

Abstract
A miniaturized, disposable microbial culture chip has been fabricated by microengineering a highly porous ceramic sheet with up to one million growth compartments. This versatile culture format, with discrete compartments as small as 7 × 7 μm, allowed the growth of segregated microbial samples at an unprecedented density. The chip has been used for four complementary applications in microbiology. (i) As a fast viable counting system that showed a dynamic range of over 10,000, a low degree of bias, and a high culturing efficiency. (ii) In high-throughput screening, with the recovery of 1 fluorescent microcolony in 10,000. (iii) In screening for an enzyme-based, nondominant phenotype by the targeted recovery of Escherichia coli transformed with the plasmid pUC18, based on expression of the lacZ reporter gene without antibiotic-resistance selection. The ease of rapid, successive changes in the environment of the organisms on the chip, needed for detection of β-galactosidase activity, highlights an advantageous feature that was also used to screen a metagenomic library for the same activity. (iv) In high-throughput screening of >200,000 isolates from Rhine water based on metabolism of a fluorogenic organophosphate compound, resulting in the recovery of 22 microcolonies with the desired phenotype. These isolates were predicted, on the basis of rRNA sequence, to include six new species. These four applications suggest that the potential for such simple, readily manufactured chips to impact microbial culture is extensive and may facilitate the full automation and multiplexing of microbial culturing, screening, counting, and selection.
Keywords: microdish, microcolony, cellular assay, nanoporous aluminum oxide
The cultivation of bacteria and fungi has hardly changed in centuries. Culture on gel surfaces, such as agar in a Petri dish, and the liquid growth media used in most microbiology laboratories would be recognized by Robert Koch, who first described the growth of bacteria into a colony in the 19th century (1). Postgenomic sciences (2, 3), the need for industrial strain improvement, and the high level of threat posed by pathogens demand versatile, automatable microbial culture formats that can be widely used (4, 5). Recent microbial cultivation technologies include highly subdivided multiwell plates (6) and capillaries (7), as well as methods of sorting encapsulated microcolonies (8). These systems require expensive and sophisticated hardware and are often specialized in application. Most parallel, miniaturized culture methods are based on growth in liquid media and are variants on the multiwell plate (6). Multiwell formats have problems of oxygen transfer by shaking, the accumulation of waste products, and poor image resolution, and if miniaturized too far suffer from rapid evaporation. Additionally, the throughput is still rather low for genuine high-throughput screening (HTS). Alternatively, in situations in which microorganisms must be detected or assayed (such as routine quality control or diagnostics), low cost is needed, and only limited laboratory facilities are available. Further miniaturization, and therefore multiplexing, of culture formats is feasible because the cells are generally only 1–10 μm in size. The challenge is how to do this in a way that will be widely used.
Centuries of experience have shown that agar-supported growth of microorganisms in the Petri dish is robust and reliable, and this is reflected in the near ubiquity of this method in microbiology. Weaknesses of the agar-containing Petri dish lie in a poor facility for automation, the requirement for user skill, limited speed to result, the generation of waste (including the enrichment of pathogenic or genetically modified organisms), a short shelf life, and limited storage time of organisms in situ after culture. Additionally, agar does not suit the growth of all microbial species. Microbial culture on flexible porous membranes, on filters, or in chambers provides valuable alternatives, permitting manipulations such as staining in situ and changes in the nutrient environment of the cells, and culturing otherwise intractable species (9, 10). However, these culture devices have never been subdivided to the point of creating sufficient growth compartments for genuine HTS. In part, this may be because flexible membranes are hard to engineer at the micrometer scale and, possibly, the perception that micro-engineered mechanical systems (MEMS) devices cannot compete on cost with a cheap plastic disposable. Porous aluminum oxide (PAO) has been recently identified as a microbial culture support (11–13). This material is exceptionally porous (40% by volume) compared with other surfaces, inert, and stable with nano-range pores (20–200 nm diameter) and retains microorganisms on the rigid planar surface while allowing nutrients to pass. Culture on PAO to date (11) has been limited to hundreds of growth areas, which is inadequate for HTS. Hence, we developed the concept to provide far more advanced culture formats fabricated by MEMS techniques. Microengineering of PAO is facilitated by the great stability, rigidity, and inertness to temperature, wetting, or solvents of the starting material. A range of disposable, surface-culture, microbial growth chips, or “micro-Petri dishes,” were therefore created by using a MEMS approach to engineer growth compartments on top of PAO, which acts as the surface on which an exceptional number of microbial samples can be grown, assayed, and recovered.
Results
Chip Manufacture, Specifications, and Physical Properties.
