

SUMMARY
Several polyketide secondary metabolites are predicted to undergo isoprenoid-like β-alkylations during biosynthesis. One such secondary metabolite is myxovirescin A1, produced by Myxococcus xanthus. Myxovirescin is of special interest in that it appears to undergo two distinct β-alkylations. Additionally, the myxovirescin biosynthetic gene cluster lacks tandem thiolation domains required in the synthesis of other β-branched secondary metabolites. To probe the origins of the β-branches in myxovirescin, we heterologously overexpressed the proteins predicted to be responsible for myxovirescin β-alkylation and reconstituted their activities in vitro on model substrates. Our results confirm that myxovirescin undergoes two isoprenoid-like β-alkylations during its biosynthesis, including an unprecedented βethylation. The study of its biosynthesis should shed light on the scope and requirements for isoprenoid-like biosynthetic logic in a polyketide context.
INTRODUCTION
Many secondary metabolites of biological interest are biosynthesized by huge protein complexes that work in a thiotemplated, assembly line-like fashion. One such class of secondary metabolites is the polyketides, which are produced by polyketide synthases (PKSs). PKSs possess a modular organization, in which each module is responsible for incorporation of a single malonyl building block. Each module is further subdivided into domains, each responsible for a single chemical manipulation. Incorporation of a malonyl unit requires a post-translationally phosphopantetheinylated thiolation (T) domain, which tethers intermediates to the synthase via a thioester linkage; an acyltransferase (AT) domain, which transfers malonyl units from coenzyme A (CoA) to one or more thiolation domains for incorporation into the polyketide; and a ketosynthase (KS) domain which catalyzes the decarboxylative Claisen condensation of a malonyl unit with the growing polyketide chain, extending the polyketide by one C2 unit. Additional domains, including ketoreductases, dehydratases, and enoyl reductases, may be present in particular modules to tailor the β-ketothioester resulting from each round of ketide extension [1].
As a consequence of this biosynthetic strategy, carbons in a polyketide chain can be assigned in an alternating pattern as having arisen from either the α- or β-carbon of intermediate β-ketothioesters. Methylation at nucleophilic α-carbons is common and arises from reaction with the electrophilic methyl source S-adenosylmethionine (SAM). Less common is methylation at β-carbons, which would formally require a nucleophilic methyl source. Recently, an example of such a β-methylation pathway was characterized in the pksX cluster of B. subtilis [2], subsequently demonstrated to be responsible for the production of bacillaene [3]. Bacillaene β-methylation requires the activity of a cassette of five proteins, including an HMG-CoA synthase homolog (HCS), a free-standing ketosynthase, a free-standing thiolation domain, and two enoyl-CoA hydratase homologs (ECH). Other biosynthetic gene clusters containing this cassette include those encoding curacin [4] and jamaicamide [5] from Lyngbya majuscula, mupirocin from Pseudomonas fluorescens [6], and pederin/onnamide from an unknown symbiont of Paederus fuscipes [7].
An additional biosynthetic gene cluster that contains this cassette produces myxovirescin A1, also known as antibiotic TA (1, Figure 1) [8]. Myxovirescin A1 is a highly reduced, hybrid polyketide/non-ribosomal peptide, with adhesive properties [9] and moderate antibiotic activity [10]. Though myxovirescin is produced by the genome-sequenced Myxococcus xanthus str. DK1622, a full annotation of the gene cluster was only recently reported [11]. Myxovirescin possesses two β-branches at C12 and C16, but inspection of the structure and biosynthetic gene cluster suggests that there are deviations from the β-methylation logic and organization characterized in bacillaene. First, though there are two putative β-branches, only a subset of the β-alkylation proteins is duplicated, suggesting that the free-standing ketosynthase TaK and the enoyl-CoA hydratase pair TaX and TaY may act on both branch incorporations (Figure 1B). Second, neither of the putative β-branches is methyl, requiring either biofunctionalization subsequent to initial β-methylation, or the incorporation of a non-methyl group by isoprenoid-like logic.
Figure 1.

β-Branches in myxovirescin A1. (A) Structure of myxovirescin A1, with C12 methoxymethyl and C16 ethyl β-branches highlighted in yellow. (Bb) Several proteins are thought to be involved in the introduction of the C12 and C16 β-branches. An acyltransferase (AT), ketosynthase (KS), HMG-CoA synthase homolog (HCS), two enoyl-CoA hydratase homologs (ECH), and free-standing donor and assembly-line acceptor thiolation domains (T) are required for each β-branch incorporation; there are two copies of the donor and acceptor thiolation domains, acyltransferase, and HMG-CoA synthase homolog encoded by the myxovirescin biosynthetic cluster. (C) Schematic of the β-methylation pathway as characterized in bacillaene [2].
Finally, myxovirescin β-alkylation at C12 and C16 is predicted to occur when the growing polyketide is tethered to the sixth and eighth thiolation domains of the megasynthase Ta1 (Ta1-T6 and Ta1-T8), respectively, both of which exist as single thiolation domains within the assembly line. In contrast, β-branch incorporation in bacillaene was reconstituted in vitro on a set of two consecutive thiolation domains [2], and runs of two or three consecutive thiolation domains appear to correspond to branch sites in other predicted β-branched secondary metabolites.
In the case of the C12 methoxymethyl branch, genetic experiments demonstrated that the terminal C12 methoxymethyl carbon is built up stepwise by hydroxylation and O-methylation of a β-methyl precursor [11]. Further, feeding studies show that the internal C12 methoxymethyl carbon arises from C2 of acetate, consistent with the β-methylation pathway characterized in bacillaene [12]. We thus predicted that the C12 methyl precursor of myxovirescin is incorporated by the following sequence: (i) malonation of phosphopantetheinylated (holo) TaB or TaE catalyzed by either the N- or C-terminal acyltransferase domain in TaV; (ii) TaK-catalyzed decarboxylation to yield Ac-S-TaB or Ac-S-TaE; (iii) nucleophilic attack of the acetyl unit on the β-ketothioester linked to Ta1-T6, catalyzed by TaC or TaF; (iv) dehydration and decarboxylation of the HMG-S-Ta1-T6 derivative by TaX and TaY to form the α,β-unsaturated, β-methylated product. (Figure 1C).
In theory, a simple extension of this pathway could lead to C16 β-ethylation. Based on feeding studies, the C16 ethyl substituent arises from the methylmalonyl/propionyl/succinyl metabolic manifold [12], and replacement of acetyl with propionyl as the proximal nucleophile would yield a parallel ethylation pathway. To probe the origins of the myxovirescin β-branches and test the ability of non-methyl β-branches to be incorporated by isoprenoid-like biosynthetic logic, we heterologously overexpressed the predicted β-branch incorporating proteins from the myxovirescin biosynthetic cluster and characterized their activities in vitro. Using this strategy, we reconstituted the formation of analogs of both the C12 β-methyl and C16 β-ethyl intermediates on model substrates. Our observations reveal that C16 β-ethylation follows a biosynthetic logic similar to the β-methylation strategy previously characterized, and provide further insight into the scope and requirements of isoprenoid-like β-alkylation in polyketide biosynthetic contexts.
