

Summary
Regulator of G-protein signaling (RGS) proteins accelerate GTP hydrolysis by Gα subunits and are thus crucial to the timing of G protein-coupled receptor (GPCR) signaling. Small molecule inhibition of RGS proteins is an attractive therapeutic approach to diseases involving dysregulated GPCR signaling. Methyl-N-[(4-chlorophenyl)sulfonyl]-4-nitrobenzenesulfinimidoate (CCG-4986) was reported as a selective RGS4 inhibitor, but with an unknown mechanism of action (Roman et al., 2007 Mol Pharmacol. 71:169−75). Here, we describe its mechanism of action as covalent modification of RGS4. Mutant RGS4 proteins devoid of surface-exposed cysteine residues were characterized using surface plasmon resonance and FRET assays of Gα binding, as well as single-turnover GTP hydrolysis assays of RGS4 GAP activity, demonstrating that cysteine-132 within RGS4 is required for sensitivity to CCG-4986 inhibition. Sensitivity to CCG-4986 can be engendered within RGS8 by replacing the wildtype residue found in this position to cysteine. Mass spectrometry analysis identified a 153 dalton fragment of CCG-4986 as being covalently attached to the surface-exposed cysteines of the RGS4 RGS domain. We conclude that the mechanism of action of the RGS protein inhibitor CCG-4986 is via covalent modification of Cys-132 of RGS4, likely causing steric hinderance with the all-helical domain of the Gα substrate.
Keywords: CCG-4986, RGS4, RGS protein inhibitor, regulator of G-protein signaling, thiol adduct
1. Introduction
The single largest class of pharmaceuticals currently prescribed are those that target G protein-coupled receptor (GPCR) signaling pathways [1, 2]. Recently, members of the “regulator of G-protein signaling” (RGS) protein superfamily have emerged as critical endogenous modulators of GPCR signal transduction (reviewed in [3, 4]). Via their conserved RGS domain that confers “GTPase-accelerating protein” (GAP) activity, RGS proteins deactivate heterotrimeric G-protein α subunits and thereby attenuate GPCR signal transduction [5, 6]. We and others have speculated that small molecule RGS protein modulators should have clinical utility in potentiating or inhibiting the actions of endogenous GPCR agonists (e.g., refs. [7, 8]); combining existing GPCR agonists with specific RGS domain inhibitors should potentiate cellular responses and could also markedly increase specificity of action of existing drugs. In particular, the diversity of RGS proteins with highly localized and dynamically regulated distributions in the human brain makes them attractive targets for pharmacotherapy of central nervous system disorders such as Parkinson's disease and opiate addiction (reviewed in [9, 10]).
Despite their obvious potential as vanguards of a novel pharmacotherapeutic strategy, few reports currently exist of small molecule inhibitors of RGS protein action. Two groups have recently described identifying inhibitors of the RGS protein/Gα interaction (BMS-195270, CCG-4986), but the specific biochemical mechanism of action for each compound remained unresolved in these initial studies [11, 12]. Roman et al. identified CCG-4986 in a flow-cytometric protein interaction assay as an inhibitor of RGS4 binding to the G-protein subunit Gαo [12]. CCG-4986 inhibits the GAP activity of RGS4 in single-turnover GTP hydrolysis and inactivates the action of recombinant RGS4 protein in inhibiting μ-opioid receptor signaling by permeabilized C6 glioma cells; in contrast, Gα binding and inhibition of μ-opioid receptor signaling by a related R4-subfamily RGS protein (RGS8) is unperturbed by the actions of CCG-4986 [12]. Here, we describe biochemical, biophysical, and mass spectrometric analyses of the interaction between CCG-4986 and RGS4, which support the conclusion that this small molecule RGS inhibitor is a reactive modifier of a solvent-exposed cysteine present in RGS4 and not RGS8, thereby explaining its in vitro RGS protein specificity.
2. Materials
2.1 Chemicals
Methyl-N-[(4-chlorophenyl)sulfonyl]-4-nitrobenzenesulfinimidoate (CCG-4986; MW 374.82) was purchased from ChemBridge (San Diego, CA); identity and purity of CCG-4986 was confirmed by electrospray mass spectrometry (ESI-MS) conducted at the UNC-Duke Michael Hooker Proteomics Core Facility. Unless otherwise noted, all reagents used were the highest grade available from Sigma Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA).
2.2 Protein expression and purification
Wildtype human RGS4 (amino acids 29−198; cloned as a hexahistidine-tagged fusion in pSGC-LIC) was obtained from the Structural Genomics Consortium (Oxford, UK); point mutations were made using QuikChange site directed mutagenesis (Stratagene, La Jolla, CA). DNA encoding wildtype human RGS4 (amino acids 50−177) and wildtype RGS8 (amino acids 62−191), and point mutants thereof, were also subcloned into a Novagen (San Diego, CA) pET vector-based prokaryotic expression construct (“pET-YFP-LIC-C”) using PCR and ligation-independent cloning [13]. The resultant constructs encoded RGS4 as C-terminal fusions to enhanced yellow fluorescent protein (hereafter described as YFP; Clontech, Mountain View, CA) with an intervening 12 amino acid linker sequence (TSRGRMYTQSNA).
