

Abstract
In addition to its role in cell adhesion, β-catenin is an important signaling molecule in the Wnt/Wingless signaling pathway. Recent studies have indicated that β-catenin is stabilized by hypertrophic stimuli and may regulate cardiac hypertrophic responses. To explore the role and requirement of β-catenin in cardiac development and hypertrophy, we deleted the β-catenin gene specifically in cardiac myocytes by crossing loxP-floxed β-catenin mice with transgenic mice expressing a Cre recombinase under the control of the α-myosin heavy chain promoter. No homozygous β-catenin deleted mice were born alive and died before embryonic day 14.5, indicating significant and irreplaceable roles of β-catenin in embryonic heart development. Heterozygous β-catenin deleted mice, however, demonstrated no structural and functional abnormality. The response of heterozygous β-catenin deleted mice to transverse aortic constriction, however, was significantly attenuated with decreased heart weight and heart weight/body weight ratio compared to controls with intact β-catenin genes. Hemodynamic analysis revealed that there was no difference in cardiac function between wild type and heterozygous β-catenin deleted mice. On the other hand, the expression of fetal genes, β-myosin heavy chain, atrial and brain natriuretic peptides was significantly higher in heterozygous β-catenin deleted mice when compared to wild type β-catenin mice. These results suggest that the cytoplasmic level of β-catenin modulates hypertrophic response and fetal gene reprogramming after pressure overload.
Keywords: catenin, hypertrophy, heart, pressure overload, cardiac remodeling, aortic constriction
Introduction
In cardiac myocytes, actin filaments anchor to adherens junctions through a catenin complex. β-Catenin directly binds to cadherin cytoplasmic domains and indirectly to actin filaments through α-catenin. In addition to this role in initiating and maintaining adherens junctions, β-catenin, is also involved in the Wnt/Wingless signal transduction [1]. In the cytoplasm, β-catenin forms a large complex including adenomatous polyposis coli (APC), glycogen synthase kinase-3β (GSK-3β), and axin. GSK-3β phosphorylates β-catenin in the complex resulting in its degradation through the ubiquitin-proteasome system, and thus lowering its cytoplasmic level [2]. In the basal state, GSK-3β is constitutively active and cytoplasmic β-catenin level is low. GSK-3ß activity is controlled by several signaling pathways and primarily regulated by inactivation. In cardiac myocytes, GSK-3ß is mainly regulated by Akt, but not Wnt/Wingless signaling pathway [3]. Akt phosphorylates GSK-3β and inhibits its activity, thereby stabilizing β-catenin and increasing its free cytoplasmic level. The inhibition of GSK-3ß activity is required for cardiac hypertrophy induced by pressure overload and calcineurin activation [3, 4]. Constitutively active GSK-3β attenuates cardiac hypertrophy by phosphorylating the nuclear factor of activated T cells (NFAT) and inhibiting its nuclear translocation. Similarly, the overexpression of wild-type GSK-3β in the heart also inhibits cardiac hypertrophy [5], most likely through the reduction of cytoplasmic β-catenin levels in cardiac myocyte [6]. Interestingly, wild-type GSK-3β also inhibits postnatal physiological growth in addition to pathological hypertrophy. It is not currently determined whether constitutively active and wild type GSK-3β have differential effects on NFAT activation or cytoplasmic β-catenin levels.
The cytoplasmic level of β-catenin is increased in cultured neonatal cardiac myocytes by hypertrophic stimuli and in adult animals after transverse aortic constriction (TAC) [3]. More importantly, adenoviral infection of adult myocardium with a stabilized form of β-catenin induces cardiac hypertrophy [3]. Thus stabilization of β-catenin in cardiac myocytes is sufficient to cause cardiac hypertrophy. More importantly, homozygous deletion of β-catenin in cardiac myocytes of maturing mice inhibits both physiological and pathological hypertrophy [7]. The knockout of β-catenin in mature mice, however, has no apparent effect on the heart weight and myocyte morphology in normal physiological condition [8]. The direct role of β-catenin in early cardiac development and postnatal physiological growth remains to be investigated.
β-catenin is required for early embryogenesis. Deletion of β-catenin disrupts axis formation. Conditional inactivation of β-catenin in endothelial cells also demonstrates that β-catenin plays a critical role during cardiac cushion development [9]. To investigate the direct role of β-catenin in cardiac development and hypertrophy, we specifically deleted β-catenin in cardiac myocytes by crossing loxP-floxed β-catenin mice [10] with transgenic mice expressing a Cre recombinase under the control of the α-myosin heavy chain promoter. No homozygous loxP-floxed β-catenin mice positive for Cre transgene were born alive, indicating that the β-catenin gene was deleted during embryonic development and required for the survival of embryos. Mice with heterozygous deletion of β-catenin gene, on the other hand, were phenotypically unremarkable under normal physiological condition. The haploinsufficiency of β-catenin, however, attenuated cardiac hypertrophy after TAC. Paradoxically, the expression of fetal genes was enhanced by the loss of one copy of the β-catenin gene. These findings also suggest that the reactivation of the fetal gene program does not always correlate with hypertrophic growth.