RESULTS
Protein overexpression
We overexpressed the putative β-branch-incorporating proteins in E. coli or Pseudomonas putida K2442, including ketosynthase TaK, HMG-CoA synthases TaC and TaF (from P. putida), and the enoyl-CoA hydratases TaX and TaY. We also expressed the free-standing thiolation domains TaB and TaE, and the sixth, seventh, and eighth assembly-line thiolation domains from Ta1 (Ta1-T6, Ta1-T7, and Ta1-T8) separately as C-His6 and N-GST fusions. We verified that the thiolation domains were active by assaying their abilities to serve as substrates for the promiscuous phosphopantetheinyl transferase Sfp from B. subtilis [13] using 2-14C-AcCoA (data not shown). Finally, we also overexpressed the C-terminal AT domain of TaV (designated TaVC) as the N-GST fusion and full-length TaV both as wild-type and the S446A mutant, in which TaVC activity is abolished. Notably, TaVC could only be expressed as the N-terminal GST fusion, whereas the N-terminal AT domain of TaV (designated TaVN) could only be expressed in the context of full-length TaV. In each case, expression yields were on the order of 3–10 mg/L, and purity was >90% by SDS-PAGE (Supplementary Information).
Formation of malonyl-S-T
Myxovirescin is biosynthesized by a “trans-AT” cluster. First reported in leinamycin biosynthetic cluster [14], individual modules in trans-AT PKS clusters do not contain dedicated acyltransferase domains. Instead, free-standing AT domains load multiple thiolation domains within the cluster in trans. In the myxovirescin cluster, there are only two acyltransferase domains, both found in TaV. We focused our initial efforts on TaVC because it is more homologous to the malonyl-loading acyltransferase PksC (46% identical, 57% similar) from the bacillaene cluster than is TaVN (18% identical, 35% similar). To test whether TaVC activates malonyl units for incorporation as the C12 branch, we incubated holo-TaB and holo-TaE with radiolabeled acetyl-, propionyl-, malonyl-, and methylmalonyl-CoA in the presence of TaVC. By gel autoradiography, we observed that TaVC catalyzed the loading of only 2-14C-malonyl units onto holo-TaB and, at higher malonyl-CoA concentrations, holo-TaE (Figure 2).
Figure 2.
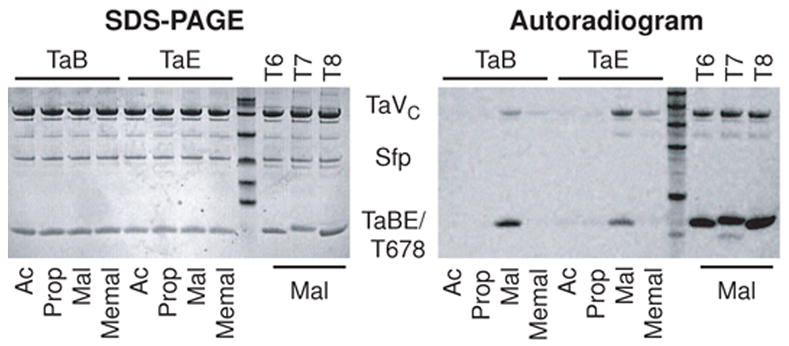
Autoradiogram of TaVC activity. TaVC was individually combined with phosphopantetheinylated TaB, TaE, Ta1-T6, Ta1-T7 and Ta1-T8 in the presence of radiolabeled acyl-CoA thioesters and subjected to denaturing gel electrophoresis. The phosphopantetheinyl transferase Sfp remaining from in situ generation of the holo-thiolation domains is visible. (Left) Coomassie-stained gel; molecular weight standards are 15 kD, 21 kD, 28 kD, 41 kD, and 58 kD. (Right) Autoradiogram; molecular weight standards are 15 kD, 21 kD, 28 kD, and 45 kD. Ac, acetyl; Prop, propionyl; Mal, malonyl; Memal, methylmalonyl.
Because there are only two acyltransferases in the myxovirescin cluster, we speculated that TaVC is responsible for malonating not only TaB or TaE for C12 β-methylation but also assembly-line thiolation domains for standard ketide extension. We therefore tested the ability of TaVC to malonate holo-Ta1-T6, Ta1-T7, and Ta1-T8. Gratifyingly, we observed TaVC-catalyzed malonyl transfer from coenzyme A to these three thiolation domains in vitro, confirming that TaVC was also the acyltransferase responsible for activation of malonyl units for ketide extension (Figure 2). The ability of TaVC to catalyze the selective transfer of malonyl from coenzyme A to holo-TaB, Ta1-T6, Ta1-T7, and Ta1-T8 was confirmed using mass spectrometry in a single-pot reaction in which only the malonated thiolation domains were observed after incubation with a mixture of acetyl, propionyl, malonyl, methylmalonyl, and succinyl coenzyme A and TaVC (data not shown).
Decarboxylation of malonyl-S-T
TaK is annotated as a ketosynthase domain, but lacks the catalytic cysteine required for Claisen condensation [15]. The ketosynthase PksF from the bacillaene cluster also lacks this cysteine but maintains decarboxylase activity, generating Ac-S-T units to serve as nucleophiles in the bacillaene β-branching sequence. We hypothesized that TaK possesses similar activity and plays an analogous role in the myxovirescin β-branching sequences. Having demonstrated that TaVC generates Mal-S-TaB and Mal-S-TaE, we next tested the ability of TaK to decarboxylate these substrates. We generated 1,3-14C-Mal-S-TaB and 1,3-14C-Mal-S-TaE by treatment of TaB or TaE with 1,3-14C-Mal-CoA and Sfp. We then subjected the Mal-S-T substrates to TaK and measured the radioactivity remaining in the protein fraction after trichloroacetic acid precipitation. We observed that TaK catalyzed rapid decarboxylation of Mal-S-TaB to form Ac-S-TaB, but showed no activity toward Mal-S-TaE (Figure 3).
Figure 3.
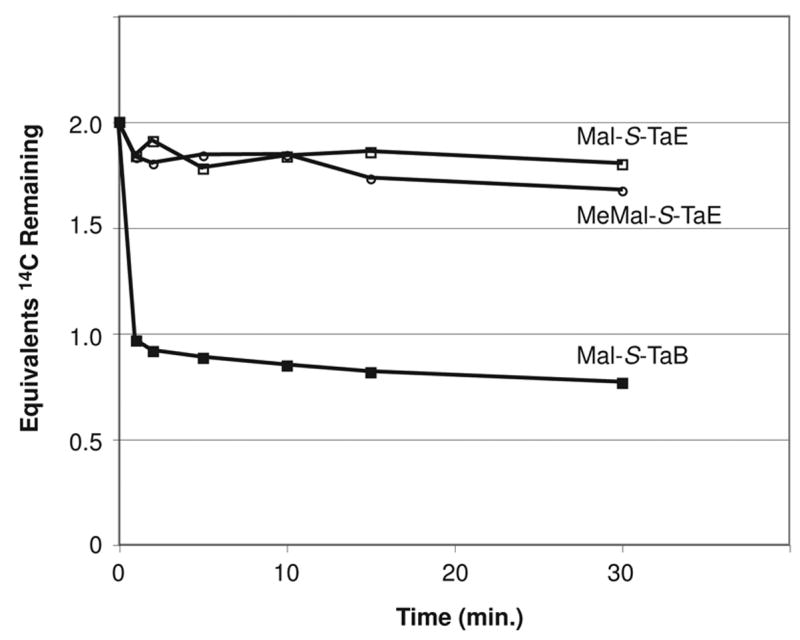
Radioassay of TaK activity. 1,3-14C-Mal-S-TaB (filled squares), 1,3-14C-Mal-S-TaE (open squares), and 1,3-14C-Memal-S-TaE (open circles) were incubated with TaK. At several time points, reaction aliquots were withdrawn and quenched by addition of trichloroacetic acid, and residual protein-linked radiolabel was measured by liquid scintillation counting. Loss of protein-linked radiolabel is associated with generation of 1-14C-Ac-S-TaB from 1,3-14C-Mal-S-TaB with concomitant evolution of 14CO2. Each point represents the average of three trials.