Reactive ion etching (RIE), a process of etching by using plasma directed by a shadow mask (14), was used to selectively remove an acrylic polymer laminated on the surface of the PAO to create areas open for growth (Fig. 1). The compartments formed (Fig. 2) were precisely defined by the photo mask, with coefficient of variation values of <4% for the X and Y dimensions of the compartments and accuracies of <2 μm across the complete chip. Some erosion of the walls occurred during etching, leading to narrowing toward the top (e.g., Fig. 2C), but breaches into adjacent compartments were rare, being found in <0.5% of cases (n = 6,840). SEM of the compartment surface suggested that the porous material forming the compartment base was undamaged and not blocked by debris, and that the walls were effectively bonded to the PAO (Fig. 2C). The completed chips were permeable to water or ethanol drawn through from the upper surface by using a vacuum manifold. The PAO was strengthened by the acrylic laminate, forming a more robust composite material. Optimization of the etching time proved critical to the manufacturing process. Too little etching time resulted in incomplete removal of the laminate, and over-etching resulted in deposition of material onto the PAO from the mask. This back-sputtering and the resultant erosion of the mask during etching was minimized by a coating of aluminum oxide on the mask. The full range of chip formats created is listed in supporting information (SI) Table 1. The maximum density of growth areas was >340,000 per cm2, permitting growth formats to be created with millions of culture areas. The smallest compartment size made was 7 × 7 μm at the base, which gave 1 million discrete growth areas on an 8 × 36 mm chip. This density allows 40 million compartments to be placed on the footprint of a standard multiwell plate. The most commonly used compartment size (8 × 36 mm chip, 20 × 20 μm growth areas, 20-μm-wide walls) had 180,000 compartments per chip. This configuration was used for experiments, unless noted otherwise. Assuming exponential growth, each compartment in the standard format allows ≈5–10 divisions of a typical rod-shaped bacterium from a single viable cell as the inoculum to a detectable monolayer of hundreds of cells or filling the compartment (thousands of bacterial cells). This gives sufficient time and biomass for culture-based screening operations to be performed.
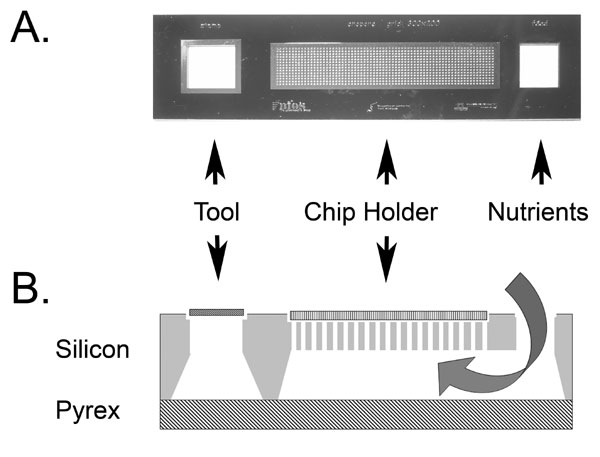
Fig. 1.

Culture chip use and manufacture. (A) Cross-section of set-up for RIE of a 36 × 8-mm strip of aluminum oxide (not to scale). (B) Cross-sectional diagram of a small part of a chip, illustrating microbial growth in the central compartment (7–150 μm wide) and supply of nutrients from beneath the 60-μm thickness of the aluminum oxide and 10-μm-high walls. (C) Photograph of a platinum-coated chip placed on sheep's blood agar in a standard Petri dish.
Fig. 2.

Images of materials, growth compartments, and microbial culture on chips. (A) SEM of aluminum oxide showing pores on average 200 nm diameter. (B) Transmission light microscopy of hundreds of 20 × 20-μm compartments viewed from above. (C) SEM of 7 × 7-μm compartments from above at a 30° angle. (D) Culture of L. plantarum in six compartments of the same dimensions as C stained with a fluorogenic dye (Syto 9) after growth and imaged from above. (E) Detection of β-galactosidase activity using the fluorogenic substrate FDG, from E. coli containing plasmid pUC18 grown in a 20 × 20-μm compartment. (F) As in E with one plasmid-containing microcolony, viewed at lower magnification. (G) View of 20 × 20-μm format chip with one area supporting a GFP-expressing strain of E. coli in a background of nonfluorescent cells. (H) Previously uncultivated oligotrophic bacterium related to Dechloromonas sp. labeled by FDP metabolism and grown in a 20 × 20-μm compartment supplied by Rhine water, before recovery and identification by PCR.
The Ordyl 314 (acrylic) material forming the grid showed a degree of autofluorescence that partially interfered with the imaging of cells by fluorescence microscopy. This autofluorescence (515–730 nm) was detectable with the 10- to 400-ms exposures typically required for imaging fluorescent cells by CCD camera. To counter this, a 20-nm-thick layer of platinum was applied by sputtering over the whole chip (Fig. 1B). This was sufficient to mask autofluorescence >20-fold (515–730 nm). The platinum did not block the pores of the aluminum oxide; when measured by SEM, the decrease in average pore area due to the platinum sputtering was negligible, measuring the face of the PAO shown in Fig. 2A (P > 0.5 by Student's t test; n = 500 for both samples). The platinum-sputtered format was therefore used for all experiments.
Growth of Test Microorganisms on Chips.
The chips were tested for the growth of microorganisms. For comparison, a description of the growth of the same organisms on unmodified PAO is included in SI Methods and SI Fig. 5. Pretreatment with ethanol (20 min with 95% vol/vol) was sufficient to sterilize the chips for most applications. The chips were stored individually in sterile tubes. Chips were deployed to agar plates directly from the tubes by tapping the tube until one end of the chip contacted the agar, then withdrawing the tube so that the base of the chip contacted the agar. Capillary action rapidly wetted the chip, which was now ready for inoculation. Chips were moved between media or staining slides by using a piece of sterile Parafilm. Effective growth and segregation of all three test organisms (Lactobacillus plantarum WCFS1, Escherichia coli XL2 Blue, and Candida albicans JBZ32) was seen in compartments of inoculated chips with culture areas from 150 × 150 μm down to 7 × 7 μm (Figs. 2 and 3A). Spread plating 20 × 20-μm-compartment chips with >10 colony-forming units (cfu) per compartment indicated that >99.95% of compartments could support growth. Culturability of L. plantarum, C. albicans, and E. coli was 49%, 96%, and 97%, respectively, comparing culture on the chip with growth on unmodified PAO.
