400 MHz) δ: 7.56 (s, 1H), 7.54 (s, 1H), 6.99−7.10 (m, 7H), 6.61 (s, 1H), 5.36 (s, 1H), 4.63 (s, 1H), 4.59 (s, 1H), 4.39 (s, 1H), 4.14−4.18 (m, 1H), 4.08 (d, J = 8.8 Hz, 1H) 3.81 (t, J = 10 Hz, 1H); ESI (-ve) m/z calcd for C20H16Na6O26S6 [(M-Na)−] 978.77, found 979.0; 8s: 1H NMR (DMSO, 400 MHz) δ: 9.76 (s, 1H), 8.94−8.96 (m, 2H), 8.62−8.68 (m, 1H), 8.09−8.14 (m, 2H), 7.72−7.75 (m, 2H), 6.92−6.95 (m, 2H); ESI (-ve) m/z calcd for C12H11NO5S [(M-pyH+)−] 200.99, found 200.8
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Preparation and characterization (NMR, mass and capillary electrophoretic data) of the molecules prepared are available free of charge via the internet at http://______________.