HMG-S-Ta1-T6 formation
Based on the above experiments, we concluded that Mal-S-TaE and Ac-S-TaB are the two potential nucleophiles generated by the action of TaVC and TaK utilized in C12 β-methylation. We hypothesized that TaC or TaF catalyzes the attack of an acetyl enolate nucleophile, generated either by deprotonation of Ac-S-TaB or decarboxylation of Mal-S-TaE, on a β-ketothioester linked to Ta1-T6. To aid in electrophoretic analysis of these experiments, we used the N-GST fusions of the acceptor thiolation domains, and chose acetoacetyl (Acac)-S-thiolation domains as model β-ketothioester substrates. We were optimistic that TaC and TaF would recognize this simplified substrate based on the reactivity of their bacillaene homolog PksG toward similar substrates [2]. We observed efficient formation of HMG-S-Ta1-T6(N-GST) by autoradiography upon incubation of 2-14C-Ac-S-TaB and Acac-S-Ta1-T6(N-GST) in the presence of TaC, but no reaction with 2-14C-Mal-S-TaE (Figure 4A). We also observed formation of a Mal-TaF acyl-enzyme intermediate, but did not observe further transfer of the malonyl. A small amount of radiolabel transfer was detected in the TaF and negative control lanes, which we attribute to a slow non-enzymatic hydrolysis/transthiolation sequence. The GST tag on the acceptor thiolation domains did not affect the reaction, as we saw similar patterns of reactivity when we swapped the GST tag to TaB and TaE (data not shown). Based on these observations, we conclude that the less-efficient malonation of holo-TaE by TaVC is unrelated to C12 β-methylation and perhaps artifactual, and that TaC and Ac-S-TaB are the catalyst and nucleophile donor in the formation of the HMG-like C12 β-methyl precursor. Additionally, an increase in mass of 60.02 Da was observed for Ta1-T6 when Acac-S-Ta1-T6 was incubated with Ac-S-TaB and TaC, consistent with the formation of HMG-S-Ta1-T6 (data not shown). The reactivity of TaC toward Ac-S-TaB is consistent with the observation that taB and taC are translationally coupled, suggesting that there is some benefit to enforcing a 1:1 stoichiometry of TaB and TaC in vivo. Also, recent in vivo experiments using the ΔtaB and ΔtaE knockout strains revealed that TaC is specific for TaB as a nucleophile donor scaffold in accord with the above observations [16].
Figure 4.
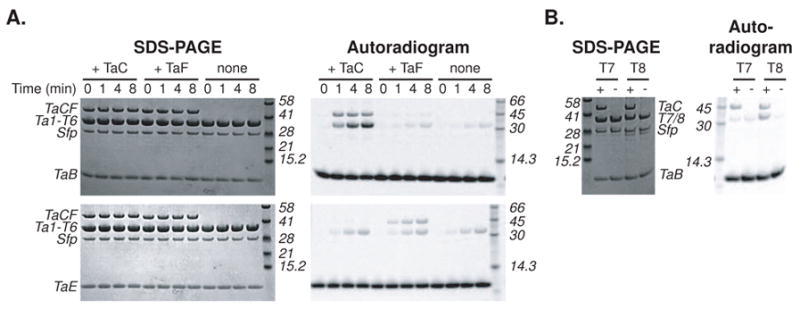
Autoradiogram of TaC and TaF activity. (A) 2-14C-Ac-S-TaB (top) or 2-14C-Mal-S-TaE (bottom) was combined with Acac-S-Ta1-T6 in the presence or absence of TaC or TaF. Reaction aliquots were withdrawn at 0, 1, 4, and 8 minutes and subjected to denaturing gel electrophoresis and autoradiography. The phosphopantetheinyl transferase Sfp remaining from in situ generation of the acyl-holo-thiolation domains is visible. (B) 2-14C-Ac-S-TaB was combined with Acac-S-Ta1-T7 (predicted not to serve as a β-branch acceptor) or Acac-S-Ta1-T8 (predicted to serve as the C12 β-ethyl branch acceptor) in the presence (+) or absence (−) of TaC for five minutes. The reaction mixtures were subjected to denaturing gel electrophoresis and autoradiography.
To ascertain whether β-branch acceptor thiolation domains could be distinguished from one another and from non-β-branch acceptor thiolation domains, we also tested Ta1-T7 (a non-β-branch acceptor) and Ta1-T8 (the predicted C16 β-branch acceptor). We observed efficient formation of HMG-S-Ta1-T8(N-GST), but not HMG-S-Ta1-T7(N-GST), from Ac-S-TaB and the respective Acac-S-thiolation domains in the presence of TaC. As no β-branch incorporation is predicted to occur when the growing polyketide is docked to Ta1-T7, this further supports the conclusion that β-methylation occurs on Ta1-T6. The reactivity of Ta1-T8 is consistent with the production of a C16 methyl myxovirescin analog by the ΔtaF strain of M. xanthus [16], and implies that T6 and T8 share some recognition element that allows interaction with TaC. The apparent recognition of Ta1-T8 by TaC is also consistent with the existence of the known myxovirescin congeners myxovirescins I1 and F1 [12], which possess C16 methyl substitution, possibly due to similar in vivo reactivity of TaC toward the appropriate Ta1-T8-linked β-ketothioester.
Dehydration and decarboxylation of HMG-S-Ta1-T6
We hypothesized that TaX and TaY dehydrate and decarboxylate the HMG-S-Ta1-T6 derivative to generate the C12 β-methyl branch precursor. We tested this hypothesis using HMG-S-Ta1-T6 as a model substrate, generated by treatment of Acac-S-Ta1-T6 with Ac-S-TaB and TaC. The resulting HMG-S-Ta1-T6 was then incubated with TaX and/or TaY followed by mass spectrometric analysis. When HMG-S-Ta1-T6 was incubated with TaX, a loss of 18.01 Da was observed, consistent with TaX serving as a dehydratase (Figure 5A–C). In contrast, when HMG-S-Ta1-T6 was incubated with TaY, no reaction was observed. When HMG-S-Ta1-T6 was incubated with both TaX and TaY, a loss of 62.00 Da was observed, consistent with TaY-catalyzed decarboxylation of the dehydrated product generated by TaX. The mass changes were further confirmed by formation of the respective phosphopantetheinyl elimination fragment ions [17] (Figure 5D–F).
Figure 5.