For expression of both hexahistidine-tagged and YFP-tagged RGS proteins, BL21(DE3) E. coli were grown to an OD600nm of 0.7−0.8 at 37°C before induction with 0.5 mM isopropyl-β-D-thiogalactopyranoside. After culture for 14−16 hours at 20°C, cells were pelleted by centrifugation and frozen at −80°C. Prior to purification, bacterial cell pellets were resuspended in N1 buffer (50 mM HEPES pH 8.0, 300 mM NaCl, 30 mM imidazole, 2.5% (w/v) glycerol). Bacteria were lysed at 10,000 kPa using pressure homogenization with an Emulsiflex (Avestin; Ottawa, Canada). Cellular lysates were centrifuged at 100,000 × g for 30 minutes at 4°C. The supernatant was applied to a nickel-nitrilotriacetic acid resin FPLC column (FF HisTrap; GE Healthcare, Piscataway, NJ), washed with 7 column volumes of N1 then 3 column volumes of 30 mM imidazole before elution of RGS proteins with 300 mM imidazole. Eluted protein was cleaved with tobacco etch virus (TEV) protease overnight at 4°C and dialyzed into low imidazole buffer (N1 plus 5 mM DTT) before being passed over a second HisTrap column to separate residual His6-RGS protein from untagged, cleaved RGS protein. The column flow-through was pooled and resolved using a calibrated 150 ml size exclusion column (Sephacryl S200; GE Healthcare) with S200 buffer (50 mM Tris pH 8.0, 250 mM NaCl, DTT 5 mM, 2.5% (w/v) glycerol). Protein was then concentrated to approximately 1 mM, as determined by A280 nm measurements upon denaturation in 8 M guanidine hydrochloride. Concentration was calculated based on predicted extinction coefficient (http://us.expasy.org/tools/protparam.html). RGS4 was prepared for MS analysis using S200 buffer without glycerol (“MS Buffer”). Human RGS8 and RGS16 constructs were also provided by the Structural Genomic Consortium and purified as described (RGS8: http://www.sgc.ox.ac.uk/structures/MM/RGS8A_2ihd_MM.html, RGS16: http://www.sgc.ox.ac.uk/structures/MM/RGS16A_2bt2_MM.html). C-terminally biotinylated Gαi1 and Gαi1-CFP fusion proteins were prepared as described previously [14, 15]. His6-GαoA was purifed as described [16].
2.3 Fluorescent and radiolabelled nucleotide single-turnover GTPase assays
BODIPYFL-GTP (Invitrogen; Carlsbad, CA) hydrolysis was measured and quantified using single nucleotide binding-and-turnover assays as previously described [16]. Single turnover [γ−32P]GTP hydrolysis assays were conducted using 100 nM Gαi1, 200 nM RGS4 protein, and 2 μM CCG-4986 as previously described [17]. Briefly, 100 nM Gαi1 was incubated for 10 minutes at 30°C with 1 × 106 cpm of [γ−32P]GTP (specific activity of 6500 dpm/Ci) in the absence of free magnesium. Reaction was then chilled on ice for 1 minute prior to the addition of 10 mM MgCl2 (final concentration) with or without added RGS protein (200 nM final) in the presence or absence of 10-fold molar excess CCG-4986. Reactions were kept on ice and 100 μl aliquots were taken at 30 second intervals, quenched in 900 μl of charcoal slurry, centrifuged, and 600 μl aliquots of supernatant counted via liquid scintillation as described [17].
2.4 Surface plasmon resonance-based binding assays
Optical detection of surface plasmon resonance (SPR) was performed using a Biacore 3000 (Biacore Inc., Piscataway, NJ). Biotinylated Gαi1 was immobilized on streptavidin sensor chips (Biacore) to densities of ∼6000 RU as previously described [15]. In pilot studies, CCG-4986 was observed to react with the biosensor surface, preventing us from obtaining high-quality protein/protein interaction data in its presence (data not shown). To obviate this problem, we removed excess CCG-4986 from RGS protein samples using rapid gel filtration. Specifically, all proteins samples were first incubated for 3 minutes at room temperature in a 50 μl volume containing 30 μM RGS protein with a 10-fold molar excess of CCG-4986 (or DMSO equivalent) in the absence or presence of 10 mM DTT. RGS protein was then separated from unbound compound and other low-molecular weight reagents by Sephadex G-25 chromatography (Illustra™ MicroSpin™ G-25 Column; GE Healthcare) via centrifugation for 1 minute at 735 × g. The flow through was then diluted in 250 μl of BIA running buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 0.05% NP40, 100 μM GDP, 5 μM EDTA, 10 mM MgCl2, 10 mM NaF, 30 μM AlCl3) for injection across biosensor surfaces. Binding curves for wildtype RGS proteins and cysteine point-mutants of RGS4 were obtained at 20°C using 200 μl injections (using the KINJECT command) with a 200 second dissociation phase at 20 μl/min. Non-specific binding to a denatured Gαi1-biotin surface was subtracted from each curve (BIAevaluation software v3.0; Biacore).
2.5 Förster resonance energy transfer (FRET)-based binding assays
Förster resonance energy transfer was used to measure binding interactions between Gαi1 and RGS proteins as previously described [14]. Briefly, FRET between Gαi1-CFP and YFP-RGS fusion proteins was measured using a SpectraMax Gemini 96-well plate fluorescence reader (Molecular Devices; Sunnyvale, CA); association of Gαi1-CFP and YFP-RGS proteins induced by the addition of aluminum tetrafluoride results in nonphotonic energy transfer and a subsequent increase in emission at 528 nm relative to the emission at 480 nm. All runs were conducted with excitation wavelength of 433 nm, a 455 nm cutoff filter, and emission scans from 474 nm to 532 nm at 2 nm intervals. FRET was calculated as the ratio of emission at 528 nm / 480 nm. Binding assays were initiated by the addition of Gαi1-CFP and fluorescence was measured within 2 minutes, given that reactions containing CCG-4986 did not appear to be stable over long time periods.
