

Abstract
Disrupted-in-schizophrenia 1 (DISC1) was initially discovered through a balanced translocation (1;11)(q42.1;q14.3) that results in loss of the C terminus of the DISC1 protein, a region that is thought to play an important role in brain development. Here, we use an inducible and reversible transgenic system to demonstrate that early postnatal, but not adult induction, of a C-terminal portion of DISC1 in mice results in a cluster of schizophrenia-related phenotypes, including reduced hippocampal dendritic complexity, depressive-like traits, abnormal spatial working memory, and reduced sociability. Accordingly, we report that individuals in a discordant twin sample with a DISC1 haplotype, associating with schizophrenia as well as working memory impairments and reduced gray matter density, were more likely to show deficits in sociability than those without the haplotype. Our findings demonstrate that alterations in DISC1 function during brain development contribute to schizophrenia pathogenesis.
Keywords: inducible, transgenic mouse, depressive, working memory, sociability
Given the natural history (1) and neuropathology of schizophrenia (2, 3), genes predisposing to the disorder are expected to play a role in brain development (4, 5). Disrupted-in-schizophrenia 1 (DISC1) is unique among the several candidate genes for schizophrenia in that its potential involvement is based on both cytogenetic (6, 7) and linkage-based evidence (8–10). Although functionally significant variants have not yet been identified, and the particular DISC1 markers and haplotypes associating with the syndrome are not consistent across studies (11–14), a developmentally regulated role of DISC1 in the pathophysiology of schizophrenia appears reasonable given that expression of DISC1 is most intense during perinatal development in rodent brain (15). DISC1 protein is known to form a functional complex with the developmentally regulated proteins Nudel and Lis1 (16–18). Interfering with the DISC1 complex has been shown to disrupt cell migration, neurite outgrowth, and synaptogenesis (16, 19–21).
Results
Deriving the Inducible DISC1 C-Terminal Fragment (DISC1-cc) Transgenic Mouse.
We derived transgenic mice expressing a DISC1-cc under the control of the α-calmodulin kinase II promoter (22), which is active only in primary neurons of the forebrain. DISC1-cc spans residues 671–852, which is the crucial portion of DISC1 for binding to NUDEL and Lis1 (16–18, 21). The DISC1-cc protein is fused to a HA-tagged mutant (G521R) estrogen receptor ligand-binding domain (LBD) (Fig. 1a). It is important to note that this mutant form of LBD is unable to bind its natural ligand (i.e., estrogen) but instead is activated specifically by tamoxifen (23). In this inducible, reversible transgenic system, the transgenic protein is sequestered by heat-shock chaperone proteins, is degraded, and therefore is nonfunctional without the inducer (tamoxifen). When tamoxifen binds the LBD, the fusion protein complex, which includes the regulated protein (i.e., DISC1-cc), undergoes a conformational switch such that the transgenic protein is freed from the chaperone proteins and can now be functional; consequently, tamoxifen is quickly metabolized, thus rendering the transgenic protein nonfunctional again (23). Western blot analyses confirmed the expression of the DISC1-cc protein in the cortex, hippocampus, striatum, and cerebellum of the transgenic mice (Fig. 1b). Immunochemistry revealed transgenic protein in both cytoplasm and branches of neurons (Fig. 1c). Transgenic mice appeared normal and did not suffer from gross morphological abnormalities. The mice showed normal activity in an open-field test and do not otherwise appear to suffer from motor deficits; behavior was also normal in the elevated plus maze, suggesting that animals do not show abnormal levels of anxiety (data not shown). To test functional induction of the DISC1-cc transgenic fusion protein, we performed an immunoprecipitation assay. Mice at postnatal day 7 received a s.c. injection of tamoxifen and were killed either 6 h or 2 days later. Protein extracted from forebrain was incubated with an anti-estrogen receptor a subunit (ERa) antibody conjugated with protein A-agarose. The immunoprecipitated proteins were separated by SDS/PAGE and subjected to Western analysis. As anticipated (23), the transgenic protein DISC1-cc binds with Nudel and Lis1 6 h after induction, but not 2 days after induction or in the absence of tamoxifen (Fig. 1d). Because DISC1-cc binds to Nudel and Lis1, it should interfere with the normal binding of the endogenous DISC1 protein (dominant-negative function). Forebrain protein extracts from postnatal day 7 mice with (+Tam 6 h) or without (−Tam) tamoxifen induction were also immunoprecipitated with an anti-Nudel antibody and tested with an anti-DISC1 N-terminal antibody, which can only identify the endogenous DISC1 protein. The data showed that induction with tamoxifen reduced endogenous DISC1 protein in DISC1/Nudel complexes [supporting information (SI) Fig. 5]. These data indicate that this inducible and reversible transgenic system can be used to study the impact of disruption of DISC1 function on specific time points during brain development.
Fig. 1.

Deriving the inducible DISC1-cc transgenic mouse. (a) Schematic of the inducible DISC1-cc transgene construct. αCaMKII, α-calmodulin kinase II. (b) Western blot showing the expression of the DISC1-cc protein in the cortex (co), hippocampus (hip), striatum (st), and cerebellum (ce) of transgenic (tg) and wild-type (wt) mice. The arrowhead indicates the DISC1-cc protein, and the lower bands are the endogenous estrogen receptor. (c) Immunocytochemical labeling showing the distribution of the transgene protein in cytoplasm and dendrites of hippocampal cells. Green indicates the transgene protein or endogenous estrogen receptor, and red indicates the neuronal nucleus. (d) Immunoprecipitation assay shows that DISC1-cc protein only binds with Nudel and Lis1 6 h after tamoxifen induction and not under other conditions. The Western blot (WB) shows expression level of DISC1-cc, Nudel, and Lis1 in transgenic (tg) or wild-type (wt) mice.