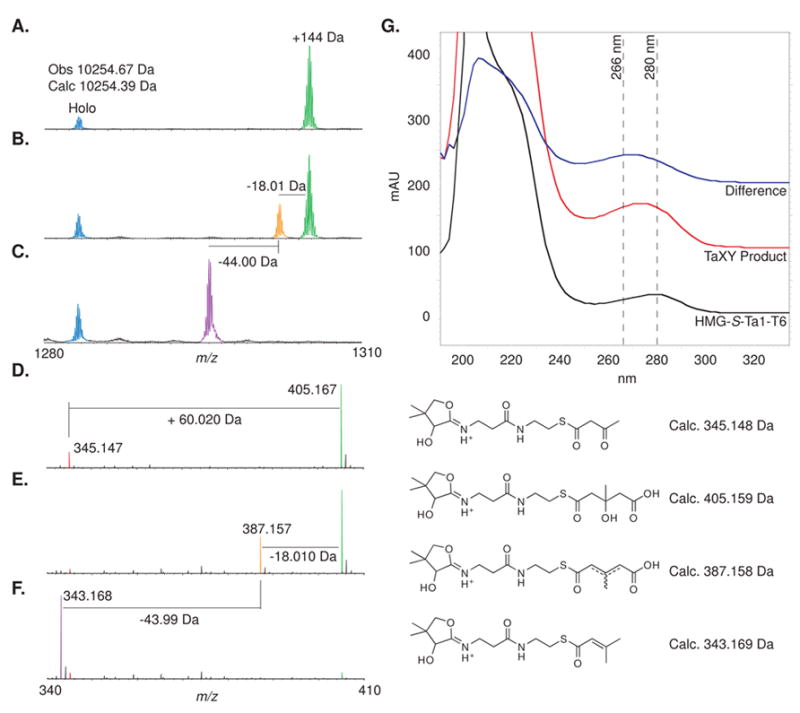
Mass spectra of TaX and TaY activity. (A–C) Broad band mass spectra of Ta1-T6 (+8 charge state) displaying holo-Ta1-T6 (blue), Acac-S-Ta1-T6 (red), HMG-S-Ta1-T6 (green), dehydrated HMG-S-Ta1-T6 (orange), and isoprenyl-S-Ta1-T6 (purple). (A) HMG-S-Ta1-T6 + TaY incubated for 15 minutes. (B) HMG-S-Ta1-T6 + TaX incubated for 15 minutes. (C) HMG-S-Ta1-T6 + TaX + TaY incubated for 15 minutes. (D–F) Mass spectra of the +1 charge state phosphopantatheinyl elimination product ions from A–C, respectively. (G) UV spectra of HMG-S-Ta1-T6 (black) and the olefin product of TaX and TaY linked to Ta1-T6 (red). The difference spectrum (blue) shows the absorbance at 266 nm of the α,β-unsaturated thioester. (Bottom) Structures of phosphopantetheinyl elimination fragment ions and their respective masses.
By querying for the presence of the α,β-unsaturated thioester chromophore at 266 nm [18], we determined that the final product is the conjugated, α,β-unsaturated olefin regioisomer (Figure 5G). Based on these experiments, it is impossible to determine the olefin regioisomer generated by TaX. In one possible reaction sequence, TaX may dehydrate α,β to the HMG thioester, followed by TaY-catalyzed direct decarboxylation. Alternatively, if TaX dehydrates β,γ to the HMG thioester, TaY may serve as an isomerase to form the α,β-unsaturated product, which could then spontaneously decarboxylate.
Formation of C16 β-ethyl branch
Though previous feeding studies demonstrated that the C16 ethyl branch arises from the succinyl/propionyl/methylmalonyl biosynthetic manifold [12], we hypothesized that propionyl was the most likely nucleophile for installation of the ethyl branch for two reasons. First, generation of the C3 enolate can occur by simple deprotonation of a propionyl unit, consistent with the precedents of TaC and PksG [2], whereas generation of the same enolate from methylmalonyl would require that the nucleophile be generated by decarboxylation. Second, processing of the HMG-like intermediate derived from attack of a succinyl enolate on a β-ketothioester would require a formal 1,2-carboxylate shift for putative TaXY-catalyzed dehydration and decarboxylation to occur. To test whether propionyl was competent to serve as a nucleophile for β-ethylation, we combined Prop-S-TaE with Acac-S-Ta1-T8 in the presence and absence of TaF. In the event, we observed an increase in mass of 74.04 Da, consistent with the formation of the HMG-like ethyl branch precursor in a TaF-dependent manner (Figure 6A,B). When the same experiment was performed with Prop-S-TaB as a nucleophile source, we saw no reaction (data not shown). We therefore conclude that TaE is the nucleophile donor scaffold cognate to TaF, consistent with the observation that taE and taF are translationally coupled as well as in vivo experiments in the ΔtaE knockout strain [16].
Figure 6.

Mass spectra of TaF, TaX, and TaY activity. (A–E) Broad band mass spectra of Ta1-T8 (+8 charge state) displaying holo-Ta1-T8 (blue), Acac-S-Ta1-T8 (red), γ-methyl-HMG-S-Ta1-T8 (green), dehydrated γ-methyl-HMG-S-Ta1-T8 (orange), and isohexenyl-S-Ta1-T8 (purple). (A) Acac-S-Ta1-T8 + PropTaE + TaF incubated for 20 minutes. (B) Acac-S-Ta1-T8 + PropTaE incubated for 20 minutes. (C) Acac-S-Ta1-T8 + PropTaE + TaF + TaY incubated for 35 minutes. (D) Acac-S-Ta1-T8 + PropTaE + TaF + TaX incubated for 35 minutes. (E) Acac-S-Ta1-T8 + PropTaE + TaF + TaX + TaY incubated for 35 minutes. (F–J) Mass spectra of the +1 charge state phosphopantatheinyl elimination product ions from A–E, respectively. (Right) Structures of phosphopantetheinyl elimination fragment ions and their respective masses.
We next attempted to identify a biosynthetic source for Prop-S-TaE within the cluster. However, under all conditions tested, we were unable to observe loading of TaE with succinyl, methylmalonyl, nor propionyl catalyzed by either acyltransferase domain in TaV (data not shown). We also tested the ability of TaK to decarboxylate 1,3-14C-Memal-S-TaE, but observed no loss of radiolabel consistent with formation of 1-14C-Prop-S-TaE (see Figure 3). Based on these observations, we propose that TaE is loaded with propionyl by an as yet unknown acyltransferase, though it is also possible that TaE is loaded with methylmalonyl and then decarboxylated by an unknown acyltransferase and decarboxylase, respectively.
Finally, we tested by mass spectrometry the ability of TaX and TaY to dehydrate and decarboxylate the HMG-like ethyl branch precursor linked to Ta1-T8. We combined Prop-S-TaE, Acac-S-Ta1-T8, and TaF with TaX and/or TaY (Figure 6C–E). When TaY only was included, we observed stable formation of the γ-methyl HMG-like thioester. However, when TaX only was added, we saw a loss of 18.01 Da consistent with dehydration, in analogy to HMG-S-Ta1-T6. When both TaX and TaY were present, we saw a loss of an additional 43.99 Da, consistent with subsequent decarboxylation to form the ethyl-branched olefin. Thus, TaX and TaY are capable of dehydrating and decarboxylating, respectively, the HMG-like ethyl branch precursor linked to Ta1-T8, and the myxovirescin biosynthetic machinery is capable of directing β-ethylation utilizing Prop-S-TaE as an ethyl source.
Biosynthetic incorporation of β-branches in myxovirescin A1
Based on the above experiments, we conclude that the following sequence of reactions introduces the methyl precursor to the C12 β-methoxymethyl branch (see Figure 1C). First, holo-TaB is malonated by action of TaVC. The resulting Mal-S-TaB is then decarboxylated by TaK to yield Ac-S-TaB. TaC catalyzes the nucleophilic attack of the acetyl enolate on a β-ketothioester linked to Ta1-T6 to form HMG-S-Ta1-T6, via an Ac-TaC intermediate. Finally, TaX dehydrates the HMG derivative, either α,β or β,γ with respect to the thioester, and TaY catalyzes a transformation that leads to overall decarboxylation yielding the Δ2 β-methylated product linked to Ta1-T6. The β-methyl group then ultimately undergoes hydroxylation and TaQ-catalyzed O-methylation, either while the intermediate is tethered to Ta1-T6 or later in the biosynthesis to yield the final C12 β-methoxymethyl branch [11].
The C16 β-ethylation pathway follows a route parallel to that of the C12 β-methylation sequence. We propose that holo-TaE is first loaded with propionyl either via direct propionation or methylmalonation followed by decarboxylation. TaF catalyzes the ensuing condensation of the propionyl enolate and the Ta1-T8 β-ketothioester to form the γ-methylated HMG-S-Ta1-T8 derivative. The γ-methylated HMG-S-Ta1-T8 derivative then undergoes dehydration and decarboxylation catalyzed by TaX and TaY, respectively, to yield the final C16 β-ethyl branch.