2.6 Mass spectrometry
Sample Preparation:
39 nmol of RGS4 proteins were incubated in MS Buffer with a 15-fold molar excess of CCG-4986 (30 mM in DMSO) for 5 minutes at room temperature and then filtered on a 5 ml sephadex G-25 column (HiTrap Desalting Column; GE Healthcare) in order to remove DMSO and excess CCG-4896.
Sample Analysis:
Prior to mass spectrometric analyses, RGS4 protein samples were applied to C4 ZipTip columns (Millipore, Billerica, MA) and eluted using 50% acetonitrile/2% acetic acid. For intact molecular weight determination, 1 μl of each sample was analyzed by electrospray ionization mass spectrometry (ESI-MS) on an ABI QSTAR-Pulsar QTOF mass spectrometer fitted with a nanoelectrospray source (Proxeon Biosistems A/S, Odense, Denmark). To determine the labeled sites of RGS4, tryptic digestions of untreated and CCG-4986-treated RGS4 were performed. Digested samples were then applied to C18 ZipTips and eluted with 50% acetonitrile/0.1% trifluoroacetic acid. 0.5 μl of each sample was mixed with 0.5 μl matrix (a saturated solution of α–cyano-4-hydroxycinnamic acid in 50% acetonitrile/0.1% trifluoroacetic acid) and analyzed by matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS and MALDI-MS/MS fragmentation) on a Bruker Ultraflex I mass spectrometer (Bruker Daltonics, Billerica, MA, USA). All mass spectrometric data were gathered at the UNC-Duke Michael Hooker Proteomics Center (Chapel Hill, NC).
2.7 Molecular modeling
Model building using the RGS4/Gαi1 complex coordinates (PDB id 1AGR; ref. [18]) was performed in the program O [19]. Structural images were made with PyMol (DeLano Scientific, South San Francisco, CA).
3. Results and Discussion
3.1 CCG-4986 is a cysteine-dependent RGS4 inhibitor
In a desire to establish the structural determinants of CCG-4986 function as an RGS protein inhibitor, we initiated crystallization trials towards obtaining a high-resolution structure of a RGS4/CCG-4986 complex by x-ray diffraction. However, we found that admixture of CCG-4986 with purified RGS4 protein solutions containing reducing agents (e.g., dithiothreitol [DTT]) led to an immediate generation of a bright yellow substance (data not shown); this proved problematic to continued crystallization trials, as the purification of RGS4 in the absence of reducing agent resulted in a heterogenous mixture of monomer and dimers (data not shown). Formation of this bright yellow reaction product, not reported in the original paper by Roman and colleagues [12], was reproduced by exposing CCG-4986 to DTT. As CCG-4986 contains two sulfur atoms, we hypothesized that its mechanism of action could be one of thiol reactivity and, hence, covalent modification of cysteine residue(s) in RGS4.
To highlight solvent-exposed residues within RGS4, a multiple sequence alignment of RGS domains was created in combination with the GETAREA 1.1 algorithm for calculating accessible surface area [20] as applied to the NMR structural coordinates of uncomplexed RGS4 (PDB id 1EZT; ref. [21]). This alignment revealed two solvent-exposed cysteines present within the RGS domain of RGS4 (Cys-71 and Cys-132, Fig. 1A). These two cysteines within RGS4 are not conserved among other R4-subfamily RGS proteins; for example, the corresponding residues in RGS8 are Tyr-65 and Gln-126 and in RGS16 are Asn-74 and Glu-135 (Fig. 1A). To determine if the inhibitory activity of CCG-4986 was dependent on the Cys-71 and/or Cys-132 residues unique to RGS4, mutant RGS4 proteins were purified (Fig. 1B) bearing Cys-71-to-Asn and/or Cys-132-to-Glu point mutations (based on the closely-related RGS16 sequence; Fig. 1A) and subjected to biochemical analyses of RGS protein function.
Fig. 1.

Multiple sequence alignment of human R4-subfamily RGS proteins RGS4, −8, −16, and the RZ-subfamily member RGS19. (A) Solvent-accessible residues within RGS4 are demarcated by “o” (outside), while internal residues are noted by “i” (inside) and partially solvent-exposed residues are unlabeled, as predicted by the GETAREA 1.1 algorithm [20] using a solvent probe of 1.40 Å as applied to the high-resolution structure of free RGS4 [21]. Position of Cys-71 and Cys-132, found uniquely within RGS4, are indicated by arrowheads. Alpha-helices observed within the NMR structures of RGS4 and RGS19 [21, 25] are numbered in Roman numerals. (B) Equivalent purification of wildtype and cysteine point-mutant RGS4 and RGS8 proteins for biochemical and mass spectrometry analyses is highlighted by coomassie blue staining of SDS-PAGE resolved proteins.