It is unclear when the critical physiological changes that lead to schizophrenia take place, although there is general agreement that this pathology is initiated, at least in part, as early as the prenatal and early postnatal periods (3–5, 24). Because the inducible transgenic system we used allows temporal control over the function of the mutant protein, we tested whether induction of the DISC1-cc dominant negative impacted prefrontal and hippocampal function in a developmentally specific manner. Based on the report that transient inactivation of ventral hippocampal activity with tetrodotoxin on postnatal day 7 produces lasting behavioral changes related to schizophrenia in young adult rat (25), we decided to contrast the effects of induction of the dominant-negative DISC1-cc construct on postnatal day 7 vs. adulthood.
Spatial Working Memory Deficits.
Working memory deficits, especially in the spatial domain, have been shown to correlate with genetic susceptibility for schizophrenia in studies of twins and families (26–28), including studies of DISC1 haplotypes (29, 30). Therefore, we tested the DISC1-cc mice in a widely used spatial working memory test [the delayed nonmatched to place (DNMTP) task]. Mice were trained in the DNMTP task to a criterion of 75% correct choices over a consecutive 3-day period, after which they were tested by using 1-, 5-, 10-, and 20-s delay intervals (31). We used the percent correct DNMTP choices as the primary behavioral measure. The adult DISC1-cc transgenic mice with induction at postnatal day 7 (tg/tam/7) showed decreased percentage of correct DNMTP choices compared with the other three control groups, including adult WT mice with tamoxifen injection at postnatal day 7 (wt/tam/7), adult DISC1-cc transgenic mice (tg/veh/7), and adult WT mice (wt/veh/7) with vehicle (control) injection at postnatal day 7 (repeated ANOVA, F(3,29) > 2.366, P = 0.0192; mice number: tg/tam/7, 10; wt/tam7, 10; tg/veh/7, 7; wt/veh/7, 6), particularly at 10 s [ANOVA, F(3,29) > 4.018, P = 0.0166; Fisher's protected least significant difference (PLSD), P (tg/tam/7,tg/veh/7) = 0.0424, P (tg/tam/7,wt/tam/7) = 0.0027, P (tg/tam/7,wt/veh/7) = 0.025] and 20 s [ANOVA, F(3,29) > 2.971, P = 0.0481; Fisher's PLSD, P (tg/tam/7,tg/veh/7) = 0.0407, P (tg/tam/7, wt/tam/7) = 0.0148, P (tg/tam/7,wt/veh/7) = 0.0326], and there were no differences among the three control groups. In addition, there were no differences between DISC1-cc transgenic mice (tg/tam/A) and WT mice (wt/tam/A; ANOVA, F(1,13) > 0.677, P = 0.5713; tg/tam/A, 9; wt/tam/A, 6) when tamoxifen was injected in adulthood (6 h before test). Additionally, these two groups did not differ from the developmental induction control groups described above (Fig. 2a). These data indicate that disruption of DISC1 function early in development, at postnatal day 7, results in deficits in spatial working memory. In contrast, there is no acute effect of DISC1 disruption in adulthood.
Fig. 2.

Behavioral studies of the inducible DISC1-cc transgenic mouse. (a) DNMTP task. Adult DISC1-cc transgenic mice with induction at postnatal day 7 (tg/tam/7) have a significantly decreased ratio of correct DNMTP choices compared with all other groups. (b) Forced swimming test. The tg/tam/7 mice cease swimming significantly sooner than all other groups. (c) Sociability test. The tg/tam/7 mice spent equal amounts of time in the stimulus mouse chamber and the empty chamber away from the stimulus mouse. All other groups spent significantly more time in the stimulus mouse chamber than in the empty chamber.
Depressive-Like Traits.
Recent evidence suggests that major depression and bipolar disorder may share some susceptibility genes with schizophrenia, including DISC1 (32). In addition to schizophrenia, depression and bipolar disorder also segregated with a balanced translocation (1;11)(q42.1;q14.3) in a Scottish kindred (7). The forced swimming test is a validated behavioral model of depressive-like behaviors in rodents (33). Mice were placed in a cylindrical container filled to 20 cm with water at 25°C, and behavior was recorded for 5 min. The latency until the first floating episode >3 s was measured. Shorter latencies are thought to reflect depressive-like behavior (34, 35). The tg/tam/7 mice showed shorter latencies to the first floating reaction compared with wt/tam7, tg/veh/7, wt/veh/7 [ANOVA, F(3,57) > 3.87, P = 0.0137; Fisher's PLSD, P (tg/tam/7,tg/veh/7) = 0.025, P (tg/tam/7,wt/tam/7) = 0.0162, P (tg/tam/7,wt/veh/7) = 0.0023; mice number: tg/tam/7, 16; wt/tam/7, 14; tg/veh/7, 15; wt/veh/7, 16]. In contrast, there were no differences between the tg/tam/A and wt/tam/A mice (ANOVA, F(1,15) > 0.065, P = 0.803; mice number: tg/tam/A, 8; wt/tam/A, 9) when tamoxifen was injected in adulthood (6 h before test), and these two groups did not differ from the developmental induction control groups described above (Fig. 2b). These data indicate that the disruption of DISC1 early in development, at postnatal day 7, results in increased depressive-like traits paralleling those seen in patients with a mutated form of DISC1. As with working memory, there is also no acute effect of DISC1 disruption in adulthood.