DISCUSSION
Both isoprene biosynthesis and polyketide biosynthesis are based on the chemistry of β-ketothioesters. In polyketide biosynthesis, β-ketothioesters are formed by the decarboxylative Claisen condensation of a malonyl unit with a thioester; in isoprene biosynthesis, an analogous coupling of two molecules of acetyl-CoA yields the β-ketothioester acetoacetyl-CoA. Isoprene biosynthesis diverges from polyketide biosynthesis at this stage. In isoprene biosynthesis, the β-carbon undergoes attack by the enolate of a third molecule of Ac-CoA to generate a branched β-hydroxy-bis-thioester, which undergoes a single thioester hydrolysis to yield the β-branched HMG-CoA monothioester. In canonical polyketide biosynthetic logic, the β-carbon does not serve as an electrophile for carbon-carbon bond formation but as an oxidation substrate, potentially undergoing ketoreduction, dehydration and reduction to form β-hydroxythioesters, α,β-unsaturated thioesters, or saturated thioesters, respectively. Thus, the net effect of the polyketide β-branching sequence described here is the utilization of isoprenoid biosynthetic logic as an alternative β-tailoring; in addition to ubiquitous polyketide β-oxidation-state adjustments, the polyketide intermediate can also undergo a formal sequence of oxygen to methylene exchange followed by tautomerization to yield the Δ2 isoprene.
Both in canonical isoprene biosynthesis as well as in the biosynthesis of the β-branched polyketide bacillaene, β-branches are limited to methyl units and do not undergo further elaboration. However, there are several examples of putative β-branched polyketides where the β-branches are non-methyl, including curacin (cyclopropane), jamaicamide (vinyl chloride), pederin/onnamide (exo-methylene) and myxovirescin (methoxymethyl and ethyl). Just as β-branching in polyketides represents a convergence with isoprene biosynthetic logic, incorporation of non-methyl β-branches would represent an extension of isoprene biosynthetic logic. This extension could occur either by β-methylation followed by subsequent branch functionalization, or by direct introduction of non-methyl β-branches. The two β-branches in myxovirescin represent examples of each of these β-alkylation strategies.
The first strategy (β-methylation followed by elaboration) is exploited in the biosynthesis of the C12 methoxymethyl branch of myxovirescin, where a β-methylated precursor is hydroxylated and O-methylated. This strategy is also likely utilized in the biosynthesis of the cyanobacterial secondary metabolite curacin. Homologs of TaX and TaY from the curacin biosynthetic cluster have been demonstrated to process HMG-CoA and HMG-S-CurB (the curacin homolog to TaB and TaE) to isoprenyl-S-CoA and isoprenyl-S-CurB, respectively, in analogy to TaX and TaY, though the generation of the HMG-S-CurB precursor has not been demonstrated [19]. It has been predicted that the thus generated isoprene is further processed to a methylcyclopropane moiety by halogenation and cyclopropanation [4], though there has not yet been an experimental validation of this hypothesis.
A more subtle example of β-branch tailoring occurs during incorporation of the C16 β-branch, likely while the intermediate is still tethered to Ta1-T8. Though we did not explicitly undertake to determine the olefin regiochemistry of the TaY-catalyzed decarboxylation during the C16 β-ethylation, we suggest based on its activity during C12 β-methyl incorporation product that it is the Δ2 isomer that is formed. In addition, generation of the α,β-unsaturated, β-ethyl derivative would then allow olefin reduction to be catalyzed by the N-terminal enoyl reductase domain of TaO, generating the saturated C16–C17 bond observed in the final product. This implies that at least in the case of the TaO enoyl reductase domain, ERs can tolerate β-substitution of the α,β-unsaturated thioester substrate and represents an additional example of β-branched substrate tailoring.
The second strategy for incorporation of non-methyl β-branches (direct incorporation) is utilized during the introduction of the C16 ethyl branch in myxovirescin. Feeding studies had suggested that the C16 ethyl arises from methylmalonyl, propionyl, or succinyl, and we demonstrated that TaF, TaX, and TaY can generate a β-ethylated substrate tethered to Ta1-T8 utilizing Prop-S-TaE as a nucleophile donor.
The structures of branches incorporated by this strategy are determined by C2-functionalization of the malonyl or acetyl branch precursors. Several C2-substituted malonyl units have been shown to be incorporated into polyketides by standard ketide extension to yield the respective α-branched products, including methylmalonyl (yielding α-methyl branches; for example, in epothilone [20]), ethylmalonyl (α-ethyl; for example, in concanamycin A [21]), and hydroxymalonyl and aminomalonyl (α-hydroxy and α-amino; both in zwittermicin [22]). Utilization of these substituted malonyl units in β-branching pathways would yield ethyl, propyl, hydroxymethyl, and aminomethyl β-branches, respectively, and thus may represent a potentially powerful, though only recently recognized, biosynthetic strategy to access β-functionalized polyketides. Notably, methoxymalonyl-CoA has also been reported as a polyketide precursor in FK520 (yielding α-methoxy branches) [23]. Utilization of methoxymalonyl-CoA in a hypothetical β-branch incorporation sequence would generate a β-methoxymethyl branch and represent an alternative β-methoxymethylation logic to that observed in myxovirescin biosynthesis. The C16 β-ethylation in myxovirescin represents the first characterized example of direct incorporation of a non-methyl branch by this strategy.
Recently, several biosynthetic gene clusters that appear to direct variations of the above β-alkylation strategies were reported. (Figure 7) In bryostatin [24], two methoxyacylidenes are proposed to be incorporated by nucleophilic attack of an acetyl enolate on a thiolation domain-linked β-ketothioester, as in bacillaene and myxovirescin. However, an alternative processing strategy, in which the HMG derivative in bryostatin is proposed to undergo β,γ-dehydration and methylation to generate the conjugated oxoester, has been proposed to account for the more complex branch structure, though there is no obvious dehydratase candidate in the cluster. Alternatively, in rhizoxin [25], a δ-lactone arises from a β-carboxymethyl branch, the biosynthesis of which diverges from the bacillaene/myxovirescin pathway by variation of the electrophile structure. Instead of utilizing a β-ketothioester, the rhizoxin biosynthetic machinery is proposed to generate an α,β-unsaturated thioester by a standard ketoreduction/dehydration sequence. This thioester can subsequently serve as a Michael-type acceptor for an acetyl enolate during the β-carbon-carbon bond formation step. In this case, because dehydration occurred prior to nucleophilic attack, decarboxylation would be impossible, as α,β-unsaturation is necessary to enable γ-decarboxylation. Finally, a biosynthesis of andrimid from Pantoea agglomerans has been proposed in which dehydration and decarboxylation of a β-hydroxy-γ-carboxylated substrate yields an exo-methylene unit [26]. The HMG-like dehydration/decarboxylation substrate, in turn, is generated by intramolecular attack of a β-ketoamide enolate on a β-ketothioester; this proposed biosynthesis diverges from the bacillaene/myxovirescin pathway by its choice of nucleophile. It is also noteworthy that there are no enoyl-CoA hydratase homologs in the andrimid cluster, suggesting that the dehydration/decarboxylation may be spontaneous. Thus, based on these examples, β-alkylation may be possible by several variants of a common core strategy of nucleophilic attack on a β-electrophile. Relative to the strategy delineated in the case of myxovirescin A1, the natures of both the nucleophile (as in andrimid) and electrophile (as in rhizoxin) may be varied, as well as HMG-processing strategies (as in bryostatin).
Figure 7.