Assays of RGS4 GAP activity were initially performed using the fluorescent nucleotide analog BODIPYFL-GTP [16]. Wildtype RGS4 stimulated the intrinsic GTPase activity of GαoA in a dose-dependent manner (Fig. 2A). GAP activity was substantially diminished by preincubation of wildtype RGS4 with 30 μM CCG-4986 (Fig. 2A). The double point-mutant RGS4(C71N/C132E) also had potent GAP activity toward GαoA, but this activity was not inhibited by preincubation with 30 μM CCG-4986 (Fig. 2B), thus demonstrating the requirement of a cysteine residue in RGS4 for CCG-4986 bioactivity.
Fig. 2.
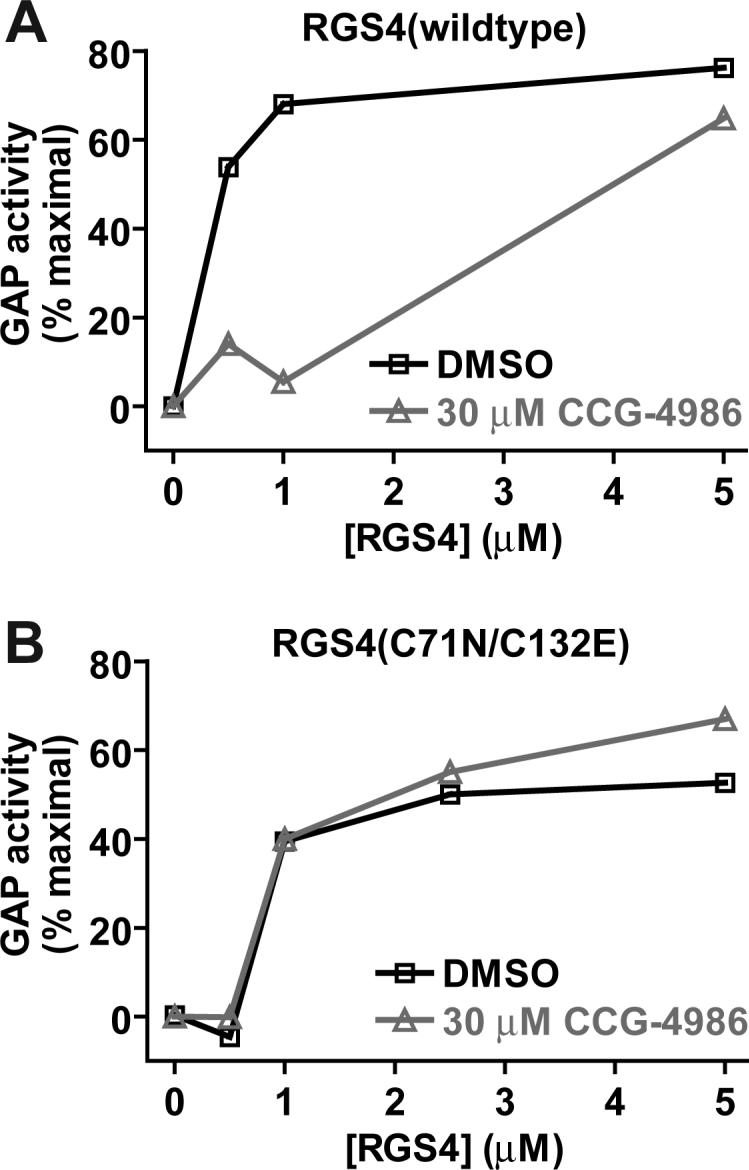
In vitro assays of RGS4 GAP activity. Single nucleotide binding-and-hydrolysis assays were conducted to measure acceleration of GαoA GTP hydrolysis rate by (A) wildtype and (B) cysteine-substituted (C71N/C132E) forms of RGS4. The fluorescence of 50 nM BODIPYFL-GTP was measured at 20 °C in 1 ml of buffer containing various concentrations of RGS4 protein (0 to 5 μM) previously incubated for 2 minutes with 30 μM CCG-4986 or DMSO vehicle only. Fluorescence measurements were initiated and, at 60 seconds, GαoA (100 nM) was added to cuvettes. Normalized GAP activity was then calculated as described [16] and plotted on the ordinate versus RGS4 concentration on the abscissa.
Surface plasmon resonance (SPR) was used to measure the ability of wildtype and mutant RGS4 proteins to bind immobilized Gαi1 in its GDP/aluminum tetrafluoride-bound transition state (the Gα nucleotide state bound most avidly by RGS proteins; ref. [22]). Addition of a 10-fold molar excess of CCG-4986 completely abolished wildtype RGS4 binding to Gαi1·GDP·AlF4- (Fig. 3A), but had no significant effect on Gαi1 binding by RGS8 and RGS16 proteins (Fig. 3B-C). Preincubation of CCG-4986 with DTT blocked its inhibitory action on the RGS4/Gαi1 interaction (Fig. 3A); we believe this loss of inhibitory action is the result of a DTT-induced reduction of CCG-4986. Mutation of the two solvent-exposed cysteines of RGS4 also resulted in a dramatic reduction in the inhibitory effect of CCG-4986 on the RGS4/Gαi1 interaction (Fig. 3D). To determine the individual roles of Cys-71 and Cys-132 in CCG-4986 activity, the single point-mutants of RGS4 were also profiled for Gαi1 binding using SPR. The Cys-71-to-Asn mutant remained sensitive to inhibition by CCG-4986; as with wildtype RGS4, this inhibitory effect was lost upon treatment of CCG-4986 with DTT (Fig. 3E). In contrast, Gαi1 binding by the Cys-132-to-Glu mutant of RGS4 was unaffected by CCG-4986 (Fig. 3F). These results suggest that Cys-132 is required for CCG-4986-mediated inhibition of Gαi1 binding by RGS4.