Fig. 3.

Example of bias and distribution of culture on chips. (A) Original image (inverted transmission light microscopy image; lighter areas show growth of L. plantarum) of a single field of view of 1,170 20 × 20-μm compartments. (B) Heat-density map of local colony density, computed by using a Gaussian smoothing kernel used, to construct statistical models of chip performance and assess bias.
Inoculation with L. plantarum was used to assess biases in culturability across the chips (Fig. 3). A statistical model was used to assess whether inoculation in one compartment lead to an unusually high probability of growth in an adjacent compartment (described in detail in SI Methods). A plot of the Poisson probability distribution function of the local density of compartment occupancy with an expected density equal to the global density showed no indications of extreme deviations. In addition, a χ2 goodness-of-fit test on 5 × 5 quadrats gave no indications that the occupation of compartments deviated from a uniform Poisson point process: in a series of 12 samples with three different dilutions of bacteria, the lowest P value was 0.37. Taken together, this suggests that the manufacture process created culture areas that were highly consistent and that biases—e.g., due to differences in the etching process across the chips or between chips, or because of differences in inoculation—were minimal.
A test for local bias, in particular for the detection of an influence of the occupation of a compartment on the occupation of compartments in the immediate neighborhood, indicated only significant deviations from a random model at only the highest inoculation densities (P < 0.05 above 10% occupancy). This was due to limited spill-over of bacteria from one compartment to the other, which may happen at high densities and when the colony volume exceeds the compartment volume. Microscopic examination of chips suggested that there was no association of local bias with incomplete wall manufacture. With overnight growth of E. coli, greater invasion of orthogonally adjacent compartments was observed, increasing this bias (data not shown). Modifying the compartment size had the expected effect, altering overgrowth by an amount consistent with the time to fill the new volume, a few hours difference for most organisms. The culturability of the organisms in the test panel was not greatly affected by compartment size (data not shown). All experiments described were conducted under conditions in which spill-over was not a significant bias.
A dilution series of L. plantarum was used to assess the ability of the chip to act as a counter for cfu. By scoring growth as present or absent in a total of 150,000 compartments (a total area of <3 cm2), a linear relationship was obtained between the number of cfu in the inoculum and the percentage of compartments supporting growth over four orders of magnitude (Figs. 3A and 4). This was also found to be the case for E. coli and C. albicans (Fig. 4). These data show that viable counts can be made on these chips for very different organisms. For comparison, a conventional viable count would require multiple Petri dishes and a 10-fold dilution series of the microbial culture, with only plates containing ≈30–300 colonies acceptable for counting. Culture on the chips required 0.5 ml of nutrient agar; assuming duplicate plating, the miniaturization factor for the chip is approximately three orders of magnitude.
Fig. 4.

Culture and viable counting of microorganisms on chips. Dilution series of microorganisms plated on a series of 20 × 20-μm format chips. The number of deposited cfu per chip was calculated from the fraction of compartments supporting growth, as scored by microscopy, with the numbers corrected for inoculation of compartments with multiple cfu. Open triangles, E. coli; filled squares, C. albicans; filled diamonds, L. plantarum. Error bars show SD with lower limits not shown at lowest dilution factor because of log10 transformation of the data.
Screening and Recovery of a Variant on the Basis of Fluorescence.
To demonstrate a simple screening application, a green fluorescent protein (GFP)-expressing, ampicillin-resistant strain of E. coli (15) was diluted 1:10,000 into cells of the same strain carrying pUC18 to confer ampicillin resistance but not expressing GFP. This mixture was used to inoculate chips by spread plating and incubated on Luria agar (L-agar) for 5 h at 37°C. Inoculation at a cell density supporting growth in 8.1% of compartments across six chips (a total of 1,080,000 compartments with 87,480 supporting growth) yielded five instances where GFP-expressing cells were detected by fluorescence microscopy after systematic scanning of the chips (e.g., Fig. 2G). By manual use of a sterile toothpick, targeted by microscopy, GFP-expressing strains could be recovered, and any contaminating nonfluorescent clones were removed by subculture on agar. Contamination with nontargeted cells, when it occurred (< 50% of cases in GFP experiments), had two causes: (i) occupancy of a compartment by two clones, as seen by segregation of fluorescence in a compartment, and (ii) imprecise recovery caused by scraping the toothpick against an adjacent compartment in addition to the target, which could be deduced by damage to the platinum coat and the disturbance of microorganisms. However, these problems were not a significant obstacle to screening. A simple purification by restreaking on a recovery plate was sufficient to purify an isolate and was not always necessary. These data suggest that screening operations and targeted strain recovery can be conducted effectively with only minimal secondary purification required to achieve a purified isolate with the desired properties or phenotype.
Screening for E. coli Transformants on the Basis of β-Galactosidase Activity.