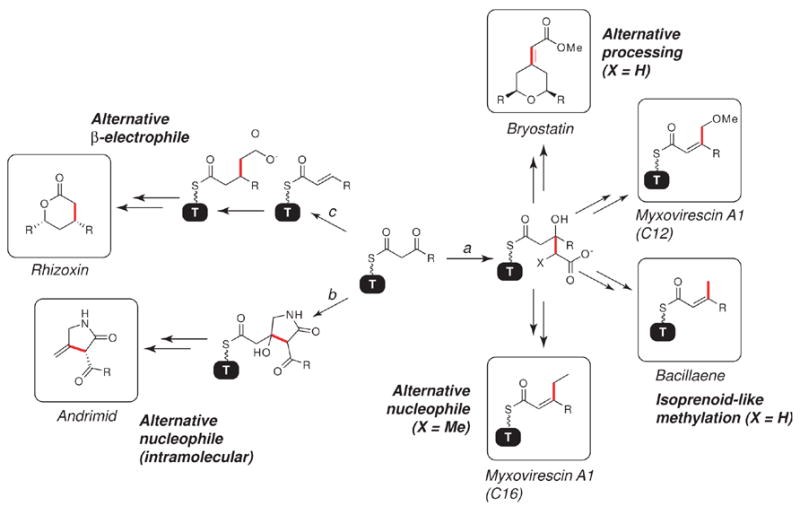
Alternative β-functionalization strategies. Variations in β-branch structure can occur by varying the branch processing sequence, the nature of the nucleophile, or the nature of the β-electrophile. In bacillaene, bryostatin, and the C12 branch of myxovirescin A1 (pathway a), differential processing of the HMG-like thioester yields either a β-methyl branch (bacillaene), a β-methoxymethyl branch (myxovirescin A1), or a methoxyacylidene branch (bryostatin). The nature of the nucleophile can also be varied; instead of an acetyl enolate, intermolecular attack of a propionyl enolate yields a β-ethyl branch in the C16 branch of myxovirescin A1. Alternatively, intramolecular nucleophilic attack by a β-ketoamide can occur to generate a five-membered ring (andrimid, pathway b). Finally, the β-electrophile can take the form of an α,β-unsaturated thioester to yield a δ-lactone (rhizoxin, pathway c). Bonds formed to the β-carbons are shown in red; the myxovirescin A1 and bacillaene β-branched products are shown linked to the respective thiolation domains to emphasize their isoprenyl-like structures.
Though the collection of β-branched natural products continues to grow, myxovirescin is one of only a small number of secondary metabolites possessing multiple β-branches with differing structures (another example is the cyanobacterial product phormidolide [27]). Thus, myxovirescin provides a unique opportunity to examine the selectivity of β-branch-incorporating enzymes toward one another. In particular, the myxovirescin biosynthetic cluster possesses two copies of the free-standing thiolation domain (TaB and TaE) and HMG-CoA synthase homologs (TaC and TaF), but only a single ketosynthase (TaK) and set of enoyl-CoA hydratases (TaX and TaY). As expected, TaX and TaY are able to act on models of both β-branch substrates, recognizing substrates that vary both in the thiolation domain scaffold as well as in the identity of the acyl group. In contrast, though we had expected TaK to recognize and decarboxylate malonyl derivatives linked to both TaB and TaE, we found that TaE was not a reactive scaffold with respect to TaK. However, TaE was not completely orthogonal to TaB, as TaVC apparently was able to recognize both thiolation domains (though less efficiently in the case of TaE). Further, TaC appears to recognize both Ta1-T6 and Ta1-T8 as acceptor scaffolds. This apparent substrate flexibility among the β-branch incorporating enzymes may have physiological relevance, and suggests that branch identity is determined by the aggregate of partial selectivities among multiple enzymes in the pathway. Several myxovirescin derivatives have been identified which vary in their C12 and C16 substituents, suggesting that the myxovirescin β-branch-incorporating enzymes show some degree of promiscuity in vivo as well. Current studies are directed at rigorously characterizing the recognition of these enzymes for one another in vitro. Identifying and characterizing the substrate selectivity of the acyltransferase responsible for propionylation of TaE will also provide insight on the means by which branch identity is determined.
Also of interest in the myxovirescin biosynthesis is the lack of tandem thiolation domains. Though there appears to be a bias toward utilizing tandem thiolation domains as scaffolds in other β-branch-containing secondary metabolites, several lines of evidence suggest that the correlation is accidental. An analysis of the tandem thiolation domains in the mupirocin cluster suggested that they act to increase biosynthetic throughput, and could act in series or in parallel [28]. To date, however, no convincing evidence has been put forward to eliminate the possibility that they act in a putative β-methylation sequence during the mupirocin biosynthesis, and we note that a run of three consecutive thiolation domains is present in the module predicted to account for β-methylation [6]. In the bacillaene cluster, the T-T regions present in PksJ and PksL were separately tested in vitro for their abilities to serve as β-methylation scaffolds, and only the tandem thiolation domains in PksL were active (C.T.C., P.C.D., N.L.K. and C.T.W., unpublished results).
Further, when site-directed mutants of the PksL-T2 region were generated, individually eliminating the phosphopantetheinylated serine residue in each thiolation domain, both domains were shown to be competent as β-methylation scaffolds [2]. Nor is it clear what factors determine whether two or three consecutive thiolation domains are present in different clusters; in many cases, the presence of a third thiolation domain seems to correlate with subsequent processing of the β-methyl unit (cyclopropanation in curacin, halogenation in jamaicamide, and olefin isomerization in pederin/onnamide), though this does not appear to hold for mupirocin. Finally, runs of up to nine consecutive thiolation domains have been observed in the PKS-like biosynthetic machinery for polyunsaturated fatty acids in marine bacteria and protists, though their biochemical roles remain unknown [29]. Thus, though there appears to be an empirical correlation between the presence of tandem thiolation domains and biosynthetic β-alkylation, it appears that in clusters containing the β-alkylation cassette, such enzymatic organization is neither necessary (e.g., myxovirescin) nor sufficient (e.g., PksJ in the bacillaene cluster) for β-methylation to occur.
In sum, the biosynthesis of myxovirescin represents an additional example of isoprene-like logic in a polyketide context. Uniquely among other β-branch-containing secondary metabolites, myxovirescin does not require tandem thiolation domains. Further, myxovirescin biosynthesis includes two separate β-branching sequences, at C12 and C16, requiring selective recognition of trans-acting domains for their respective reaction partners. Finally, the C16 ethyl arises from a direct β-ethylation pathway, an unprecedented functionalization strategy in both isoprene and polyketide biosynthesis.
Each of these facets of myxovirescin biosynthesis is the subject of ongoing investigations and will provide insight into the logic and scope of isoprene/polyketide biosynthetic convergence.
SIGNIFICANCE
A growing number of polyketide secondary metabolites appear to undergo tailoring via an isoprenoid-like biosynthetic logic. In particular, the intermediate β-ketothioesters that are generated by the iterative Claisen coupling of malonyl subunits during ketide extension can serve as electrophiles for isoprenoid-like attack by acetyl enolates. Such biosynthetic logic corresponds to an alternative β-tailoring strategy for polyketides, allowing methylation in addition to standard oxidation state adjustments. Myxovirescin is of special interest among β-alkylated secondary metabolites in that it contains two distinct β-branches. However, though myxovirescin undergoes two β-alkylations, only a partial second set of β-alkylating proteins is present in the biosynthetic machinery. Further, the myxovirescin biosynthetic gene cluster encodes only single thiolation domains, whereas other β-branch-incorporating gene clusters encode tandem thiolation domains at the sites corresponding to β-alkylation. Finally, myxovirescin contains methoxymethyl and ethyl β-branches, each of which is incorporated by a distinct extension of isoprenoid-like logic: the methoxymethyl is built up by hydroxylation and methylation of a methyl precursor, whereas the ethyl is incorporated directly. Thus, myxovirescin provides an ideal system for probing the recognition requirements of β-branch-incorporating proteins for one another, as well as the scope of isoprenoid-like biosynthetic logic in a polyketide context.