Fig. 3.

Surface plasmon resonance-based Gα-binding assays analyzing the inhibitory properties of CCG-4986 on wildtype RGS4 (A), RGS8 (B), RGS16 (C), and indicated cysteine mutants of RGS4 (panels D-F). 6000 resonance units (RU) of biotin-Gαi1 protein was immobilized on a streptavidin biosensor surface. A 200 μl aliquot of 5 μM RGS protein, previously incubated with either DMSO vehicle (black), CCG-4986 (gray), or DTT-reduced CCG-4986 (dotted gray), was injected at 20 μl/min (0 to 600 seconds) with a follow-up 200 seconds of dissociation time in running buffer only (600 to 800 seconds).
To confirm these SPR findings, we used an independent experimental approach of in vitro FRET between CFP- and YFP-labeled fusion proteins to quantify RGS4/Gαi1 binding in the presence of CCG-4986. We previously reported [14] that, upon interaction between YFP-RGS4 and Gαi1-CFP (the latter in its GDP/aluminum tetrafluoride-bound transition state), excitation of CFP at 433 nm results in an increase in acceptor (YFP) emission at 528 nm and a corresponding decrease in donor (CFP) emission at 480 nm. The ratio of emissions at 528 nm and 480 nm can thus be used to quantify binding between RGS4 and Gαi1 [14]. We observed a concentration-dependent reduction in FRET between YFP-RGS4 and transition-state Gαi1-CFP upon the addition of CCG-4986 (Fig. 4A). Similarly, addition of CCG-4986 to YFP-RGS4(C71N) reduced observed FRET (Fig. 4B), while the addition of solvent alone (DMSO) had no inhibitory effect on FRET from either protein pairing (Fig. 4A,B). In contrast, no reduction in FRET was seen upon addition of CCG-4986 to YFP-RGS4(C132E) or YFP-RGS4(C71N, C132E) proteins (Fig. 4C,D). To confirm that the interaction between transition state Gαi1-CFP and YFP-RGS4 was reversible, unlabeled RGS4 was used as a positive control for inhibition (Fig. 4C,D).
Fig. 4.

Competition FRET assays of the Gαi1-CFP /YFP-RGS4 interaction. (A) Addition of increasing concentrations of CCG-4986 inhibitor to 200 nM Gαi1-CFP and 280 nM YFP-RGS4, in buffer containing aluminum tetrafluoride (AMF), decreased the 528 nm/ 480 nm emission ratio, indicating a decrease in the RGS4/Gαi1 interaction. Neither mock treatment with DMSO vehicle alone nor addition of CCG-4986 in GDP-containing buffer (i.e., lacking aluminum tetrafluoride) resulted in a significant change in the FRET ratio, indicating that competition is caused specifically by CCG-4986. While a covalent reaction cannot be characterized by traditional pharmacological analysis (i.e., IC50 or Ki calculations), the signal was seen to be reduced by 50% at 18 μM CCG-4986. (B) Analogous to wildtype YFP-RGS4, the YFP-RGS4(C71N) mutant displayed a dose-dependent decrease in the emission ratio, indicating the ability of CCG-4986 to act as an inhibitor of its association with Gαi1-CFP. 50% inhibition was achieved at 4 μM CCG-4986. No inhibitory effect was seen as the result of DMSO treatment alone. (C) Increasing amounts of CCG-4986 had no inhibitory effects on the interaction between the C71N/C132E double mutant of YFP-RGS4 (560 nM) and 400 nM Gαi1-CFP in its aluminum tetrafluoride-loaded form. Unlabeled RGS4, added as a positive control for competitive inhibition of YFP-RGS4 binding, was able to decrease the emission ratio as expected (IC50 value of 375 nM; 95% confidence interval of 300−470 nM). (D) As with the double mutant, the inhibitory effects of CCG-4986 were abolished upon mutating solely the cysteine-132 of YFP-RGS4. To confirm that this single cysteine mutation did not alter the sensitivity of the assay to detect inhibition, unlabeled RGS4 was used as a competitor and found to have an IC50 of 284 nM (95% C.I. of 200−410 nM). All samples in all panels were performed in triplicate, with error bars representing the mean ± SEM.
Our BODIPYFL-GTP hydrolysis, SPR, and FRET data all confirmed previously published findings describing CCG-4986 as a selective inhibitor of the RGS4/Gα binding interaction and of RGS4 GAP activity on Gαo [12]; our analysis of cysteine point-mutants of RGS4 further demonstrated that Cys-132 is necessary for the inhibitory activity of CCG-4986. To confirm that CCG-4986 inhibition of RGS4-mediated GAP activity required the Cys-132 residue within RGS4, we repeated in vitro GAP assays using radiolabeled GTP as previously described [17]. Calculated initial rates of [γ−32P]GTP hydrolysis from these single-turnover assays were as follows: Gα alone, 0.011 s−1; Gα + RGS4(wildtype), 0.21 s−1; Gα + RGS4(wildtype) + CCG-4986, 0.074 s−1; Gα + RGS4(C132E), 0.044 s−1; Gα + RGS4(C132E) + CCG-4986, 0.036 s−1. The Cys-132-to-Glu mutation reduced the GAP activity of RGS4 (4-fold increase in initial GTP hydrolysis rate vs 19-fold for wildtype RGS4); this reduced GAP activity could be the result of some degree of steric hindrance to the binding of Gα substrate given the replacement of cysteine with a charged, more bulky residue of glutamate. However, this mutant RGS protein was clearly still active as an accelerator of Gα GTP hydrolysis and not significantly inhibited by preincubation with a 10-fold molar excess CCG-4986 (18% reduction in GAP activity vs 65% reduction of wildtype RGS4 GAP activity). These results again highlight the requirement of the Cys-132 residue in RGS4 to CCG-4986 inhibitory activity.