Materials and methods
Animals
Mice with two loxP sites inserted in introns 1 and 6 of the β-catenin gene were generously donated to the research community by Dr. Rolf Kemler (Max-Planck Institute of Immunology, Germany) [10] and maintained at The Jackson Laboratory (Bar Harbor, ME). Transgenic mice expressing Cre recombinase under the control of the α-myosin heavy chain promoter (αMyHC-Cre) were created as described previously [11]. Homozygous β-catenin loxP-floxed (β-cateninfl/fl) mice were crossed with αMyHC-Cre mice to generate heterozygous β-catenin loxP-floxed (β-cateninfl/wt) negative for αMyHC-Cre or heterozygous β-catenin deleted (β-catenindel/wt) positive for αMyHC-Cre. Mice with β-catenindel/wt genotype were bred with β-cateninfl/fl or β-cateninfl/wt to produce homozygous β-catenin loxP-floxed or deleted mice. DNA was isolated from tail or toe biopsy of neonates following proteinase K digestion as described [10]. αMyHC-Cre transgene, loxP-floxed and wild type β-catenin genes were amplified by PCR as described previously [10, 11]. A pilot study confirmed that recombination of β-catenin gene only occurred in atrial and ventricular tissues. All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (US Department of Health, Education, and Welfare, Department of Health and Human Services, NIH Publication 85-23), and approved by the University of South Dakota Animal Care and Use Committee.
Transverse aortic constriction (TAC)
Eighty 3- to 4-month old β-cateninfl/wt and β-catenindel/wt mice were randomly subjected to TAC or sham surgery. Wild type β-catenin mice with Cre transgene were included in the preliminary study and demonstrated similar change to β-cateninfl/wt mice. Midline thoracotomy was performed with sterile technique under endotracheal intubation and isoflurane anesthesia. The aortic arch was ligated with a 7-0 silk suture against a 27-gauge needle between the origin of the innominate and left common carotid arteries. The needle was quickly removed and the incision was closed with sutures. The sham operation was performed identically except that the aorta was not ligated. Mice were allowed to recover on a warming pad until they were fully awake.
Echocardiography and hemodynamics
Four weeks after the surgery, transthoracic echocardiography was performed using a high-resolution Vevo660 echocardiogram system with a 30MHz transducer (Visual Sonics, Toronto, Canada). The animals were lightly anesthetized with 1-2% isoflurane via a nose cone during echocardiography. Two-dimensional parasternal short axis images of the left ventricle (LV) were acquired at mid ventricle between the papillary muscles with guided M-mode recordings. Measurements of diastolic and systolic wall thicknesses and left ventricular end-diastolic and end-systolic chamber dimensions were made from leading edge to leading edge of the tracings. Ejection fraction (EF) and percentage fractional shortening (FS) were calculated with the accompanying software.
After echocardiography, mice were endotracheally intubated and anesthetized with isoflurane. The right carotid artery was isolated and cannulated with a 1.4F Millar micro-tip catheter transducer (model SPR-835, Millar Instruments, TX). After carotid arterial pressure was recorded, the transducer was advanced to the LV cavity. Heart rate (HR) and LV pressures were measured respectively after 15 minutes stabilization. The first derivatives of rising and declining LV pressure (+dP/dtmax and -dP/dtmin) were recorded using a PowerLab data acquisition system (AD Instruments, Colorado Springs, CO).
Myocyte isolation and Morphometry
Cardiac myocytes were enzymatically isolated from 8 hearts in each group by retrograde coronary perfusion described previously [12, 13]. The dissociated cells were filtered through 200-μm nylon mesh and fixed in 1.5% glutaraldehyde in 80 mM phosphate buffer. Optimal samples with more than 90% rod-shape cardiac myocytes were subjected to morphometric measurements. Cell volume was determined with a Coulter Channelyzer system using a 100-μm aperture [12]. Cell length was measured directly with NIH image analysis software. Forty myocytes from each heart were measured. Average myocyte cross sectional area was derived from cell volume/cell length, while cell width was derived from cross-sectional area (assuming a circular profile) [12].
RNA isolation and dot blot analysis
Total RNA was isolated from the LV of four mice from each group using the Trizol reagent (Invitrogen, Carlsbad, CA) following manufacturer's instruction. Four μg of total RNA was loaded to each well of the dot blot apparatus (Whatman Biometra, 96-3 mm). The membranes were then baked at 80 °C for 2 hours in a vacuum for adequate binding. Transcript-specific oligonucleotides (oligos) for atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), ß-myosin heavy chain (MHC), skeletal α-actin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), sarcoplasm reticulum Ca2+-ATPase 2a (SERCA), and phospholamban (PLN), were used as probes [14, 15]. The oligos were labeled by γ32P ATP via the polynucleotide kinase reaction and then hybridized with the membranes as described previously [14] [15]. The hybridized membranes were then exposed to a phosphor screen, and detected with the Personal Molecular Imager FX (BioRad Laboratories, Hercules, CA) and quantitated with associated Quantity-One software. The GAPDH density of each sample was used as the loading control for normalization.
Protein separation and Western blot analyses
LV tissues from 5 mice in each group were homogenized using a PT1200C Polytron Homogenizer in ice-cold Tris/Triton extraction buffer (10mM Tris-HCl, 50mM NaCl, 5mM EGTA, 1%Triton X-100, pH 7.4) with phosphatase inhibitors (0.1mM Na3VO4, 30mM Na4P2O7, 50mM NaF) and proteinase inhibitors (10mg/ml PMSF, 1μg/ml aprotinin) [16]. The homogenates were centrifuged at 15,000 g for 15 minutes and separated into Triton soluble supernatant and insoluble pellet. The pellets were suspended, vortexed, and then boiled in Laemmli SDS sample buffer for 5 minutes. Equal amount of proteins were separated by Laemmli SDS-PAGE after the quantification with BCA Protein Assay (Pierce Biotechnology, Rockford, IL) and subsequently transferred to nitrocellulose membrane. Western blots were performed with polyclonal anti-β-catenin (Sigma-Aldrich, St Louis, MO), and detected with ECL detection reagents (Amersham Bioscience, Piscataway, NJ). A VersaDoc imaging system (model 3000, Bio-Rad, Hercules, CA) was used to digitize Western blot images. The density of protein bands with each antibody was quantified with NIH image software. The same-lane actin level was used as an internal control to insure equal loading [17].