Sociability Deficits.
Social withdrawal is one of the most disabling symptoms in schizophrenia and related psychiatric disorders (36). To test whether induction of DISC1-cc affected sociability, we used a social choice paradigm (37, 38). This test is conducted in a three-chambered apparatus in which the test mouse could move freely between a chamber including an unfamiliar 6-week-old C57BL/6 stimulus mouse enclosed in a ventilated container, another chamber including an empty container, and a middle chamber, also empty, connecting the other two. The time that the test mouse spent in the chamber containing the stimulus mouse vs. the time in the empty chamber was measured as sociability. The tg/tam/7 mice spent equivalent time in the stimulus mouse chamber and in the empty chamber away from the stimulus mouse (n = 18; t test, P = 0.384). In contrast, all control group mice, including wt/tam/7 mice(n = 17; t test, P = 0.0002), tg/veh/7 mice (n = 10; t test, P < 0.0001), and wt/veh/7 mice (n = 10; t test, P = 0.0009), spent significantly more time in the stimulus mouse chamber than in the empty chamber away from the stimulus mouse. The tg/tam/7 mice spent a shorter time in the mouse chamber compared with wt/tam7, tg/veh/7, wt/veh/7 [ANOVA, F(3,51) > 3.26, P = 0.029; Fisher's PLSD, P (tg/tam/7,tg/veh/7) = 0.0378, P (tg/tam/7, wt/tam/7) = 0.0068, P (tg/tam/7,wt/veh/7) = 0.0378; mice number: tg/tam/7, 18; wt/tam/7, 17; tg/veh/7, 10; wt/veh/7, 10] and a longer time in the empty chamber [ANOVA, F(3,51) > 3.68, P = 0.0179; Fisher's PLSD, P (tg/tam/7,tg/veh/7) = 0.0169, P (tg/tam/7, wt/tam/7) = 0.0074, P (tg/tam/7,wt/veh/7) = 0.0194]. In addition, there were no differences between the tg/tam/A and wt/tam/A mice (time in mouse chamber, ANOVA, F(1,14) > 0.087, P = 0.773; time in empty chamber, ANOVA, F(1,14) > 0.530, P = 0.479) when the tamoxifen injection is given in adulthood 6 h before test. Both tg/tam/A mice (n = 8; t test, P = 0.0153) and wt/tam/A mice (n = 8; t test, P = 0.0181) spent significantly more time in the stimulus mouse chamber, and these two groups did not differ from the developmental induction control groups described above (Fig. 2c). These data indicate that disruption of DISC1 function early in development, at postnatal day 7, results in reduced sociability. In contrast, there is no acute effect of DISC1 disruption in adulthood.
Social and Working Memory Deficits Associated with DISC1 SNPs in Humans.
Because the DISC1-cc mice were observed to have sociability deficits, we sought to determine whether inherited variations in DISC1 in humans are associated with social impairment. Functional variants of DISC1 in humans have not yet been definitively isolated, and there is no direct analog of the induced rodent DISC1-cc mutation in humans. Nevertheless, two studies of family and twin samples from the Finnish population have observed associations between haplotypes of DISC1, risk for schizophrenia, and working memory (29, 30). In addition, in the study of Finnish twins discordant for schizophrenia, the same haplotypes were associated with reduced gray matter density in the prefrontal cortex and hippocampus (29). We thus evaluated a rating scale of sociability derived from structured psychiatric interviews in relation to the DISC1 haplotype; this haplotype is associated with risk for schizophrenia, impairment in working memory, and reduced gray matter density in the prefrontal cortex and hippocampus in the same discordant twin sample from Finland (29). Individuals with this haplotype were four times more likely to show deficits in sociability than those without the haplotype (i.e., 16.6 vs. 4%; odds ratio, 3.9; 95% confidence interval, 1.3–11.6; P = 0.016). Although both the DISC1 haplotype and sociability deficits are strongly associated with schizophrenia, their association is detectable even among the healthy comparison twins (odds ratio, 6.2; 95% confidence interval, 1.4–28.0; P = 0.018), indicating a general effect of DISC1 on social behavior similar to its effects on cognition and gray matter (29). The test for association of this DISC1 haplotype with social impairment in the human sample was the only such test conducted for the present report.
Reduced Dendritic Complexity.