3.2 CCG-4986 covalently modifies RGS4 cysteine residues
To unambiguously determine if CCG-4986 is a covalent modifier of RGS4, intact molecular weight determinations of unreacted and CCG-4986-treated RGS4 protein samples were performed by nano-ESI-MS. Compared to unreacted RGS4 protein (Fig. 5A), CCG-4986-treated RGS4 protein revealed three prominent forms (Fig. 5B), with molecular weights that correspond to wildtype RGS4 (19,743 Da), RGS4 plus one 153 Da substituent (19,896 Da), and RGS4 plus two 153 Da substituents (20,050 Da). To identify specific reaction sites, tryptic digestion of unreacted and CCG-4986-treated RGS4 protein samples was performed, followed by MALDI-MS detection of tryptic peptide fragments. Two tryp tic peptides, T13 (amino acids 59-WAESLENLISHECGLAAFK-77) and T23 (amino acids 126-EVNLDSCTR-134), that contain Cys-71 and Cys-132, respectively, were found to have an increased mass of 153 Da in the CCG-4986 treated sample (data not shown). MS/MS data from peptide fragmentation confirmed that the molecular mass modification reaction arising from CCG-4986 treatment occurred specifically on Cys-71 and Cys-132 residues (data not shown). While such MS/MS analysis does not define the structure of the attached moiety, it most likely represents a fragment of CCG-4986 covalently bonded to a cysteine via a disulfide bond (Fig. 5C). We hypothesize that the attached fragment is a 4-nitrobenzenethiol radical (MW 154.17) derived from breakdown of CCG-4986. Formation of a disulfide bond by this reacting fragment of CCG-4986 would result in the loss of a hydrogen atom from the thiol of cysteine (1.0 Da) and account for the observed 153 Da moiety observed as a covalent substituent on Cys-71 and Cys-132 residues.
Fig. 5.

Intact molecular weight determination of unreacted and CCG-4986 treated RGS4. (A) Untreated RGS4 was found to exist in a single dominant form corresponding to its predicted molecular weight. (B) RGS4 preincubated with CCG-4986 was found in to consist of three major forms. The three peaks obtained by nano-ESI-MS correspond to the molecular weight of RGS4 (19,743 Da), RGS4 + 153 Da (19,896 Da), and RGS4 + 2×(153 Da) (20,050 Da). (C) CCG-4986 (methyl-N-[(4-chlorophenyl)sulfonyl]-4-nitrobenzenesulfinimidoate) has two sulfur atoms that potentially could react with thiol groups of solvent-exposed cysteine residues. Based on the mass spectrometry data from CCG-4986-treated RGS4, we propose that the 4-nitrobenzenethiol group is covalently attached to the two surface-exposed cysteines Cys-71 and Cys-132 in RGS4. The mass of the disulfide-bonded adduct derived from CCG-4986 would correspond precisely with the MS peaks at 19,896 Da (RGS4 + one 4-nitrobenzenethiol group) and 20,050 Da (RGS4 + two 4-nitrobenzenethiol groups).
3.3 RGS8 inhibition by CCG-4986 upon to mutation of glutamine-126 to cysteine
To test whether a cysteine residue at position 132 of RGS4 was sufficient for CCG-4986 sensitivity, we mutated the analogous position within RGS8 (glutamine-126; Fig. 1A) to cysteine in the context of the YFP-RGS protein fusion and tested both wildtype and RGS8(Q126C) proteins using the in vitro FRET assay for Gα binding. While wildtype YFP-RGS8 was insensitive to CCG-4986 inhibition, as previously described (Fig. 3 and ref. [12]), the YFP-RGS8(Q126C) fusion protein was inhibited in a dose-dependent fashion by CCG-4986 (Fig. 6).
Fig. 6.

Competition FRET assays of the Gαi1-CFP / YFP-RGS8 interaction. (A) Addition of increasing concentrations of CCG-4986 inhibitor to 400 nM Gαi1-CFP and 560 nM YFP-RGS4, in buffer containing aluminum tetrafluoride (AMF), did not decrease the 528 nm/ 480 nm emission ratio, indicating that CCG-4986 does not decrease the RGS8/Gαi1 interaction. Unlabeled RGS8 was added as a positive control for competitive inhibition of YFP-RGS8 binding and was able to decrease the emission ratio as expected (IC50 value of 352 nM; 95% confidence interval of 307−403 nM). Addition of CCG-4986 in GDP-containing buffer did not result in a significant change of FRET. (B) The YFP-RGS8(Q126C) / Gαi1-CFP interaction was decreased in a dose-dependent manner upon the addition of CCG-4986. 50% inhibition was observed at 1.9 μM. Neither treatment with DMSO vehicle alone nor addition of CCG-4986 in GDP-containing buffer resulted in significant change in the FRET ratio, indicating that RGS8(Q126C) was inhibited specifically by CCG-4986. All samples in all panels were performed in triplicate, with error bars representing the mean ± SEM.