Statistics
Data are expressed as mean ± SD and analyzed by two-way analysis of variances. The Turkey test for multiple group comparisons was performed to determine the statistical significance. A P value smaller than 0.05 was regarded as statistically significant.
Result
Homozygous deletion of β-catenin in cardiac myocytes was embryonically lethal
If β-catenin loxP-floxed mice are positive for αMyHC-Cre, the sequence between two loxP sites will be deleted in the heart to inactivate β-catenin gene expression. Between the crossing of heterozygous loxP-floxed β-catenin mice positive (β-catenindel/wt) and negative (β-cateninfl/wt) for αMyHC-Cre, no homozygous loxP-floxed β-catenin mice positive for αMyHC-Cre (β-catenindel/del) were detected at birth (Table 1). All other genotypes are present in normal number according to the Mendelian inheritance. To confirm the lethality of homozygous β-catenin deletion, we also crossed homozygous loxP-floxed β-catenin mice negative for αMyHC-Cre (β-cateninfl/fl) with β-catenindel/wt mice. Again, no β-catenindel/del mice were present at birth (Table 1). Our preliminary observation revealed that β-catenindel/del mice died before post coitus day (PCD) 14.5, the earliest time points so far investigated. Embryos of each expected genotypes are present in normal ratio according to the Mendelian inheritance at PCD 14.5. Homozygous β-catenin deleted embryos at PCD 14.5, however, were grossly pale, small, and developmentally delayed (Figure 1). Under microscope, β-catenindel/del embryos at PCD 14.5 demonstrated diffuse necrosis with inflammatory infiltrates. Based on the gross morphology of the deceased embryos, we estimated that the β-catenindel/del embryos developed up to post coitus day 11.5 to 12.5. The cause and mechanism of fetal demise in homozygous knockout mice requires further investigation.
Table 1.
Genotype of offspring between β-catenin loxP floxed mice negative and positive for the Cre
| Genotype Cross | Age | Catdel/del/Cre+ | Catdel/wt/Cre+ | Catwt/wt/Cre+ | Catfl/fl/Cre− | Catfl/wt/Cre− | Catwt/wt/Cre− | Total |
|---|---|---|---|---|---|---|---|---|
| Catfl/wt/Cre− × Catdel/wt/Cre+ | birth | 0 | 19 | 10 | 8 | 15 | 8 | 60 |
| Catfl/fl/Cre− × Catdel/wt/Cre+ | birth | 0 | 37 | 0 | 31 | 31 | 0 | 99 |
| Catfl/fl/Cre− × Catdel/wt/Cre+ | PCD 14.5 | 12 | 14 | 0 | 14 | 13 | 0 | 53 |
Cat, β-catenin; fl, loxP-floxed β-catenin; del, deleted β-catenin; wt, wild type β-catenin; Cre+, positive for αMyHC-Cre transgene; Cre−, negative for αMyHC-Cre transgene; PCD, post coitus day.
Figure 1.
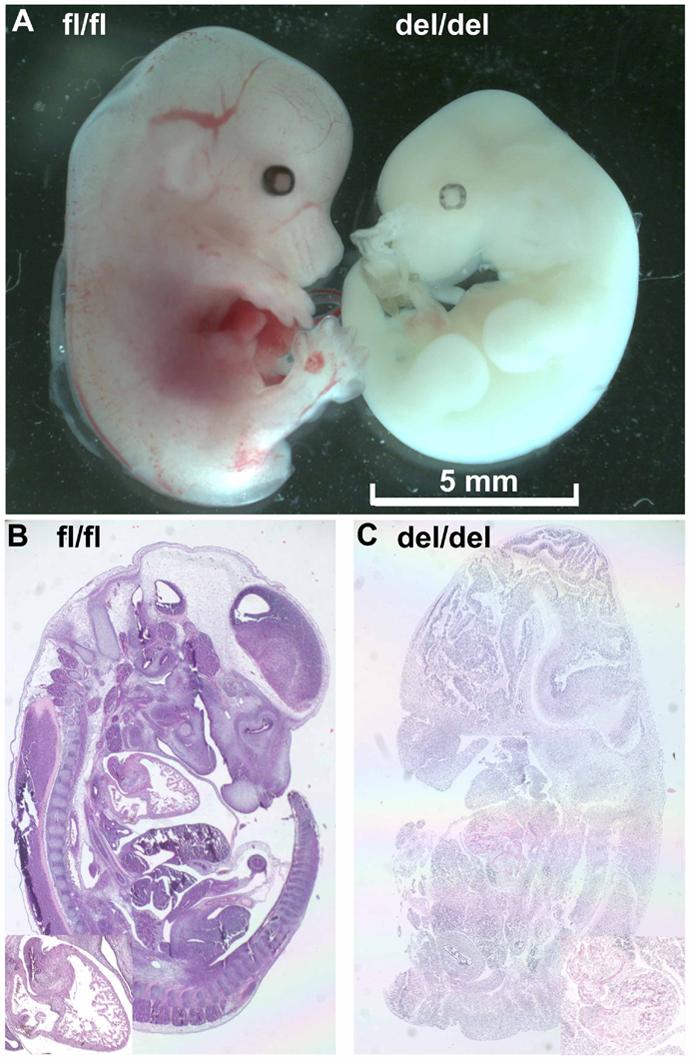
Gross (upper, A) and microscopic (bottom, B and C) morphology of β-catenin knockout embryos at post coitus day 14.5. Homozygous β-catenin deleted embryos (right embryo in A) were pale and smaller compared to that with intact β-catenin genes (left embryo in A). Under microscope, mutant embryos demonstrated diffuse necrosis with inflammatory infiltrates (C). Inserts in B and C, enlargement of the thoracic region with the heart. fl/fl, β-cateninfl/fl mouse; del/del, β-catenindel/del mouse.