The studies reported above demonstrate that interfering with DISC1 function in a very specific developmental window (postnatal day 7) leads to a cluster of schizophrenia-related behavioral phenotypes. In human studies, schizophrenia-related SNP markers of DISC1 were found to associate with reduced gray matter density in the prefrontal cortex and hippocampus (29, 39). Possible alterations in neuronal structure and function during development could underlie the behavioral deficits described above. Thus, we investigated whether the activation of DISC1-cc at postnatal day 7 affected hippocampal dendritic complexity in adulthood (≥3 months). Because the DISC1 protein is most richly expressed in the dentate gyrus of the hippocampus (15, 40), we tested the dendritic complexity of granule cells; we used the DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine) fluoresce rapid neuronal labeling method (41). The numbers of dendritic branching points and intersections were counted in successive radial segments of 25-μm distances by considering the center of the soma as a reference point (Sholl's analysis) (42). The points where the dendrites cross the lines of concentric rings were taken as intersecting points. The data showed that the tg/tam/7 mice had reduced dendritic complexity when compared with the wt/tam/7, tg/veh/7, and wt/veh/7 mice (repeated ANOVA, F(3,51) > 2.076, P = 0.0115; neuron number: tg/tam/7, 14; wt/tam/7, 14; tg/veh/7, 17; wt/veh/7, 10) especially at 150-μm distance from soma [ANOVA, F(3,51) > 3.113, P = 0.0342; Fisher's PLSD, P (tg/tam/7,tg/veh/7) = 0.0095, P (tg/tam/7,wt/tam/7) = 0.0452, P (tg/tam/7,wt/veh/7) = 0.0155] (Fig. 3). These data indicate that disruption of DISC1 function early in development, at postnatal day 7, results in reduced dendritic complexity, perhaps the underlying cause for the reduction in gray matter density associated with SNP markers of DISC1 (29, 39).
Fig. 3.

Reduced dendritic complexity. The DiI fluorescent rapid neuronal labeling study showed that the tg/tam/7 mice had a reduced number of dendritic branches of granule cell in dentate gyrus compared with all other groups. Branching appears normal toward the soma but is deficient at distances beyond 150 μm from the soma.
Reduced Hippocampal Synaptic Transmission but Normal Long-Term Potentiation (LTP) in DISC1 Mice.
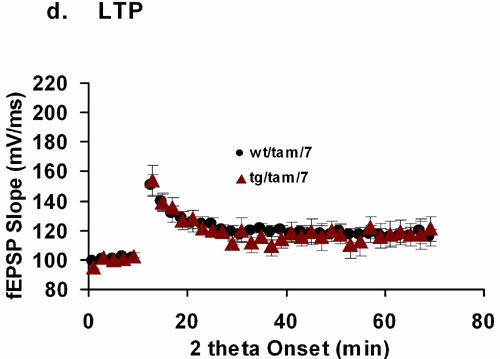
The hippocampal reduction in dendritic complexity observed in the tg/tam/7 group could result in changes in hippocampal synaptic function. Therefore, next, we determined whether the induction of the DISC1-cc at postnatal day 7 affected hippocampal synaptic transmission and short- or long-term synaptic plasticity (LTP) in adulthood (≥3 months). Hippocampal CA1 synaptic transmission was evaluated by measuring slope of field excitatory postsynaptic potentials (fEPSPs) after stimulation with a range of stimulus intensities (0–100 μA). To control for the differential recruitment of presynaptic axons in different slices, we plotted fEPSPs against presynaptic fiber volley amplitudes (Fig. 4). Analysis of the synaptic input/output (I/O) relationship showed that tg/tam/7 mice had reduced synaptic I/O function compared with the other three groups [average slope: tg/tam/7, 3.459 ± 0.287; tg/veh/7, 5.359 ± 0.365; wt/tam/7, 5.103 ± 0.236; wt/veh/7 = tg/veh, 5.789 ± 0.278; ANOVA, F(3,133) = 11.394, P < 0.0001; Fisher's PLSD, P (tg/tam/7,tg/veh/7) < 0.0001, P (tg/tam/7,wt/tam/7) = 0.0001, P (tg/tam/7,wt/veh/7) < 0.0001]. There was no slope difference among three control groups. A study of paired-pulse facilitation across different interstimulus intervals did not reveal differences among genotype/treatment groups, suggesting that activation of the DISC1-cc protein does not affect presynaptic function (SI Fig. 6a). In contrast with the reduction in basal synaptic transmission, CA1 LTP was not impaired in the developmentally induced DISC1-cc animals with either a 100-Hz tetanus, or theta burst protocols (SI Fig. 6 b–d). Together, these data demonstrate that disruption of DISC1 early in development, at postnatal day 7, is associated with reduced hippocampal synaptic transmission, a result consistent with the reduction in dendritic complexity described above. Additionally, these synaptic changes were specific because paired-pulse facilitation and LTP tested with a variety of parameters were intact.
Fig. 4.

Reduced hippocampal synaptic transmission in DISC1 mice. I/O relationship between the fiber volley amplitude and fEPSP slope was determined over a range of stimulus intensities (0–100 μA). Each point represents the mean of all slices tested for certain stimulus intensities. Error bars illustrate standard error for both axes. ANOVA analysis showed that the tg/tam/7 mice had reduced synaptic I/O function compared with the other three groups.
Discussion
Animal models are crucial to the validation of susceptibility genes in polygenic inherited disorders such as schizophrenia because the small magnitude of effect of any one locus on the disease phenotype makes it statistically difficult to isolate causative sequence variations even in relatively large samples of human populations (13, 43). In the case of DISC1, whereas functional polymorphisms have not yet been isolated by using linkage or association methods, the rare cytogenetic anomaly that led to the discovery of this gene strongly implicates DISC1 as a likely source of the linkage/association signals on 1q42 (12–14). In the absence of a known functional target, we sought to model disruption of DISC1 in relation to the functional complexes it forms with Nudel and Lis1, genes that regulate several aspects of brain development and that bind with the C terminus region of DISC1 (16–18, 20, 21), the region that is altered by the (1;11)(q42.1;q14.3) translocation (6). Our findings show that induction of a mutant C-terminal fragment of the DISC1 protein early in postnatal development results in a number of phenotypic changes that parallel changes associated with schizophrenia-related DISC1 sequence variations in humans, including impaired spatial working memory and depressive-like traits. These phenotypic effects were not observed with induction of the DISC1-cc protein in adulthood. Our animal studies demonstrate that disruption of DISC1 during development impacts on adult social behaviors in mice. Motivated by these findings, we subsequently tested this hypothesis in humans and found an association between DISC1 haplotypes and social impairment.