A screening was performed to recover E. coli transformed with plasmid pUC18 without using a growth-based selection marker (such as an antibiotic-resistance gene) by means of a plasmid-dependent enzyme activity; i.e., β-galactosidase expression (16). The plasmid (10 ng) was transformed into chemically competent cells of E. coli strain OmniMax (Invitrogen) according to the manufacturer's protocol. An aliquot of the transformation mixture was used for conventional plating, resulting in 1.1 × 109 ampicillin-resistant transformants per μg pUC18 DNA with 1 in 2,000 cells in the original mixture gaining ampicillin resistance. Other aliquots were analyzed on the chips for α-complementation and the production of a functional β-galactosidase. This was done by using the fluorogenic substrate fluorescein di-β-d-galactopyranoside (FDG) with a competitive inhibitor of β-galactosidase to regulate the rate of FDG hydrolysis (17, 18). The cells were loaded with the fluorogenic substrate by osmotic shock, by moving chips from growth medium to a hypertonic medium to a hypotonic medium containing FDG then to a recovery medium. Osmotic shock is a standard method of loading FDG (18), and this demonstrates an important advantage of the chip: effectively a series of changes in cell environment were made for a large number of samples. By screening six chips (a total of 113,000 microcolonies) for the most highly fluorescent microcolonies (e.g., Fig. 2 E and F), using customized GridFinder software and an automated XY table to locate and center “hits” (see SI Methods) for recovery, seven transformants were recovered successfully in 12 attempts. This was confirmed by restriction digestion of the isolated plasmids after further culture and DNA purification. No successful recoveries were made with the same number of recovery attempts of the most fluorescent microcolonies in control experiments using cells transformed with a noncomplementing plasmid (pUC18 containing an insert in the polylinker) or in mock transformations. Additionally, a metagenomic fosmid library of DNA isolated from human ileum effluent constructed in E. coli (E. Zoetendal, C. Booijnk, M. Kleerebezem, and W.M.d.V., unpublished work) was screened by the same procedure. This resulted in the detection and isolation of four β-galactosidase-producing microcolonies from 12,000 clones (data not shown).
When exchanging medium with the bulk phase, a distance of only 60 μm had to be traversed from the base of the chips to the cells on the upper surface. Therefore, the rapid exchange of salt and FDG, required for the osmotic shock experiments as described above, was effective. Control experiments were performed by using fluorescein to follow the kinetics of small-molecule exchange between the chips and the bulk medium (method described in SI Methods). In loading the chip with small molecules from beneath, an influx of fluorescein equivalent to >90% of the concentration of the medium below was achieved in <10 s. In terms of efflux from a chip loaded with fluorescein, dilution of more than two orders of magnitude occurred in <25 s.
Screening Oligotrophic Bacteria on the Basis of Organic Phosphate Metabolism.
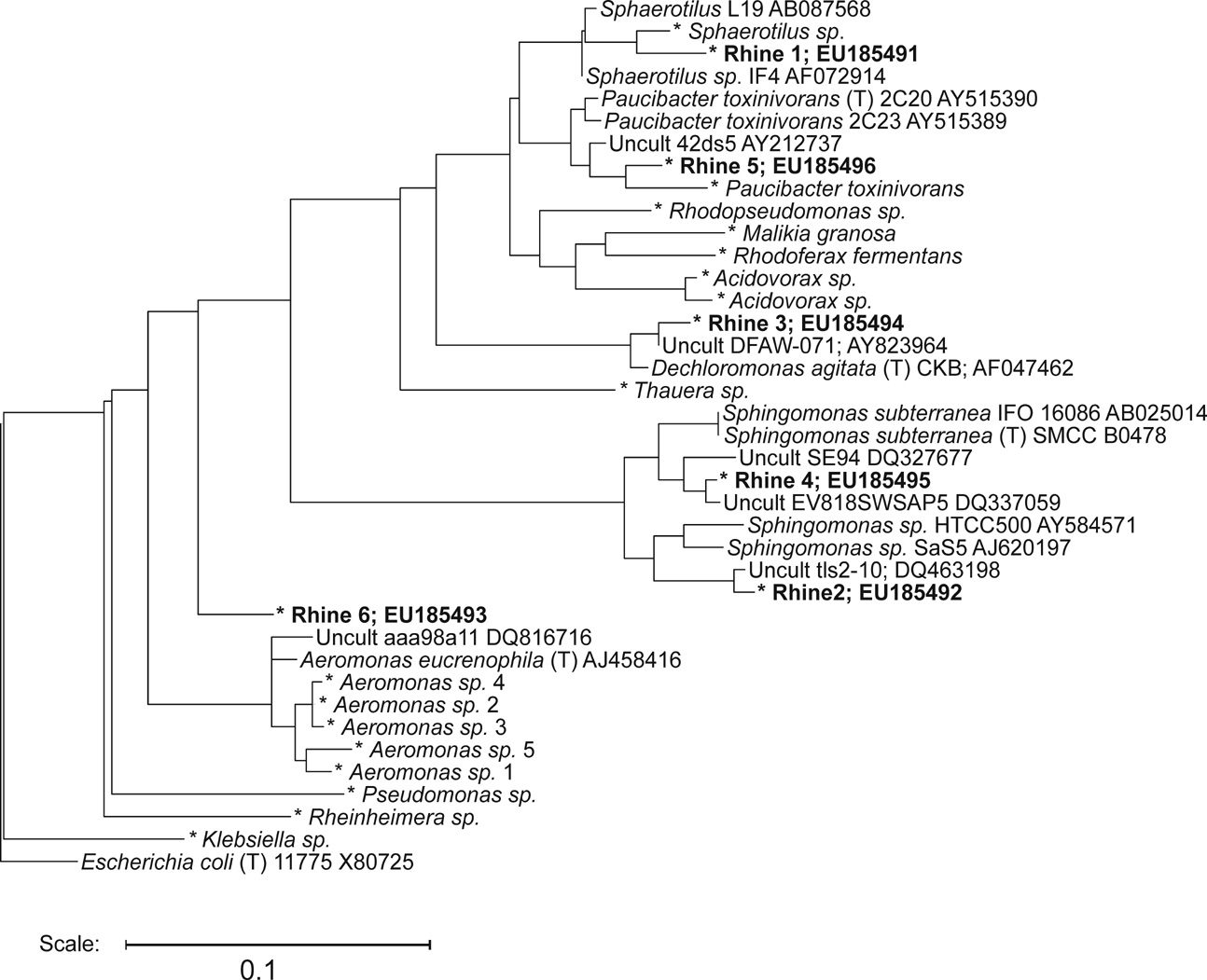
A screen of microbial samples isolated from a low-nutrient environment (the Dutch region of the Rhine River at Wageningen, The Netherlands) was undertaken on the chips. The Rhine water that was the source of the inoculum was also filter-sterilized and used as the growth medium; this was only supplemented with 0.3% (wt/vol) Gelrite (Merck) as a minimal gelling agent. The total cell count by microscopy was 8.7 × 105 cells per ml of Rhine water. After 6 days of incubation under aerobic conditions at 15°C, the viable count on Rhine water Gelrite was 4.4 × 103 cfu/ml (0.5% culturability). By counting microcolonies on chips placed on the same medium, the viable count was 6.9 × 104 cfu/ml, a >10-fold increase in culturability. These data are consistent with viable counts of oligotrophs obtained from similar environments (19). Higher viable counts are known to be observed when assessing microcolonies grown on porous supports than visible colonies on agar (20). Chips (n = 25, average compartment occupancy of 4.6%) were screened after culture for 6 days (207,000 microcolonies). Selection of microcolonies for recovery was on the basis of uptake of fluorescein 3,6-diphosphate (FDP) and rapid conversion to fluorescein (21) (Fig. 2H). The criteria for recovery were the conversion of FDP and microcolony labeling >100-fold more intense than the mean, and a cell morphology likely to be prokaryotic. Less than 1 in 2,000 cfu fit these criteria. The fluorescence of FDP-converting cells was sufficiently intense that it was clear from imaging the toothpick during recovery that the intended