3.4 Structural basis of RGS4 inhibition by CCG-4986
From the atomic resolution structure of the RGS4/Gαi1 complex [18], it is apparent that covalent addition of a 153 Da 4-nitrobenzenethiol moiety to the Cys-132 residue of RGS4 would cause significant steric hindrance to the binding of Gα. Specifically, the addition of a CCG-4986 fragment is predicted to result in van der Waals collisions with Arg-86 and Arg-90 residues of the all-helical domain of Gαi1 (Fig. 7). Hence, from the biochemical and mass spectrometry data presented above, our conclusion is that the most likely mechanism for the inhibitory properties of CCG-4986 is non-specific modification of surface-exposed cysteine residues in RGS4 causing a steric inhibition of the RGS4/Gαi1 interaction. While reactive inhibitors have made widely successful drugs (e.g., aspirin, clopidogrel; refs. [23, 24]), the lack of selectivity of CCG-4986 for cysteines on RGS4 suggests that CCG-4986 will react with surface-exposed cysteines in a myriad of proteins beyond its intended RGS protein target. Furthermore, although Roman and colleagues have speculated that the lack of CCG-4986 activity on intact RGS4-transfected cells is a result of poor membrane permeability [12], it is more likely that the requirement for cell membrane permeabilization to observe the effects of CCG-4986 reflects sensitivity to a reducing environment such as that found inside intact cells. These predictions as to the labile and reactive nature of CCG-4986 bode ill for its further development as a lead chemical entity for RGS protein-directed pharmacotherapy.
Fig. 7.
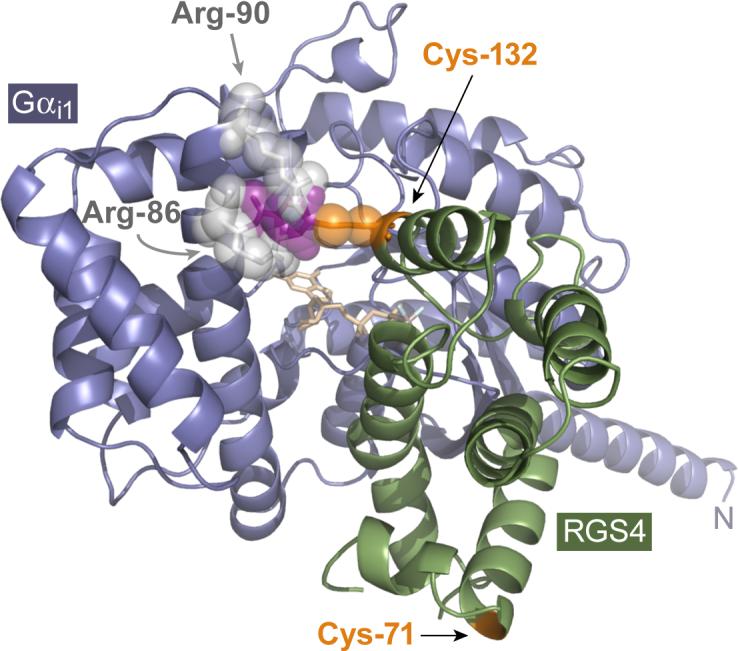
Proposed model for RGS4 inhibition by CCG-4986. The presence of the cysteine-132 residue of RGS4 is clearly necessary for the inhibitory action of CCG-4986. While cysteine-132 is not critical for the RGS4/Gα interaction per se (i.e., its conversion to glutamate does not eliminate GAP activity [Fig. 2] nor Gα association [Figs. 3 & 4]), the known high-resolution structure of the RGS4/Gαi1 complex [18] suggests that a small moiety (purple) covalently coupled to Cys-132 (orange) would result in steric hindrance with the Arg-86 and Arg-90 (light gray) of the all-helical domain of Gαi1 (steel blue). The Cα ribbon trace of RGS4 is illustrated in green; GDP bound within the G-protein is colored in wheat, with the aluminum tetrafluoride ion in light blue and gray.
Acknowledgements
We thank Drs. Declan Doyle and Meera Soundararajan (SGC Oxford) for RGS protein expression vectors, and Dr. Dmitriy Gremyachinskiy (UNC) for his chemistry advice. A.J.K. gratefully acknowledges prior support of the UNC MD/PhD program (T32 GM008719) and current predoctoral support from National Institute of Mental Health (F30 MH074266). C.A.J. and D.P.S. were supported by National Institutes of Health grants F32 GM076944 and R03 NS053754, respectively.