What is the mechanism responsible for this cluster of schizophrenia-like phenotypes? Truncation of the DISC1 protein results in reduced interaction of DISC1 with NUDEL and in impoverished neurite outgrowth in vitro (16, 21). Induction of our transgenic construct is likely to have a similar effect, because our data indicate that DISC-cc sequesters Nudel and Lis1 away from the normal endogenous DISC1. Indeed, our findings confirm that induction of the DISC1-cc transgene at day 7 interferes with normal DISC1 function and results in pronounced hippocampal synaptic, structural, and physiological deficits likely underlying the behavioral phenotypes of these mutant mice. Consistent with this perspective, previous studies showed that disrupting the interaction between the hippocampus and prefrontal cortex in development models key aspects of schizophrenia (3). These structures interact in the mediation of short- and long-term memory (24, 44) as well as sociability (45). Sequence variations in DISC1 are associated with altered physiological activity in the hippocampus during memory processing (39). Furthermore, studies in patients suggested that developmental changes in gray matter, perhaps reflecting changes in neuronal structure, may contribute to the complex behavioral phenotypes associated with schizophrenia (2, 46).
In agreement with our findings, recently mice with an N-ethyl-N-nitrosourea-induced mutation in exon 2 of mouse Disc1 were shown to have deficits in the forced swim, prepulse inhibition, and latent inhibition tests. Together with our DISC1 inducible studies, these studies demonstrate a role for DISC1 in schizophrenia (47). Additionally, a deletion variant in mDisc1 specific to the 129S6/SvEv strain has been reported (48). Disc1 was reported to terminate at exon 7 in those mice, abolishing production of the full-length protein. The mutation impaired working memory performance when transferred to the C57BL/6J genetic background (48). Subsequent work has examined expression of DISC1 protein in the 129S6/SvEv strain, and systematically compared expression with that in the C57BL/6J strain by using antibodies against >10 independent epitopes that were generated from nine independent groups. The robust detection of DISC1 in the 129S6/SvEv strain by 9 of the 10 antibodies suggests that the strain has functional DISC1 protein (49).
Remarkably, our studies demonstrated that disruption of DISC1 function early in development results in reduced dentate gyrus dendritic complexity. These changes appear to affect mostly the most distal segments of granule cell dendritic arbors, an area that receives predominantly entorhinal inputs. Thus, these results suggest that in the DISC 1 mice the hippocampus has been partially disconnected from its entorhinal input, a finding that could account for the behavioral deficits described above. Additionally, it is possible that the synaptic deficits observed in CA1, the main output of the hippocampus, reflect morphological changes similar to those that we found in the dentate gyrus. Therefore, DISC1 mutations may affect not only the input but also the output of the hippocampus.
Our findings also provide a critical functional link between the histological ramifications of altered DISC1 and the reduced gray matter density in schizophrenia that is known to vary with genetic proximity to affected individuals in monozygotic and dizygotic twins discordant for this disorder (50, 51) and to be associated with schizophrenia-related haplotypes of DISC1 (29, 39).
It is important to emphasize that the DISC1-cc mutation evaluated in this study is not a direct analog of a known functional inherited sequence variation of DISC1 in humans. Rather, it is a mutation of a region of DISC1 known to regulate neurodevelopmental processes through interactions with NUDEL and LIS. In complexly inherited neurobehavioral syndromes such as schizophrenia, it is likely that iterative genetic analyses translating between mice and humans will be necessary to resolve the functional variants contributing to disease susceptibility in humans. Mice can be used to nominate regions of the gene that have critical roles in processes associated with the pathophysiology of the disorder, and subsequent work in humans can target these regions for more dense typing and subsequent functional analysis.
Methods
Transgene Construction and Generation of DISC1-cc Mice.
The transgene (in the pMM-LBDG521R-DISC1-cc plasmid) used in these studies was designed according to a procedure previously described (23): it contains an α-calmodulin kinase II promoter, a hybrid intron in the 5′ untranslated leader, a HA virus-tag sequence, and a LBDG521R cDNA fused 5′ to the DISC1-cc cDNA (encoding protein residues 671–852, C-terminal portion of DISC1 protein), as well as a polyadenylation signal. The pMM-LBDG521R-DISC1-cc plasmid was digested with SfiI, and transgenic mice were generated by injecting the purified insert into pronuclei of C57BL/6 zygotes. Founders were backcrossed into C57BL/6N mice (Taconic Farms, Germantown, NY). All procedures used were approved by the Animal Research Committee of University of California at Los Angeles.
Functional Induction of DISC1-cc Transgenic Protein.