sample was being picked up. Hundreds of cells were recovered from each target strain. These were identified after DNA extraction by PCR amplification, sequencing of the variable regions of DNA encoding 16S rRNA, and comparative analysis with the RDP II ribosomal database (22, 23). Of 23 recovered strains, 22 amplified and gave good quality sequence. This suggests that recovery was from a single bacterial strain. A more detailed phylogenic analysis of the recovered strains is available in SI Fig. 6. Of the previously cultured organisms, several were from recently identified groups known to be involved in organic phosphate degradation, including Paucibacter toxinivorans and Malikia granulosa (23–26). Of the six previously uncultured organisms, the nearest relatives were P. toxinivorans, and members of the genera Dechloromonas, Sphaerotilus, Aeromonas, and Sphingomonas. At least one newly cultured isolate (Rhine 6) may be a member of an uncultured genus. The remainder of strains were identified to the genus level and included Aeromonas, Acidovorax, Klebsiella, and Rhodobacter spp. Attempts to perform a similar screening on PAO without the compartment structure failed because of cross-contamination by microorganisms capable of spreading across the surface over the 6- to 9-day incubation period. Taken together, these data suggest that HTS for a phenotype, followed by recovery, from a complex microbial population was possible on the chips.
Discussion
A microengineering method has been used to create a series of simple but effective microbial growth chips based around a nanoporous support. The manufacturing method was reliable; compartments on the micrometer scale could be constructed with precise dimensions that were consistent and homogeneous with respect to microbial growth. Almost every compartment appeared to be available for culture, showing that selective removal of the laminate without degrading the functional properties of the PAO beneath was efficient. An advantage of the RIE method is that the spacing of the compartments directed by the shadow mask is extremely precise, which is an important feature when other tools (e.g., printing, inoculation, or recovery systems) need to be aligned with the growth chip.
Culture and containment of three very different microorganisms was demonstrated. The culturability of these organisms on the chips was similar to that on unmodified PAO. If necessary, improved containment was possible by increasing wall height. However, given that microcolonies of hundreds of cells can be contained in the standard format (SI Table 1), the current height was sufficient for most detection and assay methods. Manual recovery, or with a basic micromanipulator, of variant cells was possible at a greater dilution than could have been performed by any conventional multiwell plate. In the micrometer-scale wells, the microcolonies are efficiently supplied with nutrients from below and oxygen from above by diffusion.
The ready recovery of organisms from these chips, and that 7–40 million compartments can be placed on a 96-well footprint, suggests that the chips are advantageous for the detection of rare variants and mutations in HTS. The assay for β-galactosidase activity demonstrates potential in enzyme detection and therefore applicability in industrial strain improvement. Additionally, this screen required a succession of changes in cell environment at high throughput, achieved by simply moving the chip and taking advantage of the short diffusion distance through the PAO. This illustrates the versatility of the chips; there are many contexts where changing the nutrients, removing waste products, or changing other effectors is advantageous. Furthermore, organisms were isolated from the environment and screened on the basis of uptake and conversion of the organophosphate dye FDP (21). A diverse collection of species, including previously uncultured organisms, could therefore be grown to microcolonies of hundreds of cells and screened on the basis of a phenotype. This is an additional illustration of HTS (>200,000 samples), one that opens the way to the creation of strategies for culture of new organisms or the creation of metagenomic libraries biased toward expressing desired phenotypes. Additionally, the same river water that was the source of the organisms screened was used on the chips as a growth medium. This approach may be usable in other situations, such as soil samples or food, where the environment can be kept close to the natural one for long enough to purify, identify, or obtain genes from otherwise intractable strains. The barriers formed in the chip appear to help in this, reducing cross-contamination from spreading organisms or from differential growth rates, which are likely in any complex microbiological sample.