Heterozygous deletion of β-catenin in cardiac myocytes had no effect on normal development or physiological growth
The deletion of one copy of β-catenin genes in the heart reduced β-catenin levels in Triton X-100 soluble, but not insoluble fraction (Figure 2). The Triton X-100 fraction contains cytoplasmic and membrane associated β-catenin. The reduction of β-catenin in this fraction indicates that there is less β-catenin available for signaling to the nucleus. Echocardiography revealed that the reduction of β-catenin expression in the heart had no detectable effect on cardiac dimension and function (Table 2). Hemodynamic measurements further confirmed that β-catenindel/wt mice had normal cardiac function (Table 3). No difference in body weight, heart weight, ventricular weight, or heart weight/body weight and ventricular weight/body weight ratio was detected among heterozygous β-catenin loxP-floxed and deleted mice (Table 3). More importantly, myocyte volume and cell dimension remained identical between two genotypes (Table 4).
Figure 2.
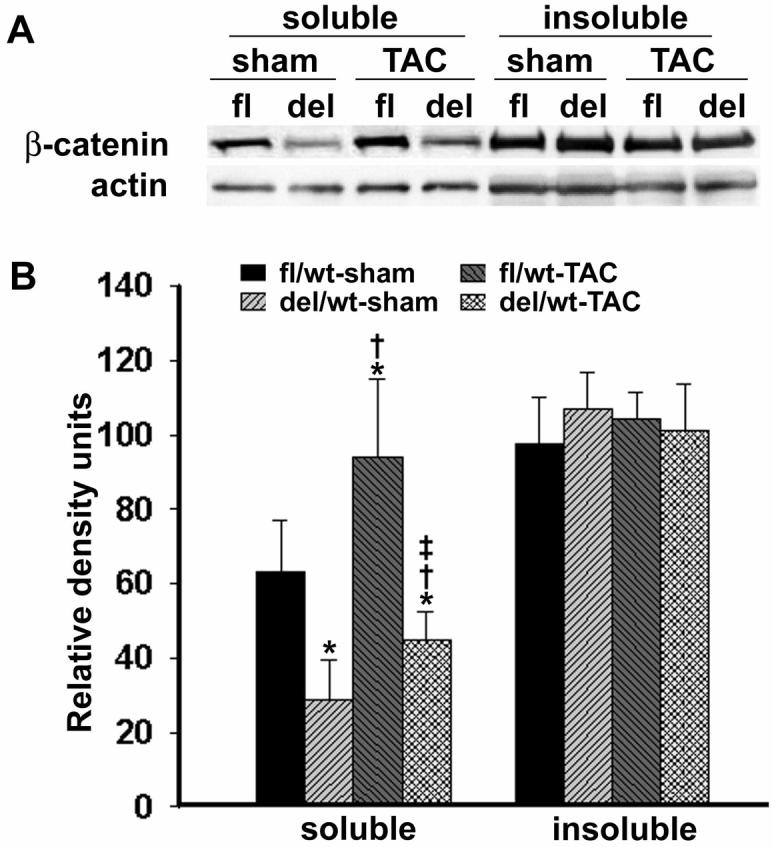
β-Catenin expression levels in left ventricles (LVs) of β-catenindel/wt (del in A or del/wt in B) and β-cateninfl/wt (fl in A or fl/wt in B) mice. LV whole tissue lysates were separated into Triton X-100 soluble and insoluble fractions and probed with polyclonal anti-β-catenin antibody. Representative Western blots (N=5) were shown in A (upper). The band density of Western blots was quantified and graphed in B (bottom). At baseline, β-catenin level in soluble fraction was significantly decreased in β-catenindel/wt mice. After transverse aortic constriction (TAC), β-catenin in soluble fraction increased in both β-catenindel/wt and β-cateninfl/wt mice, but was significantly lower in β-catenindel/wt mice. There was no difference in β-catenin levels in insoluble fraction among all four groups. *, †, ‡, P<0.05 when comparing statistically with fl/wt sham (*), del/wt sham (†), and fl/wt TAC (‡), respectively.
Table 2.