Mice were housed two to five per cage on a 12-h light–dark cycle. The mice received s.c. injections of tamoxifen (Sigma, St. Louis, MO; 20 mg per kg body weight) or DMSO vehicle either at postnatal day 7 or in adulthood (3 months old). The manipulated pups continued to grow into adulthood and were analyzed as described.
Immunochemistry.
Forty-micrometer sections were incubated in 1:50 rabbit antibody to ERa (sc-543; Santa Cruz, Santa Cruz, CA) and 1:200 mouse antibody to NeuN (Chemicon, Temecula, CA) overnight at 4°C. Immunoreactivity was visualized by using fluorescent secondary antibodies, Alexa Fluor 594 donkey anti-mouse IgG, and Alexa Fluor 488 donkey anti-rabbit IgG (Molecular Probes, Eugene, OR). The images were analyzed on a Zeiss (Oberkochen, Germany) LSM 510 Meta confocal microscope.
Western Blotting and Immunoprecipitation.
Samples of the hippocampus, cortex, striatum, and cerebellum of DISC1-cc and WT littermate control mice were isolated and homogenized in protein extraction buffer. Supernatants were analyzed by Western blotting. The blotting filter was probed with the ERa antibody or HA antibody (Covance, Berkeley, CA).
For the immunoprecipitation study, mice received an injection of tamoxifen on postnatal day 7 and were killed either 6 h or 2 days later. The forebrain was isolated and homogenized in protein extraction buffer. The supernatant (500 μg total protein) was incubated with anti-ERa or anti-Nudel (from L.-H. Tsai, Harvard University, Cambridge, MA and Abcam, Cambridge, MA) antibody conjugated to protein A-agarose at 4°C overnight or room temperature for 2 h. The immunoprecipitated proteins were detected by antibodies including anti-Nudel antibody and anti-Lis antibody (Abcam) and anti-DISC1 N-terminal antibody.
Sociability.
The approach of a “test” mouse toward a novel (unfamiliar) “stimulus” mouse was measured in a three-chamber apparatus (37, 38). Animals were habituated to the apparatus by placing them in the middle (empty) chamber and allowing them to explore for 5 min, with the doorways into the two side chambers open. In the test phase, an unfamiliar mouse (a 6-week-old C57BL/6J female) was enclosed in a cylinder and placed in one of the side chambers. The test mouse was allowed to explore the entire test apparatus for a 5-min session.
Spatial Working Memory Task.
Mice were food-deprived and maintained at 85% of normal body weight throughout the duration of the experiment. Spatial working memory was assessed by using a DNMTP task on a modified automated eight-arm radial maze (31). After visiting an arm for a reward, the mouse was confined in the dark on the center platform for a delay period, after which the visited and a baited adjacent arm were opened. A correct choice was recorded when the animal chose the baited arm.
Forced Swimming Test.
Mice were tested in a 1-day modified version of the forced swim test: mice were placed in a cylindrical container filled to 20 cm with water at 25°C, and behavior was recorded for 5 min. Floating was defined as immobility with only occasional slight movements required to keep the body balanced and the nose above water.
Dendritic Morphology.
Sagittal brain slices (400 μm thick) were prepared from adult mice (3–4 months old) labeled with DiI (41). Granule cells in dentate gyrus in hippocampus were examined at ×40 by using a Leica (Deerfield, IL) confocal microscope and Leica confocal software. At the same magnification, concentric circles were drawn at 25-μm equivalent intervals with the aid of a stage micrometer. The number of dendritic branching points and intersections were counted by using Sholl's analysis. Two to three neurons were traced from each animal, and five to seven mice were used per group.
Electrophysiology.
Sagittal hippocampal slices (400 μm thick) were prepared from adult mice (3–4 months old). Hippocampi were dissected in ice-cold artificial cerebrospinal fluid and recovered in artificial cerebrospinal fluid at room temperature for at least 1 h before the start of the experiment. Extracellular recordings of fEPSP were made in the stratum radiatum of CA1. fEPSPs were evoked in two pathways by stimulating the Schaffer afferent fibers at one-half to two-thirds maximum. LTP was induced by using 100 Hz per 1 s and theta-burst stimulation protocols. Statistical comparisons were made between groups with Fisher's PLSD t tests on the last 10 min of data (averaged).
Human Samples.
Two hundred thirty-two subjects were drawn from a Finnish database. Details of selection criteria, procedures, and measures are included in SI Methods because of length restrictions.
Supplementary Material
Acknowledgments
We thank Li-Huei Tsai for the anti-Nudel antibody and Rafal Czajkowski, Anna Matynia, Carrie Shyliansky, Akira Sawa, and Brian Wiltgen for critical discussions. This research was supported by National Institute of Mental Health Grant MH52857, a National Alliance for Research on Schizophrenia and Depression (NARSAD) Distinguished Investigator Award (to T.D.C.), a gift from the Staglin Music Festival for Mental Health (to T.D.C.), a NARSAD Young Investigator Award (to W.L.), and the Tennenbaum Creativity Chair (to A.J.S.). W.L. is a NARSAD Daniel X. Freedman Investigator. J.K. and L.P. were supported by the Academy of Finland Centre of Excellence in Complex Disease Genetics.
Abbreviations
- DISC1
disrupted-in-schizophrenia 1
- DISC1-cc
DISC1 C-terminal fragment
- LBD
ligand-binding domain
- ERa
estrogen receptor a subunit
- DNMTP
delayed nonmatched to place
- PLSD
protected least significant difference
- tam
tamoxifen
- LTP
long-term potentiation
- fEPSP
field excitatory postsynaptic potential
- I/O
input/output.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0706900104/DC1.