Abbreviations
- CCG-4986
methyl-N-[(4-chlorophenyl)sulfonyl]-4-nitrobenzenesulfinimidoate
- DTT
dithiothreitol (Cleland's reagent)
- ESI-MS
electrospray ionization mass spectrometry
- GAP
GTPase-accelerating protein
- FRET
Förster resonance energy transfer
- GDP
guanosine diphosphate
- GPCR
G protein-coupled receptor
- G protein
guanine nucleotide-binding protein
- GTP
guanosine triphosphate
- GTPase
GTP hydrolysis activity
- PCR
polymerase chain reaction
- RGS
regulator of G-protein signaling
- SPR
surface plasmon resonance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.McCudden CR, Hains MD, Kimple RJ, Siderovski DP, Willard FS. G-protein signaling: back to the future. Cell Mol Life Sci. 2005;62:551–77. doi: 10.1007/s00018-004-4462-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–6. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 3.Ross EM, Wilkie TM. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem. 2000;69:795–827. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- 4.Siderovski DP, Willard FS. The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci. 2005;1:51–66. doi: 10.7150/ijbs.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berman DM, Wilkie TM, Gilman AG. GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell. 1996;86:445–52. doi: 10.1016/s0092-8674(00)80117-8. [DOI] [PubMed] [Google Scholar]
- 6.Siderovski DP, Hessel A, Chung S, Mak TW, Tyers M. A new family of regulators of G-protein-coupled receptors. Curr Biol. 1996;6:211–2. doi: 10.1016/s0960-9822(02)00454-2. [DOI] [PubMed] [Google Scholar]
- 7.Chasse SA, Dohlman HG. RGS proteins: G protein-coupled receptors meet their match. Assay Drug Dev Technol. 2003;1:357–64. doi: 10.1089/154065803764958649. [DOI] [PubMed] [Google Scholar]
- 8.Neubig RR, Siderovski DP. Regulators of G-protein signalling as new central nervous system drug targets. Nat Rev Drug Discov. 2002;1:187–97. doi: 10.1038/nrd747. [DOI] [PubMed] [Google Scholar]
- 9.Jones MB, Siderovski DP, Hooks SB. The Gbetagamma dimer as a novel source of selectivity in G-protein signaling: GGL-ing at convention. Mol Interv. 2004;4:200–214. doi: 10.1124/mi.4.4.4. [DOI] [PubMed] [Google Scholar]
- 10.Traynor JR, Neubig RR. Regulators of G protein signaling & drugs of abuse. Mol Interv. 2005;5:30–41. doi: 10.1124/mi.5.1.7. [DOI] [PubMed] [Google Scholar]
- 11.Fitzgerald K, Tertyshnikova S, Moore L, Bjerke L, Burley B, Cao J, Carroll P, Choy R, Doberstein S, Dubaquie Y, Franke Y, Kopczynski J, Korswagen H, Krystek SR, Lodge NJ, Plasterk R, Starrett J, Stouch T, Thalody G, Wayne H, van der Linden A, Zhang Y, Walker SG, Cockett M, Wardwell-Swanson J, Ross-Macdonald P, Kindt RM. Chemical genetics reveals an RGS/G-protein role in the action of a compound. PLoS Genet. 2006;2:e57. doi: 10.1371/journal.pgen.0020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roman DL, Talbot JN, Roof RA, Sunahara RK, Traynor JR, Neubig RR. Identification of small-molecule inhibitors of RGS4 using a high-throughput flow cytometry protein interaction assay. Mol Pharmacol. 2007;71:169–75. doi: 10.1124/mol.106.028670. [DOI] [PubMed] [Google Scholar]
- 13.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 14.Willard FS, Kimple RJ, Kimple AJ, Johnston CA, Siderovski DP. Fluorescence-based assays for RGS box function. Methods Enzymol. 2004;389:56–71. doi: 10.1016/S0076-6879(04)89004-9. [DOI] [PubMed] [Google Scholar]
- 15.Willard FS, Low AB, McCudden CR, Siderovski DP. Differential G-alpha interaction capacities of the GoLoco motifs in Rap GTPase activating proteins. Cell Signal. 2007;19:428–38. doi: 10.1016/j.cellsig.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 16.Willard FS, Kimple AJ, Johnston CA, Siderovski DP. A direct fluorescence-based assay for RGS domain GTPase accelerating activity. Anal Biochem. 2005;340:341–51. doi: 10.1016/j.ab.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 17.Afshar K, Willard FS, Colombo K, Johnston CA, McCudden CR, Siderovski DP, Gonczy P. RIC-8 is required for GPR-1/2-dependent Galpha function during asymmetric division of C. elegans embryos. Cell. 2004;119:219–30. doi: 10.1016/j.cell.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 18.Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4--activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell. 1997;89:251–61. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 19.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 20.Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem. 1998;19:319–333. [Google Scholar]
- 21.Moy FJ, Chanda PK, Cockett MI, Edris W, Jones PG, Mason K, Semus S, Powers R. NMR structure of free RGS4 reveals an induced conformational change upon binding Galpha. Biochemistry. 2000;39:7063–73. doi: 10.1021/bi992760w. [DOI] [PubMed] [Google Scholar]
- 22.Berman DM, Kozasa T, Gilman AG. The GTPase-activating protein RGS4 stabilizes the transition state for nucleotide hydrolysis. J Biol Chem. 1996;271:27209–12. doi: 10.1074/jbc.271.44.27209. [DOI] [PubMed] [Google Scholar]
- 23.Patrono C. Aspirin and human platelets: from clinical trials to acetylation of cyclooxygenase and back. Trends Pharmacol Sci. 1989;10:453–8. doi: 10.1016/S0165-6147(89)80010-0. [DOI] [PubMed] [Google Scholar]
- 24.Savi P, Herbert JM. Clopidogrel and ticlopidine: P2Y12 adenosine diphosphate-receptor antagonists for the prevention of atherothrombosis. Semin Thromb Hemost. 2005;31:174–83. doi: 10.1055/s-2005-869523. [DOI] [PubMed] [Google Scholar]
- 25.de Alba E, De Vries L, Farquhar MG, Tjandra N. Solution structure of human GAIP (Galpha interacting protein): a regulator of G protein signaling. J Mol Biol. 1999;291:927–39. doi: 10.1006/jmbi.1999.2989. [DOI] [PubMed] [Google Scholar]