Echocardiography of β-catenin deleted and floxed mice 4 weeks after transaortic constriction
| Group | IVSDT(mm) | IVSST(mm) | LVPWDT(mm) | LVPWST(mm) | LVIDD(mm) | LVISD(mm) | EF | FS |
|---|---|---|---|---|---|---|---|---|
| fl/wt sham | 0.93±0.14 | 1.28±0.12 | 0.91±0.14 | 1.27±0.11 | 4.08±0.23 | 2.45±0.28 | 70.8±5.1 | 40.2±5.1 |
| del/wt sham | 0.96±0.11 | 1.38±0.15 | 0.90±0.15 | 1.28±0.12 | 3.89±0.14 | 2.31±0.14 | 72.6±4.0 | 40.9±3.4 |
| fl/wt TAC | 1.43±0.10*† | 1.84±0.11*† | 1.33±0.18*† | 1.68±0.17*† | 4.55±0.49*† | 3.12±0.55*† | 57.9±10.7*† | 30.7±7.4*† |
| del/wt TAC | 1.20±0.14*†‡ | 1.58±0.19*†‡ | 1.16±0.17*†‡ | 1.50±0.24*†‡ | 4.70±0.58*† | 3.47±0.67*† | 52.9±10.7*† | 26.7±7.7*† |
fl/wt, mice with one loxP-floxed and one wild type β-catenin gene; del/wt, mice with one deleted and one wild type β-catenin gene; TAC, transaortic constriction; sham, mice without TAC; IVSDT, interventricular septum diastolic thickness; IVSST, interventricular septum systolic thickness; LVPWDT, left ventricular posterior wall diastolic thickness; LVPWST, left ventricular posterior wall systolic thickness; LVIDD, left ventricular internal diastolic dimension; LVISD, left ventricular internal systolic dimension; EF, ejection fraction; FS, fractional shortening; numbers of mice in each group = 10
P<0.05 when comparing statistically with fl/wt sham(*).
P<0.05 when comparing statistically with del/wt sham(†).
P<0.05 when comparing statistically with fl/wt TAC(‡).
Table 3.
Hemodynamic and gravimetric data of □-catenin deleted and loxP-floxed mice 4 weeks after transaortic constriction
| Group | ESP (mm Hg) |
EDP (mm Hg) |
+dP/dt (mm Hg/sec) |
−dP/dt (mm Hg/sec) |
HR (beats/min) |
BW (g) |
HW (mg) |
VW (mg) |
HW/BW | VW/BW |
|---|---|---|---|---|---|---|---|---|---|---|
| fl/wt sham | 112.4±11.0 | 7.4±3.8 | 6460±635 | 4356±802 | 420±84 | 31.0±4.0 | 145.2±23.1 | 97.5±13.4 | 4.68±0.48 | 3.1±0.2 |
| del/wt sham | 109.5±13.6 | 7.3±3.0 | 7120±1841 | 4998±1095 | 413±68 | 30.7±4.1 | 144.8±14.3 | 97.2±9.4 | 4.76±0.62 | 3.2±0.4 |
| fl/wt TAC | 175.5±15.0*† | 19.8±5.9*† | 7260±1156 | 6262±1828* | 434±88 | 28.5±4.1 | 196.3±36.0*† | 141.5±27.9*† | 7.03±1.10*† | 5.1±0.9*† |
| del/wt TAC | 174.6±19.1*† | 16.8±5.8*† | 7880±1884 | 6340±2174* | 461±83 | 29.6±4.1 | 176.6±20.1*†‡ | 125.6±20.1*†‡ | 5.87±1.11*†‡ | 4.3±1.0*†‡ |
fl/wt, mice with one loxP-floxed and one wild type β-catenin gene; del/wt, mice with one deleted and one wild type β-catenin gene; TAC, transaortic constriction; sham, mice without TAC; ESP, left ventricular end systolic pressure; EDP, left ventricular end diastolic pressure; +dP/dt, the first derivative of left ventricular rising pressure; −dP/dt, the first derivative of left ventricular declining pressure; HR, heart rate; BW, body weight; HW, heart weight; VW, left ventricular weight; HW/BW, HW and BW ratio; VW/BW, VW and BW ratio; numbers of mice in each group = 10
P<0.05 when comparing statistically with fl/wt sham(*).
P<0.05 when comparing statistically with del/wt sham(†).
P<0.05 when comparing statistically with fl/wt TAC(‡).
Table 4.
Morphometry of isolated cardiocytes from □-catenin deleted and floxed mice 4 weeks after TAC
| Groups | N | Volume (μm3) | Length (μm) | Diameter (μm) | CSA (μm2) |
|---|---|---|---|---|---|
| fl/wt sham | 7 | 29138.9±4329.0 | 143.9±9.2 | 16.0±1.1 | 202.6±28.9 |
| del/wt sham | 6 | 29537.8±5462.2 | 147.1±14.9 | 15.9±0.8 | 199.6±20.0 |
| fl/wt TAC | 6 | 37672.9±3385.7*† | 153.4±15.8 | 17.7±0.6*† | 246.3±16.3*† |
| del/wt TAC | 7 | 31580.4±4628.9‡ | 156.4±4.3 | 16.0±1.4‡ | 202.5±33.3‡ |
fl/wt, mice with one loxP-floxed and one wild type β-catenin gene; del/wt, mice with one deleted and one wild type β-catenin gene; TAC, transaortic constriction; sham, mice without TAC; CSA, cross section area; N = numbers of mice in each group
P<0.05 when comparing statistically with floxed sham(*).
P<0.05 when comparing statistically with deleted sham(†).
P<0.05 when comparing statistically with floxed TAC(‡).
Heterozygous deletion of β-catenin in cardiac myocytes inhibited cardiac hypertrophy after TAC
The constriction of the aorta created a similar LV pressure increase of 60 to 65mmHg in both β-catenindel/wt and β-cateninfl/wt mice 4 weeks after TAC (Table 3). Hemodynamic changes in β-catenindel/wt mice 4 weeks after TAC were similar to that in β-cateninfl/wt mice (Table 3). β-Catenin in Triton X-100 soluble, but not insoluble fraction, was increased in β-cateninfl/wt and β-catenindel/wt mice after TAC (Figure 2). The percentage increase was similar in β-cateninfl/wt (48%) and β-catenindel/wt (56%) mice. Heterozygous β-catenin deleted mice, however, contained significantly less β-catenin in Triton X-100 fraction after TAC since they had a marked lower baseline level (Figure 2).