References
- 1.Cannon TD, Bearden CE, Hollister JM, Rosso IM, Sanchez LE, Hadley T. Schizophr Bull. 2000;26:379–393. doi: 10.1093/oxfordjournals.schbul.a033460. [DOI] [PubMed] [Google Scholar]
- 2.Selemon LD, Goldman-Rakic PS. Biol Psychiatry. 1999;45:17–25. doi: 10.1016/s0006-3223(98)00281-9. [DOI] [PubMed] [Google Scholar]
- 3.Lipska BK, Weinberger DR. Neurotox Res. 2002;4:469–475. doi: 10.1080/1029842021000022089. [DOI] [PubMed] [Google Scholar]
- 4.Arnold SE, Talbot K, Hahn CG. Prog Brain Res. 2005;147:319–345. doi: 10.1016/S0079-6123(04)47023-X. [DOI] [PubMed] [Google Scholar]
- 5.Weinberger DR. Arch Gen Psychiatry. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- 6.Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, Devon RS, Clair DM, Muir WJ, Blackwood DH, et al. Hum Mol Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- 7.St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, Gosden C, Evans HJ. Lancet. 1990;336:13–16. doi: 10.1016/0140-6736(90)91520-k. [DOI] [PubMed] [Google Scholar]
- 8.Hennah W, Varilo T, Kestila M, Paunio T, Arajarvi R, Haukka J, Parker A, Martin R, Levitzky S, Partonen T, et al. Hum Mol Genet. 2003;12:3151–3159. doi: 10.1093/hmg/ddg341. [DOI] [PubMed] [Google Scholar]
- 9.Hodgkinson CA, Goldman D, Jaeger J, Persaud S, Kane JM, Lipsky RH, Malhotra AK. Am J Hum Genet. 2004;75:862–872. doi: 10.1086/425586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hwu HG, Liu CM, Fann CS, Ou-Yang WC, Lee SF. Mol Psychiatry. 2003;8:445–452. doi: 10.1038/sj.mp.4001235. [DOI] [PubMed] [Google Scholar]
- 11.Harrison PJ, Weinberger DR. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- 12.Porteous DJ, Thomson P, Brandon NJ, Millar JK. Biol Psychiatry. 2006;60:123–131. doi: 10.1016/j.biopsych.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Ross CA, Margolis RL, Reading SA, Pletnikov M, Coyle JT. Neuron. 2006;52:139–153. doi: 10.1016/j.neuron.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 14.Ishizuka K, Paek M, Kamiya A, Sawa A. Biol Psychiatry. 2006;59:1189–1197. doi: 10.1016/j.biopsych.2006.03.065. [DOI] [PubMed] [Google Scholar]
- 15.Austin CP, Ky B, Ma L, Morris JA, Shughrue PJ. Neuroscience. 2004;124:3–10. doi: 10.1016/j.neuroscience.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 16.Ozeki Y, Tomoda T, Kleiderlein J, Kamiya A, Bord L, Fujii K, Okawa M, Yamada N, Hatten ME, Snyder SH, et al. Proc Natl Acad Sci USA. 2003;100:289–294. doi: 10.1073/pnas.0136913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brandon NJ, Handford EJ, Schurov I, Rain JC, Pelling M, Duran-Jimeniz B, Camargo LM, Oliver KR, Beher D, Shearman MS, et al. Mol Cell Neurosci. 2004;25:42–55. doi: 10.1016/j.mcn.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 18.Morris JA, Kandpal G, Ma L, Austin CP. Hum Mol Genet. 2003;12:1591–1608. doi: 10.1093/hmg/ddg162. [DOI] [PubMed] [Google Scholar]
- 19.Kamiya A, Kubo K, Tomoda T, Takaki M, Youn R, Ozeki Y, Sawamura N, Park U, Kudo C, Okawa M, et al. Nat Cell Biol. 2005;7:1167–1178. doi: 10.1038/ncb1328. [DOI] [PubMed] [Google Scholar]
- 20.Millar JK, Christie S, Porteous DJ. Biochem Biophys Res Commun. 2003;311:1019–1025. doi: 10.1016/j.bbrc.2003.10.101. [DOI] [PubMed] [Google Scholar]
- 21.Kamiya A, Tomoda T, Chang J, Takaki M, Zhan C, Morita M, Cascio MB, Elashvili S, Koizumi H, Takanezawa Y, et al. Hum Mol Genet. 2006;15:3313–3323. doi: 10.1093/hmg/ddl407. [DOI] [PubMed] [Google Scholar]
- 22.Mayford M, Wang J, Kandel ER, O'Dell TJ. Cell. 1995;81:891–904. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 23.Kida S, Josselyn SA, de Ortiz SP, Kogan JH, Chevere I, Masushige S, Silva AJ. Nat Neurosci. 2002;5:348–355. doi: 10.1038/nn819. [DOI] [PubMed] [Google Scholar]
- 24.Lipska BK, Aultman JM, Verma A, Weinberger DR, Moghaddam B. Neuropsychopharmacology. 2002;27:47–54. doi: 10.1016/S0893-133X(02)00282-8. [DOI] [PubMed] [Google Scholar]
- 25.Lipska BK, Halim ND, Segal PN, Weinberger DR. J Neurosci. 2002;22:2835–2842. doi: 10.1523/JNEUROSCI.22-07-02835.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cannon TD. Curr Opin Psychiatry. 2005;18:135–140. doi: 10.1097/00001504-200503000-00005. [DOI] [PubMed] [Google Scholar]