EXPERIMENTAL PROCEDURES
General experimental methods
Radiochemicals were obtained from American Radiolabeled Chemicals (St. Louis, MO) and other chemicals were obtained from Sigma (St. Louis, MO). Molecular biological manipulations were performed according to standard methods. DNA was propagated in TOP10 E. coli, and heterologous expression in E. coli was performed in the BL21*(DE3) strain. Heterologous expression in P. putida was performed in the rifampicin-resistant K2442 strain [30].
Protein expression
Genes encoding proteins of interest were amplified from M. xanthus str. DK1622 genomic DNA using Phusion polymerase (New England Biolabs, Beverly, MA) and primers incorporating convenient restriction sites (see Supplementary Material for sequences). Amplicons were digested and ligated into appropriately digested expression vectors (pET-29a for C-His6-tagged fusions; pET-41a for N-GST/C-His8-tagged fusions; pET-28a for N-His6-tagged fusions), and sequences were confirmed by sequencing (Dana-Farber Molecular Biology Core Facility, Boston, MA). Vectors for the expression of TaC (N-His6) and TaF (N-His6) were obtained by digesting pET-41a containing sequence-confirmed taC or taF with AvrII and XbaI and ligating into the XbaI site of pVLT33 [30]. The presence and orientation of the inserts were verified by PCR. TaV(S446A) was generated by QuikChange (Stratagene, La Jolla, CA) according to the manufacturer’s protocol. Proteins were expressed as C-His6-tagged fusions [TaB, TaE, Ta1-T6, Ta1-T7, Ta1-T8, TaV(S446A), TaK, TaX and TaY], N-His6-tagged fusions (TaC and TaF), or N-GST/C-His8 fusions (TaVC, TaB, TaE, Ta1-T6, Ta1-T7, and Ta1-T8)
For expression, cells were grown in LB medium containing 50 μg/mL kanamycin slowly cooled from 37 C to 15 C until OD600 ≈ 0.5 (for P. putida expression incubation was started at 30 C). Cells were induced by addition of 400 μM IPTG, and incubation was continued at 15 C for 16 h. Cells were harvested by centrifugation, resuspended in lysis buffer (20 mM Tris, 500 mM NaCl, 10% glycerol pH 8.0 with 1 mM MgCl2 and 1 mM CaCl2) containing 10 μg/mL DNaseI and lysed by passage through a French press. The lysate was clarified by ultracentrifugation and batch-bound to Ni-NTA-agarose resin 90 min at 4 C. His6-tagged protein was isolated by step-gradient elution with lysis buffer containing 25–200 mM imidizole. Fractions containing protein were identified by SDS-PAGE. Appropriate fractions were combined and either dialyzed against 20 mM Tris, 50 mM NaCl, 10% glycerol pH 8.0 (2 L for 2 h at 4 C followed by 12 h against 2 L fresh buffer) or subjected to gel filtration chromatography using the same solvent system. Proteins were flash frozen in liquid nitrogen and stored at −80°C until use.
Characterization of TaVC activity
TaB, TaE, Ta1-T6, Ta1-T7, and Ta1-T8 were first phosphopantetheinylated using Sfp and coenzyme A. In a typical experiment, 5 μL of TaE (794 μM stock; 133 μM final) was combined in 30 μL total volume with 500 μM CoA, 30 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM TCEP, and 4 μM Sfp and incubated at room temperature 60 min. The holo-T domain was then added without purification to the assay mixture (12 μL total volume): 20 μM holo-T, 5 μM TaVC, 50 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM TCEP at room temperature. Reactions were initiated by adding 20 μM radiolabeled acyl-CoA and terminated by addition of denaturing SDS-PAGE loading buffer, followed by gel electrophoresis.
Characterization of TaV activity by mass spectrometry
holo-TaB, holo-TaE, holo-Ta1-T6, holo-Ta1-T7, and holo-Ta1-T8 were prepared as described above. The reactions contained all five thiolation domains (50 μM each) in a final volume of 50 μL and were initiated by the addition of TaV, TaVC, or TaVS446A (6 μM final) and a coenzyme A pool (acetyl, malonyl, methylmalonyl, propionyl, and succinyl, each 630 μM final). After 8 minutes, the reactions were terminated by the addition of an equal volume of 10% formic acid. The samples were subsequently mass analyzed via on-line LC/MS with a 7 Tesla LTQ-FTMS (Thermo Finnigan, San Jose, CA). Samples were injected onto an Agilent Zorbax 300SB-C8 2.1 × 100 mm column and were eluted using an increasing gradient of 0.1% formic acid in acetonitrile. Mass analysis was performed in positive ion mode with three repeating events: an intact low resolution ion trap scan; an intact high resolution FT scan; and a source induced dissociation/high resolution intact FT scan.
Characterization of TaK activity
Acylated TaB and TaE were generated using 1,3-14C-MalCoA or 1,3-14C-MemalCoA as above. The acyl-holo-T domains (50 μM final) were then individually combined with 50 mM HEPES pH 7.5, 1 mM TCEP, and 2 μM TaK at room temperature. Periodically, reaction aliquots corresponding to 20000 cpm were withdrawn and added to 200 μL 10% trichloroacetic acid, followed by 5 μL 20 mg/mL BSA. Precipitated protein was isolated by microcentrifugation, washed twice with 200 μL 10% TCA, and subjected to liquid scintillation counting.
Characterization of TaC and TaF activity
Acyl-holo-T domains were obtained as above. The labeled T domains were then combined as follows: 25 μM acyl donor, 8.3 μM acyl acceptor, 50 mM HEPES pH 7.5, 1 mM TCEP, 2 μM TaC/TaF. Reactions were terminated after 5 minutes by addition of denaturing SDS-PAGE loading buffer, followed by gel electrophoresis. For FTMS analysis, reactions contained in a final volume of 100 μL: 75 mM Tris, pH 7.6, 1 mM TCEP, 150 μM acyl donor, 50 μM acyl acceptor, and 4 μM TaC/TaF. Reactions were terminated after 15 minutes by the addition of an equal volume of 10% formic acid and subsequently mass analyzed as described above.
Characterization of TaX and TaY activity
HMG-S-Ta1-T6 and γ-methyl-HMG-S-Ta1-T8 were prepared by combining 150 μM acyl donor (Ac-S-TaB or Prop-S-TaE), 50 μM acyl acceptor (Acac-S-Ta1-T6 or Acac-S-Ta1-T8) and 4 μM TaC/TaF, and incubating for 20 minutes. The reactions were initiated by the addition of TaX and/or TaY (2 μM final), terminated after 15 minutes by the addition of an equal volume of 10% formic acid, and subjected to mass analysis as described above. To determine the olefin regiochemistry of the TaY-catalyzed reaction, D,L-HMG-S-Ta1-T6 was generated as above using D,L-HMG-CoA and Sfp. It was then incubated (25 μM) with 2 μM TaX and 2μM TaY in 50 mM HEPES pH 7.5, 1 mM TCEP. After 30 min, the reaction was terminated by addition of 0.1% trifluoroacetic acid and 20% acetonitrile, followed by HPLC analysis (20%–80% acetonitrile in 0.1% TFA over 30 min). A difference spectrum was then generated using the UV spectrum corresponding to the unreacted HMG-S-Ta1-T6 peak (identified by comparison with authentic HMG-S-Ta1-T6) and the UV spectrum corresponding to the product olefin peak (identified by comparison with authentic Δ2 isoprenyl-S-Ta1-T8), where the spectra were normalized to the highest-absorbing peak in each. We note that the corresponding Δ3 olefin-S-Ta1-T6 coeluted with the Δ2 product under these HPLC conditions.