To facilitate the automation and throughput of the system required for HTS, we have integrated additional elements. These include a chip holder with a microscope slide footprint (SI Fig. 7). This holder can be autoclaved, permits growth in situ, and facilitates positioning of the chip on an automated XY table. Additionally, software enhances the ability to locate a specific growth area, which aids recovery of organisms of interest. To our knowledge, this is the only method that can offer millions of microbial culture areas with such a small footprint, almost 348,000 per cm2 in the 7 × 7-μm format and 62,500 per cm2 for the 20 × 20-μm (20-μm-wide wall) configurations. Other growth formats, such as microcapillary arrays and highly subdivided multiwell plates (up to 1,000 compartments per cm2), with a lower sample density have been described (6, 7). Conceptually, the culture chip we describe is more closely related to multiwell plates that have a filter base (e.g., Nunc and ref. 13) but with a far higher sample number. Subdivided, polystyrene-based “living” arrays have also been described for the analysis of single mammalian cells. These were manufactured with up to 200,000 compartments per chip (27). However, these arrays were not designed for cell division and do not involve a porous support. Finally, systems based around fluorescence-activated cell sorters are sometimes perceived as the only systems with sufficient throughput for enzyme-based strain improvement (28). The chips described here challenge this view and offer advantages over cell sorters in allowing rapid changes in the environment and analysis under a wide variety of culture conditions. This opens up possibilities such as screening a chemical library or in recovery from drug treatments.
We show that, in addition to HTS, more basic microbiological culture operations can be undertaken because the chips are versatile. For example, simply by spread plating such a chip with a suspension of cells, a viable cell count can be done that reduces the need for a dilution series. Dilution of microorganisms is a barrier to automating microbiology. The culture chips were used for quantification of cfu over four orders of magnitude range, something that normally requires a stack of agar plates; this suggests applications in environmental monitoring and quality control. The chip presents culture in a regular grid format that is more amenable to interpretation by software than colonies distributed on an unstructured surface; this makes the position of colony growth predictable and therefore easily scored, and the large number of compartments support good statistical analysis. While agar can be used as a matrix to supply nutrients to cells, there is also the possibility of providing nutrients and test compounds by other means. Non-agar matrices or liquid medium supplied beneath the chip are alternatives. The growth medium can also be a complex or turbid one yet still allow imaging of organisms. Alternative nutrient sources have uses within screening but also may have ecological applications (e.g., in the optimization of the growth of organisms not readily cultivated on agar or typing organisms on the basis of nutritional requirements) and diagnostics (e.g., antibiotic sensitivity testing of pathogens). Additionally, the chip is small enough not to require a large incubator increasing potential uses outside of the laboratory. PAO has also been used as scaffolds for molecular assays, such as flow-through microarrays capable of rapidly detecting the interaction of biomolecules (29, 30). Through control of individual microwells, the micro-Petri dish may be integrated in a more complex lab-on-a-chip system, with opportunities for a variety of applications in, e.g., drug discovery and HTS (31–33). There is therefore the potential for the direct integration of culture-based assays and enrichment with molecular detection and other technologies.
Materials and Methods
Chip and Chip Holder Manufacture.
Chips were created by using 36 × 8-mm strips of PAO as a starting point (sold as Anopore, manufactured by Whatman). Briefly, the process was one of lamination of the upper surface with an Ordyl 314 acrylic film (Elga Europe) and then removal of the laminate from defined areas by using RIE directed by means of a shadow mask. The result was a grid of walls delineating discrete culture areas. After etching a 20-nm-thick layer of platinum was sputtered over the upper surface of the chip. Additionally, a process of photolithography and etching was used to create holders for the chip with the footprint of a microscope slide. The chip manufacture process is illustrated in Fig. 1A, and the construction of both chip and chip holder is described in detail in SI Methods.
Culture of Microorganisms on Chips and PAO.
A test panel comprising a Gram-positive bacterium (L. plantarum WCFS1), a Gram-negative bacterium (E. coli XL2 Blue), and a yeast (C. albicans JBZ32) (11) was used to assess the growth of microorganisms on the chips. Ethanol-sterilized chips were placed on the appropriate (prewarmed) agar plate and inoculated with up to 10 μl of culture. After waiting 10 min for the inoculations to dry onto the chip, the plates were incubated inverted and subsequently imaged by transmission light microscopy through the agar plate and chip. Unmodified PAO strips and agar without this material were also inoculated to assess relative culturability (described more fully in SI Methods). For the growth of L. plantarum, PAO strips or microbial chips were placed on MRS agar (Oxoid) and incubated under anaerobic conditions at 37°C. Culture of C. albicans was on chips or unmodified PAO placed on Sabouraud agar with incubation at 37°C under aerobic conditions. Culture of E. coli was on L-agar with incubation at 37°C under aerobic conditions. In counting experiments, compartments were scored for growth manually by using transmission light microscopy after 5 h (E. coli), 9 h (L. plantarum), and 6 h (C. albicans) of incubation. Where necessary, bacteria were stained with Syto 9 (Invitrogen) in situ through the pores of the chip from beneath (11).