Echocardiography demonstrated that there was more increase in the wall thickness after TAC in β-cateninfl/wt than in β-catenindel/wt mice (Table 2). The chamber dimension was slightly enlarged in both β-catenindel/wt and β-cateninfl/wt mice after TAC, but no difference was detected between two genotypes. Cardiac function was depressed in both β-catenindel/wt and β-cateninfl/wt mice with reduced EF and FS. The EF and FS was slight lower in β-catenindel/wt mice than in β-cateninfl/wt mice, but no statistical difference was present between two groups. There was significantly more increase in heart and ventricular weight in β-cateninfl/wt than in β-catenindel/wt mice after TAC. Consequently, there was much less increase in heart weight and body weight ratio, and ventricular weight and body weight ratio in β-catenindel/wt mice after TAC. Histologically, there was no significant fibrosis in either type of animals after TAC. To determine the impairment of hypertrophic response in knockout mice, we isolated cardiac myocytes and performed morphometric measurement. Myocyte volume increase was significantly attenuated in β-catenindel/wt mice after TAC. More dramatically, there was basically no change in cell diameter in β-catenindel/wt mice while the change in cell length was identical in both genotypes after TAC (Table 4). These findings suggest that deficiency of β-catenin in cardiac myocytes mainly affects the cross-sectional growth after pressure overload.
Heterozygous deletion of β-catenin in cardiac myocytes differentially regulated hypertrophic gene expression
To assess the expression of fetal gene program, RNA dot blot analysis was performed on LV tissues 4 weeks after TAC. The transcript levels of ANF, BNP, ß-MHC, skeletal α-actin were significantly increased in both β-catenindel/wt and β-cateninfl/wt mice 4 weeks after TAC (Figure 3). Interestingly, the upregulation of ANF, BNP, and ß-MHC was more pronounced in β-catenindel/wt mice that in β-cateninfl/wt mice. On the other hand, the increase of skeletal α-actin after TAC was blunted in β-catenindel/wt mice. SERCA was decreased in both groups after TAC, but more change was detected in β-catenindel/wt mice despite the similar basal level (Figure 3). Another Ca2+ regulator, PLN, was lower in β-catenindel/wt mice compared to β-cateninfl/wt mice both in basal state and after TAC (Figure 3). The percentage of decrease after TAC was also less in β-catenindel/wt mice (10%) than in β-cateninfl/wt mice (18%).
Figure 3.

Dot blot analysis of hypertrophic gene expression normalized to GAPDH. As shown by autoradiographs (A, upper) and histograms (B, bottom), steady mRNA levels of atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), ß-myosin heavy chain (MHC), skeletal (sk) α-actin were upregulated in both β-cateninfl/wt (fl/wt) and β-catenindel/wt (del/wt) mice 4 weeks after transverse aortic constriction (TAC). The upregulation of ANF, BNP, and ß-MHC was further enhanced in β-catenindel/wt mice. Sarcoplasm reticulum Ca2+-ATPase 2a (SERCA) and phospholamban (PLN) were decreased in both β-catenindel/wt and β-cateninfl/wt mice after TAC. *, †, ‡, P<0.05 when comparing statistically with fl/wt sham (*), del/wt sham (†), and fl/wt TAC (‡), respectively.
Discussion
Mammalian cardiac myocytes rapidly lose their proliferative activity soon after birth [18]. In response to the increased workload in many different physiological and pathological conditions, cardiac myocytes mainly increase their size through building more contractile units. The change in myocyte shape and dimension reflects the type of hemodynamic stress and results in different ventricular remodeling [19]. In adults, afterload increase of the LV induced by hypertension or aortic stenosis initially induces myocyte cross-sectional growth [20]. But sustained and severe afterload increase often provokes myocyte longitudinal growth eventually leading to ventricular chamber dilatation [21]. The change in myocyte length and diameter is proportional with volume overload during postnatal physiological growth, arterial and venous fistula or hyperthyroidism [22]. On the other hand, myocardial infarction (MI) only promotes myocyte lengthening without adequate CSA growth in adjacent and remote noninfarcted region [21]. Infarct expansion and myocyte lengthening in noninfarcted region of the ventricle after MI thus causes a relatively thinner ventricle and progressive ventricular dilatation [23].
The molecular mechanisms that regulate myocyte shape and ventricular remodeling remain elusive. Recent investigations have revealed distinct pathways modulate cross-sectional and longitudinal growth of cardiac myocytes. Cardiotrophin-1 (CT-1) leads to preferential addition of sarcomeres in series rather than in parallel in cultured cardiac myocytes [24]. Mitogen-activated protein kinase (MAPK) kinase5 (MEK5), an upstream activator of MAPK/extracellular signal-regulated kinase5 (ERK5), plays a critical role in mediating CT-1 signal transduction. MEK5/ERK5 activation induces myocyte elongation in vitro. More importantly, cardiac specific overexpression of MEK5 causes eccentric cardiac hypertrophy and eventually CHF in vivo [4].
Wnt/Wingless signaling pathway controls cell shape and polarity. β-Catenin, a key signaling molecule in this pathway, is implicated in cardiac myogenesis, differentiation and hypertrophy. During early cardiogenesis in gastrulating vertebrate embryos, β-catenin signaling is inhibited [25]. Thus deletion of β-catenin in the endoderm promotes cardiac differentiation leading to ectopic heart formation [26]. Activation of β-catenin signaling, however, is required for in vitro cardiac myogenesis and differentiation [27]. More importantly, β-catenin knockout in the endothelium causes defects in cardiac septation and valve formation [9]. Our result reveal that homozygous deletion of β-catenin in cardiac myocytes is embryonically lethal, indicating that β-catenin is indispensable for late cardiac development. The exact mechanism, however, remains to be investigated.