- 27.Cannon TD, Huttunen MO, Lonnqvist J, Tuulio-Henriksson A, Pirkola T, Glahn D, Finkelstein J, Hietanen M, Kaprio J, Koskenvuo M. Am J Hum Genet. 2000;67:369–382. doi: 10.1086/303006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glahn DC, Therman S, Manninen M, Huttunen M, Kaprio J, Lonnqvist J, Cannon TD. Biol Psychiatry. 2003;53:624–626. doi: 10.1016/s0006-3223(02)01641-4. [DOI] [PubMed] [Google Scholar]
- 29.Cannon TD, Hennah W, van Erp TG, Thompson PM, Lonnqvist J, Huttunen M, Gasperoni T, Tuulio-Henriksson A, Pirkola T, Toga AW, et al. Arch Gen Psychiatry. 2005;62:1205–1213. doi: 10.1001/archpsyc.62.11.1205. [DOI] [PubMed] [Google Scholar]
- 30.Hennah W, Tuulio-Henriksson A, Paunio T, Ekelund J, Varilo T, Partonen T, Cannon TD, Lonnqvist J, Peltonen L. Mol Psychiatry. 2005;10:1097–1103. doi: 10.1038/sj.mp.4001731. [DOI] [PubMed] [Google Scholar]
- 31.Lee I, Kesner RP. J Neurosci. 2003;23:1517–1523. doi: 10.1523/JNEUROSCI.23-04-01517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Craddock N, O'Donovan MC, Owen MJ. Schizophr Bull. 2006;32:9–16. doi: 10.1093/schbul/sbj033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porsolt RD, Le Pichon M, Jalfre M. Nature. 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- 34.Strekalova T, Spanagel R, Bartsch D, Henn FA, Gass P. Neuropsychopharmacology. 2004;29:2007–2017. doi: 10.1038/sj.npp.1300532. [DOI] [PubMed] [Google Scholar]
- 35.Slawecki CJ, Thorsell AK, El Khoury A, Mathe AA, Ehlers CL. Neuropeptides. 2005;39:369–377. doi: 10.1016/j.npep.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 36.Harvey CR, Curson DA, Pantelis C, Taylor J, Barnes TR. Br J Psychiatry. 1996;168:562–570. doi: 10.1192/bjp.168.5.562. [DOI] [PubMed] [Google Scholar]
- 37.Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. Genes Brain Behav. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- 38.Sankoorikal GM, Kaercher KA, Boon CJ, Lee JK, Brodkin ES. Biol Psychiatry. 2006;59:415–423. doi: 10.1016/j.biopsych.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 39.Callicott JH, Straub RE, Pezawas L, Egan MF, Mattay VS, Hariri AR, Verchinski BA, Meyer-Lindenberg A, Balkissoon R, Kolachana B, et al. Proc Natl Acad Sci USA. 2005;102:8627–8632. doi: 10.1073/pnas.0500515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Austin CP, Ma L, Ky B, Morris JA, Shughrue PJ. NeuroReport. 2003;14:951–954. doi: 10.1097/01.wnr.0000074342.81633.63. [DOI] [PubMed] [Google Scholar]
- 41.Grutzendler J, Tsai J, Gan WB. Methods. 2003;30:79–85. doi: 10.1016/s1046-2023(03)00009-4. [DOI] [PubMed] [Google Scholar]
- 42.Sholl DA. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- 43.Gottesman II, Gould TD. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 44.Wang GW, Cai JX. Behav Brain Res. 2006;175:329–336. doi: 10.1016/j.bbr.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 45.Sams-Dodd F, Lipska BK, Weinberger DR. Psychopharmacology. 1997;132:303–310. doi: 10.1007/s002130050349. [DOI] [PubMed] [Google Scholar]
- 46.Glantz LA, Lewis DA. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- 47.Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, Lerch JP, Trimble K, Uchiyama M, Sakuraba Y, et al. Neuron. 2007;54:387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 48.Koike H, Arguello PA, Kvajo M, Karayiorgou M, Gogos JA. Proc Natl Acad Sci USA. 2006;103:3693–3697. doi: 10.1073/pnas.0511189103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishizuka K, Chen J, Taya S, Li W, Millar JK, Xu Y, Clapcote SJ, Hookway C, Morita M, Kamiya A, et al. Mol Psychiatry. 2007;12:897–899. doi: 10.1038/sj.mp.4002024. [DOI] [PubMed] [Google Scholar]
- 50.Cannon TD, Thompson PM, van Erp TG, Toga AW, Poutanen VP, Huttunen M, Lonnqvist J, Standerskjold-Nordenstam CG, Narr KL, Khaledy M, et al. Proc Natl Acad Sci USA. 2002;99:3228–3233. doi: 10.1073/pnas.052023499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Erp TG, Saleh PA, Huttunen M, Lonnqvist J, Kaprio J, Salonen O, Valanne L, Poutanen VP, Standertskjold-Nordenstam CG, Cannon TD. Arch Gen Psychiatry. 2004;61:346–353. doi: 10.1001/archpsyc.61.4.346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}