GFP Screening and Strain Recovery Experiment.
E. coli XL2 Blue was transformed with the plasmid pEGFP1 (Clontech), expressing of GFP (15, 16). As a control, the same strain was transformed with the plasmid pUC18. The GFP-expressing strain was diluted 1:10,000 with the nonfluorescent strain, spread plated on six chips, and incubated for 5 h on L-agar plates with 200 μg/ml ampicillin at 37°C. This dilution for spread plating was made with the aim that 10% of the compartments on each chip should support growth, so that in most cases only a single cfu would be inoculated per growth area. After incubation, the chips were scanned by microscopy (11) to visualize compartments supporting the growth of GFP-expressing cells. The percentage of compartments supporting growth was scored, imaging 1,170 compartments per field of view for the 20 × 20-μm configuration. Recovery of GFP-expressing cells was accomplished by hand, using a sharp, sterile toothpick (tip <60 μm in diameter) targeted by microscopy. The result of each recovery attempt was streaked on an L-agar plate with ampicillin.
Screening for Transformation Events.
Transformation and recovery of a plasmid was performed on the chips by direct screening of β-galactosidase activity, without antibiotic selection, to identify plasmid-containing clones. OmniMax-competent cells (Invitrogen) were transformed with 10 ng of plasmid pUC18 (16). As controls, cells were transformed with pUC18 containing an insert in the polylinker to inactivate α-complementation. Inoculation was by spread plating dilutions from transformation mixes directly onto chips. Incubation of chips was on L-agar plates with 100 μM IPTG and 0.5 mM d-glucose for 5 h at 37°C. An on-chip osmotic shock was used to load cells with the fluorogenic β-galactosidase substrate FDG (Sigma) (17). A 1-mm layer of L-agar with 0.3 M NaCl was poured on a sterile microscope slide and used to transfer the chips to subject the microcolonies on each chip to a hypertonic environment. After 10 min, the chips were transferred to a second set of slides coated with 1/5 strength Luria broth gelled with 1.5% agar with 100 μM FDG and 20–100 μM phenylethyl-d-thiogalactopyranoside (18). After 10 min, chips were transferred to a third set of slides covered with L-agar for cell recovery and FDG hydrolysis (15 min at 37°C). More than 110,000 microcolonies were screened by fluorescence microscopy in each experiment. Recovery of plasmid-containing clones was performed in a fashion similar to the GFP screening experiment but using L-agar plates containing X-gal and IPTG at standard concentrations (16) without antibiotics. Ten recoveries were made per experiment, targeting the most fluorescent microcolonies from six chips. Plasmid recovery was confirmed by liquid culture of blue colonies in 1.5 ml of Luria broth in the absence of antibiotics with plasmid preparation and restriction digestion (16).
Screening for Oligotrophs Metabolizing Organophosphate Dye.
Samples of water from the Rhine River (The Netherlands) were plated on plates of filter-sterilized Rhine water solidified with 0.3% (wt/vol) Gelrite (Merck) directly or on chips placed on this medium. Incubation of samples was at 15°C for 6–9 days under aerobic conditions. Additionally, filter counts were made with the Sybr II Gold strain to determine the number of microorganisms in the samples (20). After incubation, chips were transferred to slides with low-melting-point agarose containing the fluorogenic substrate FDP at 100 μM (21). After incubation for 20 min at 15°C, the chips were transferred to a second slide without dye and imaged. Recovery of selected samples was by toothpick with the criteria for selection being unusually strong FDP conversion to fluorescein (>100-fold greater than the mean for microcolonies grown from this source) and observation of cellular morphology, selecting for strains likely to be prokaryotic. Bacterial cells were harvested by using a toothpick. A few hundred cells were resuspended in 20 μl of InstaGene matrix (Bio-Rad), and the bacterial DNA was extracted. The partial DNA sequence of 16S rRNA genes of the isolates was determined after PCR amplification (22). Stain identification and phylogenetic analysis used the Ribosomal Database Project II, Release 9.52 (23).
Supplementary Material
Acknowledgments
We thank PamGene International for PAO; J. van der Oost for discussions; M. Beerthuyzen for technical assistance; T. Koonan, R. Kuiper, and E. Mols for software; J. Rademaker for rRNA analysis; and E. Zoetendal for access to a metagenomic library. This work was supported by Top Institute Food and Nutrition Project C-015.
Abbreviations
- FDG
fluorescein di-β-d-galactopyranoside
- FDP
fluorescein 3,6-diphosphate
- HTS
high-throughput screening
- L-agar
Luria agar
- PAO
porous aluminum oxide
- RIE
reactive ion etching.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0701693104/DC1.