Overexpression of constitutive β-catenin is sufficient to induce hypertrophic growth in adult cardiac myocytes [3]. On the other hand, homozygous deletion of β-catenin in cardiac myocytes of adult mice has no apparent effect on cardiac morphology and function in normal physiological condition [8]. The deletion of β-catenin in the heart of 5-week old mice, however, attenuates both physiological and pathological hypertrophy [7]. Our data indicate that even loss of one copy of β-catenin also significantly inhibits cardiac hypertrophy in response to aortic constriction. The haploinsufficiency of β-catenin, however, has no major effect on physiological cardiac hypertrophy during postnatal development. The primary defect in hypertrophic response to pressure overload is the inability to grow in cross-sectional growth. GSK-3β is a well-characterized regulator of cytoplasmic β-catenin level. Recent study has shown that cardiac overexpression of wild type GSK-3β inhibits postnatal physiological growth [6]. The deficiency in cardiac growth lies in the defect of cardiac myocytes to growth in cell diameter, while myocyte growth in length is maintained or even enhanced. Interestingly, cardiac specific overexpression of wild type GSK-3β decreases cytoplasmic levels of β-catenin in cardiac myocyte [6]. Similarly, overexpression of constitutively active GSK-3β also dampens hypertrophic response to calcineurin overexpression and pressure overload. It has been shown that GSK-3β antagonizes the actions of calcineurin by phosphorylating NFAT proteins and promoting their nuclear export [4]. Although the effect of constitutively active GSK-3β on cytoplasmic β-catenin level is not determined, it is reasonable to assume that it also reduces cytoplasmic level of β-catenin similar to wild-type GSK-3β. Furthermore, overexpression of dominant negative Lef, a functional partner of β-catenin in the nucleus, accelerates myocyte lengthening, but inhibits cross-sectional growth in cardiac myocytes [7]. These data all support the hypothesis that cytoplasmic β-catenin level regulate myocyte shape and size, especially cross-sectional growth. Thus signaling pathways lowering cytoplasmic β-catenin are likely strong inhibitors of concentric cardiac hypertrophy.
Reactivation of the fetal gene program is a critical molecular hallmark of pathological cardiac hypertrophy. The expressional changes of fetal genes usually correlate with the severity of cardiac hypertrophy. Recent studies, however, have demonstrated the discordance and disassociation of fetal gene program from cardiac hypertrophy [4, 28]. Inhibition of calcineurin by cardiac overexpression of myocyte-enriched calcineurin-interacting protein (MCIP1) enhances the expression of ANF and BNP by pressure overload despite marked attenuation of hypertrophic response [28]. GSK-3β, a key regulator of cytoplasmic β-catenin level, is inhibited by hypertrophic stimuli both in vitro and in vivo [5, 29]. Adenoviral infection of an active form of GSK-3β prevents cardiac hypertrophy and ANF expression induced by Endothelin-1 and phenylephrine in cultured neonatal cardiac myocytes [5]. Cardiac specific overexpression of constitutively active GSK-3β, also reduce cardiac hypertrophy mediated by calcineurin activation, pressure overload, or isoproterenol infusion [4] [5]. Interestingly, GSK-3β enhances the effects of calcineurin on the expression of ANF and BNP, but antagonizes on the switch from α- to β-myosin heavy chains [4]. In this study, we also demonstrated that the heterozygous deletion of β-catenin has an opposite effect on cardiac growth and fetal gene program after pressure overload: attenuation of hypertrophy and enhancement of fetal gene expression.
Acknowledgement
We thank Dr. Rolf Kemler for generously providing his mouse model to the scientific community. The project described was supported by Grant Number P20 RR017662 (F Li, and XJ Wang) from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) and “Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH.” This work was also supported by the National Institute of Health grant HL72166 (XJ Wang) and the South Dakota Health Research Foundation, which is a partnership between the University of South Dakota School of Medicine and Sanford Health. XP Yi, a visiting scholar from China, was partly supported by the National Natural Science Foundation of China, 30470696.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Miller JR, Moon RT. Signal transduction through beta-catenin and specification of cell fate during embryogenesis. Genes Dev. 1996;10(20):2527–2539. doi: 10.1101/gad.10.20.2527. [DOI] [PubMed] [Google Scholar]
- 2.Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382(6592):638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 3.Haq S, Michael A, Andreucci M, Bhattacharya K, Dotto P, Walters B, et al. Stabilization of beta-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc Natl Acad Sci U S A. 2003;100(8):4610–4615. doi: 10.1073/pnas.0835895100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, et al. Activated glycogen synthase-3 beta suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A. 2002;99(2):907–912. doi: 10.1073/pnas.231619298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haq S, Choukroun G, Kang ZB, Ranu H, Matsui T, Rosenzweig A, et al. Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J Cell Biol. 2000;151(1):117–130. doi: 10.1083/jcb.151.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michael A, Haq S, Chen X, Hsich E, Cui L, Walters B, et al. Glycogen synthase kinase-3beta regulates growth, calcium homeostasis, and diastolic function in the heart. J Biol Chem. 2004;279(20):21383–21393. doi: 10.1074/jbc.M401413200. [DOI] [PubMed] [Google Scholar]