References
- 1.Carter KC. Essays of Robert Koch. Portsmouth, NH: Greenwood; 1987. [Google Scholar]
- 2.Bochner B. Nat Rev Genet. 2003;4:309–314. doi: 10.1038/nrg1046. [DOI] [PubMed] [Google Scholar]
- 3.Tringe SG, von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW, Podar M, Short JM, Mathur EJ, Detter JC, et al. Science. 2005;308:554–557. doi: 10.1126/science.1107851. [DOI] [PubMed] [Google Scholar]
- 4.Raoult D, Fournier PE, Drancourt M. Nat Rev Microbiol. 2004;2:151–159. doi: 10.1038/nrmicro820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kell DB, Brown M, Davey HM, Dunn WB, Spasic I, Oliver SG. Nat Rev Microbiol. 2005;3:557–565. doi: 10.1038/nrmicro1177. [DOI] [PubMed] [Google Scholar]
- 6.Lafferty M, Dycaico MJ. Methods Enzymol. 2004;388:119–134. doi: 10.1016/S0076-6879(04)88011-X. [DOI] [PubMed] [Google Scholar]
- 7.Brenan CJ, Morrison T, Stone K, Heitner T, Arrin K, Kanigan TS, Hess R, Kwon S-J, Pan J-G. Proc SPIE Int Soc Opt Eng. 2002;4626:560–569. [Google Scholar]
- 8.Zengler K, Walcher M, Clark G, Haller I, Toledo G, Holland T, Mathur EJ, Woodnutt G, Short JM, Keller M. Proc Natl Acad Sci USA. 2002;99:15681–15686. doi: 10.1073/pnas.252630999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaeberlein T, Lewis K, Epstein SR. Science. 2002;296:1127–1129. doi: 10.1126/science.1070633. [DOI] [PubMed] [Google Scholar]
- 10.Binnerup SJ, Højberg O, Sørensen J. J Microbiol Methods. 1998;31:185–192. [Google Scholar]
- 11.Ingham CJ, van den Ende M, Pijnenburg D, Wever PC, Schneeberger PM. Appl Environ Microbiol. 2005;71:8978–8981. doi: 10.1128/AEM.71.12.8978-8981.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ingham CJ, van den Ende M, Wever PC, Schneeberger PM. J Med Microbiol. 2006;55:1511–1519. doi: 10.1099/jmm.0.46585-0. [DOI] [PubMed] [Google Scholar]
- 13.Ferrari BC, Binnerup SJ, Gillings M. Appl Environ Microbiol. 2005;71:8714–8720. doi: 10.1128/AEM.71.12.8714-8720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Congchum Z, Chunsheng Y, Duifu D. J Micromech Microeng. 2004;14:663–666. [Google Scholar]
- 15.Chalfie M, Kain SR, editors. Green Fluorescent Protein: Properties, Applications and Protocols. Vol 47. New York: Wiley; 2006. Methods of Biochemical Analysis. [Google Scholar]
- 16.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Lab Press; 1989. [Google Scholar]
- 17.Rotman B. Proc Natl Acad Sci USA. 1961;47:1981–1991. doi: 10.1073/pnas.47.12.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russo-Marie F, Roederer M, Sager B, Herzenberg LA, Kaiser D. Proc Natl Acad Sci USA. 1993;90:8194–8198. doi: 10.1073/pnas.90.17.8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer SP. Int Microbiol. 2000;4:203–211. [PubMed] [Google Scholar]
- 20.Ferrari BC, Binnerup SJ, Gillings M. Appl Environ Microbiol. 2005;71:8714–8720. doi: 10.1128/AEM.71.12.8714-8720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner JK, Setayeshgar S, Sharon LA, Reilley JP, Brun YV. Proc Natl Acad Sci USA. 2006;103:11772–11777. doi: 10.1073/pnas.0602047103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klijn N, Weerkamp AH, de Vos WM. Appl Environ Microbiol. 1991;57:3390–3393. doi: 10.1128/aem.57.11.3390-3393.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, Bandela AM, Cardenas E, Garrity GM, Tiedje JM. Nucleic Acids Res. 2007;35:952–956. doi: 10.1093/nar/gkl889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spring S, Wagner M, Shumann P, Kämpfer P. Int J Syst Evol Microbiol. 2005;55:621–629. doi: 10.1099/ijs.0.63356-0. [DOI] [PubMed] [Google Scholar]
- 25.Rapala J, Berg KA, Lyra C, Niema M, Manz W, Suomalainen S, Paulin L, Lahti K. Int J Syst Evol Microbiol. 2005;55:1563–1568. doi: 10.1099/ijs.0.63599-0. [DOI] [PubMed] [Google Scholar]
- 26.Sing BK, Walker A. FEMS Microbiol Rev. 2006;30:428–471. doi: 10.1111/j.1574-6976.2006.00018.x. [DOI] [PubMed] [Google Scholar]
- 27.Yamamura S, Kishi H, Tokimitsu Y, Kondo S, Honda R, Rao SR, Omori M, Tamiya E, Muraguchi A. Anal Chem. 2005;74:8050–8056. doi: 10.1021/ac0515632. [DOI] [PubMed] [Google Scholar]
- 28.Aharoni A, Griffiths AD, Tawfik DS. Curr Opin Chem Biol. 2005;9:210–216. doi: 10.1016/j.cbpa.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Wu Y, de Kievit P, Vahlkamp L, Pijnenburg D, Smit M, Dankers M, Melchers D, Stax M, Boender PJ, Ingham CJ, et al. Nucleic Acids Res. 2004;32:e123. doi: 10.1093/nar/gnh118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang M, Trofin L, Mota MO, Martin CR. Anal Chem. 2005;77:6243–6249. doi: 10.1021/ac0508907. [DOI] [PubMed] [Google Scholar]
- 31.Whitesides GM. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 32.van den Berg A, Bergveld P. Lab Chip. 2006;6:1266–1273. doi: 10.1039/b612120a. [DOI] [PubMed] [Google Scholar]
- 33.King KR, Wang S, Irima D, Jayaraman A, Toner M, Yarmush ML. Lab Chip. 2007;7:77–85. doi: 10.1039/b612516f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}