- 7.Chen X, Shevtsov SP, Hsich E, Cui L, Haq S, Aronovitz M, et al. The {beta}-Catenin/T-Cell Factor/Lymphocyte Enhancer Factor Signaling Pathway Is Required for Normal and Stress-Induced Cardiac Hypertrophy. Mol Cell Biol. 2006;26(12):4462–4473. doi: 10.1128/MCB.02157-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou J, Qu J, Yi XP, Graber K, Huber L, Wang X, et al. Upregulation of {gamma}-catenin compensates for the loss of beta-catenin in adult cardiomyocytes. Am J Physiol Heart Circ Physiol. 2007;292(1):H270–276. doi: 10.1152/ajpheart.00576.2006. [DOI] [PubMed] [Google Scholar]
- 9.Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, et al. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol. 2004;166(3):359–367. doi: 10.1083/jcb.200403050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, et al. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128(8):1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 11.Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene Recombination in Postmitotic Cells . Targeted Expression of Cre Recombinase Provokes Cardiac-restricted, Site-specific Rearrangement in Adult Ventricular Muscle In Vivo. J Clin Invest. 1997;100(1):169–179. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerdes AM, Moore JA, Hines JM, Kirkland PA, Bishop SP. Regional differences in myocyte size in normal rat heart. Anat Rec. 1986;215(4):420–426. doi: 10.1002/ar.1092150414. [DOI] [PubMed] [Google Scholar]
- 13.Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol. 1996;28(8):1737–1746. doi: 10.1006/jmcc.1996.0163. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Osinska H, Dorn GW, 2nd, Nieman M, Lorenz JN, Gerdes AM, et al. Mouse model of desmin-related cardiomyopathy. Circulation. 2001;103(19):2402–2407. doi: 10.1161/01.cir.103.19.2402. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, et al. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89(1):84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 16.Yi XP, Wang X, Gerdes AM, Li F. Subcellular redistribution of focal adhesion kinase and its related nonkinase in hypertrophic myocardium. Hypertension. 2003;41(6):1317–1323. doi: 10.1161/01.HYP.0000072772.74183.5F. [DOI] [PubMed] [Google Scholar]
- 17.Yi XP, Zhou J, Huber L, Qu J, Wang X, Gerdes AM, et al. Nuclear compartmentalization of FAK and FRNK in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2006;290(6):H2509–2515. doi: 10.1152/ajpheart.00659.2005. [DOI] [PubMed] [Google Scholar]
- 18.Bierkamp C, McLaughlin KJ, Schwarz H, Huber O, Kemler R. Embryonic heart and skin efects in mice lacking plakoglobin. Dev Biol. 1996;180(2):780–785. doi: 10.1006/dbio.1996.0346. [DOI] [PubMed] [Google Scholar]
- 19.Grossmann KS, Grund C, Huelsken J, Behrend M, Erdmann B, Franke WW, et al. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol. 2004;167(1):149–160. doi: 10.1083/jcb.200402096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Li F, Gerdes AM. Chronic pressure overload cardiac hypertrophy and failure in guinea pigs: I. Regional hemodynamics and myocyte remodeling. J Mol Cell Cardiol. 1999;31(2):307–317. doi: 10.1006/jmcc.1998.0884. [DOI] [PubMed] [Google Scholar]
- 21.Gerdes AM, Capasso JM. Structural remodeling and mechanical dysfunction of cardiac myocytes in heart failure [editorial] [see comments] J Mol Cell Cardiol. 1995;27(3):849–856. doi: 10.1016/0022-2828(95)90000-4. [DOI] [PubMed] [Google Scholar]
- 22.Gerdes AM, Campbell SE, Hilbelink DR. Structural remodeling of cardiac myocytes in rats with arteriovenous fistulas. Lab Invest. 1988;59(6):857–861. [PubMed] [Google Scholar]
- 23.Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81(4):1161–1172. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- 24.Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, Perry WL, 3rd, et al. The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell. 1997;90(1):181–192. doi: 10.1016/s0092-8674(00)80324-4. [DOI] [PubMed] [Google Scholar]
- 25.Schneider VA, Mercola M. Wnt antagonism initiates cardiogenesis in Xenopus laevis. Genes Dev. 2001;15(3):304–315. doi: 10.1101/gad.855601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lickert H, Kutsch S, Kanzler B, Tamai Y, Taketo MM, Kemler R. Formation of multiple hearts in mice following deletion of beta-catenin in the embryonic endoderm. Dev Cell. 2002;3(2):171–181. doi: 10.1016/s1534-5807(02)00206-x. [DOI] [PubMed] [Google Scholar]
- 27.Nakamura T, Sano M, Songyang Z, Schneider MD. A Wnt- and beta -catenin-dependent pathway for mammalian cardiac myogenesis. Proc Natl Acad Sci U S A. 2003;100(10):5834–5839. doi: 10.1073/pnas.0935626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hill JA, Rothermel B, Yoo KD, Cabuay B, Demetroulis E, Weiss RM, et al. Targeted inhibition of calcineurin in pressure-overload cardiac hypertrophy. Preservation of systolic function. J Biol Chem. 2002;277(12):10251–10255. doi: 10.1074/jbc.M110722200. [DOI] [PubMed] [Google Scholar]
- 29.Morisco C, Zebrowski D, Condorelli G, Tsichlis P, Vatner SF, Sadoshima J. The AktGlycogen Synthase Kinase 3beta Pathway Regulates Transcription of Atrial Natriuretic Factor Induced by beta -Adrenergic Receptor Stimulation in Cardiac Myocytes. J Biol Chem. 2000;275(19):14466–14475. doi: 10.1074/jbc.275.19.14466. [DOI] [PubMed] [Google Scholar]