Supplementary Material
Acknowledgments
This work was supported by NIH NRSA Fellowships to C.T.C. (GM081743) and P.C.D. (GM073323), NIH grants to N.L.K. (GM067725) and C.T.W. (AI042738 and GM020011), and a generous grant to N.L.K. from The David and Lucille Packard Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 2.Calderone CT, Kowtoniuk WE, Kelleher NL, Walsh CT, Dorrestein PC. Convergence of isoprene and polyketide biosynthetic machinery: Isoprenyl-S-carrier proteins in the pksX pathway of Bacillus subtilis. Proc Natl Acad Sci U S A. 2006;103:8977–8982. doi: 10.1073/pnas.0603148103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butcher RA, Schroeder FC, Fischbach MA, Straight PD, Kolter R, Walsh CT, Clardy J. The identification of bacillaene, the product of the PksX megacomplex in Bacillus subtilis. Proc Natl Acad Sci U S A. 2007;104:1506–1509. doi: 10.1073/pnas.0610503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang Z, Sitachitta N, Rossi JV, Roberts MA, Flatt PM, Jia J, Sherman DH, Gerwick WH. Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine cyanobacterium Lyngbya majuscula. J Nat Prod. 2004;67:1356–1367. doi: 10.1021/np0499261. [DOI] [PubMed] [Google Scholar]
- 5.Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 6.El-Sayed AK, Hothersall J, Cooper SM, Stephens E, Simpson TJ, Thomas CM. Characterization of the mupirocin biosynthesis gene cluster from Pseudomonas fluorescens NCIMB 10586. Chem Biol. 2003;10:419–430. doi: 10.1016/s1074-5521(03)00091-7. [DOI] [PubMed] [Google Scholar]
- 7.Piel J, Hui D, Wen G, Butzke D, Platzer M, Fusetani N, Matsunaga S. Antitumor polyketide biosynthesis by an uncultivated bacterial symbiont of the marine sponge Theonella swinhoei. Proc Natl Acad Sci U S A. 2004;101:16222–16227. doi: 10.1073/pnas.0405976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paitan Y, Orr E, Ron EZ, Rosenberg E. Genetic and functional analysis of genes required for the post-modification of the polyketide antibiotic TA of Myxococcus xanthus. Microbiology. 1999;145(Pt 11):3059–3067. doi: 10.1099/00221287-145-11-3059. [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg E, Porter JM, Nathan PN, Manor A, Varon M. Antibiotic TA: An adherent antibiotic. Bio/Technology. 1984;2:796–799. [Google Scholar]
- 10.Gerth K, Irschik H, Reichenbach H, Trowitzsch W. Antibiotics from gliding bacteria. 8. The myxovirescins, a family of antibiotics from Myxococcus virescens (Myxobacterales) Journal Of Antibiotics. 1982;35:1454–1459. doi: 10.7164/antibiotics.35.1454. [DOI] [PubMed] [Google Scholar]
- 11.Simunovic V, Zapp J, Rachid S, Krug D, Meiser P, Müller R. Myxovirescin A biosynthesis is directed by hybrid polyketide synthases/nonribosomal peptide synthetase, 3-hydroxy-3-methylglutaryl-CoA synthases, and trans-acting acyltransferases. Chembiochem. 2006;7:1206–1220. doi: 10.1002/cbic.200600075. [DOI] [PubMed] [Google Scholar]
- 12.Trowitzsch-Kienast W, Schober K, Wray V, Gerth K, Reichenbach H, Hofle G. Zur Konstitution der Myxovirescine B-T und Biogenese des Myxovirescins A. Liebigs Annalen Der Chemie. 1989;1989:345–355. [Google Scholar]
- 13.Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 14.Tang GL, Cheng YQ, Shen B. Leinamycin biosynthesis revealing unprecedented architectural complexity for a hybrid polyketide synthase and nonribosomal peptide synthetase. Chem Biol. 2004;11:33–45. doi: 10.1016/j.chembiol.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 15.Bisang C, Long PF, Cortes J, Westcott J, Crosby J, Matharu AL, Cox RJ, Simpson TJ, Staunton J, Leadlay PF. A chain initiation factor common to both modular and aromatic polyketide synthases. Nature. 1999;401:502–505. doi: 10.1038/46829. [DOI] [PubMed] [Google Scholar]
- 16.Simunovic V, Muller R. 3-hydroxy-3-methylglutaryl-CoA-like synthases direct the formation of methyl and ethyl side groups in the biosynthesis of the antibiotic myxovirescin A. Chembiochem. 2007;8:497–500. doi: 10.1002/cbic.200700017. [DOI] [PubMed] [Google Scholar]
- 17.Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL. Facile detection of acyl and peptidyl intermediates on thiotemplate carrier domains via phosphopantetheinyl elimination reactions during tandem mass spectrometry. Biochemistry. 2006;45:12756–12766. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Speir TW, Barnsley EA. The conjugation of glutathione with unsaturated acyl thiol esters and the metabolic formation of S-carboxyalkylcysteines. Biochem J. 1971;125:267–273. doi: 10.1042/bj1250267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu L, Jia J, Liu H, Hakansson K, Gerwick WH, Sherman DH. Metabolic coupling of dehydration and decarboxylation in the curacin A pathway: Functional identification of a mechanistically diverse enzyme pair. J Am Chem Soc. 2006;128:9014–9015. doi: 10.1021/ja0626382. [DOI] [PubMed] [Google Scholar]
- 20.Julien B, Shah S, Ziermann R, Goldman R, Katz L, Khosla C. Isolation and characterization of the epothilone biosynthetic gene cluster from Sorangium cellulosum. Gene. 2000;249:153–160. doi: 10.1016/s0378-1119(00)00149-9. [DOI] [PubMed] [Google Scholar]
- 21.Haydock SF, Appleyard AN, Mironenko T, Lester J, Scott N, Leadlay PF. Organization of the biosynthetic gene cluster for the macrolide concanamycin A in Streptomyces neyagawaensis ATCC 27449. Microbiology. 2005;151:3161–3169. doi: 10.1099/mic.0.28194-0. [DOI] [PubMed] [Google Scholar]
- 22.Chan YA, Boyne MT, 2nd, Podevels AM, Klimowicz AK, Handelsman J, Kelleher NL, Thomas MG. Hydroxymalonyl-acyl carrier protein (ACP) and aminomalonyl-ACP are two additional type I polyketide synthase extender units. Proc Natl Acad Sci U S A. 2006;103:14349–14354. doi: 10.1073/pnas.0603748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu K, Chung L, Revill WP, Katz L, Reeves CD. The FK520 gene cluster of Streptomyces hygroscopicus var. ascomyceticus (ATCC 14891) contains genes for biosynthesis of unusual polyketide extender units. Gene. 2000;251:81–90. doi: 10.1016/s0378-1119(00)00171-2. [DOI] [PubMed] [Google Scholar]
- 24.Sudek S, Lopanik NB, Waggoner LE, Hildebrand M, Anderson C, Liu H, Patel A, Sherman DH, Haygood MG. Identification of the putative bryostatin polyketide synthase gene cluster from “Candidatus Endobugula sertula”, the uncultivated microbial symbiont of the marine bryozoan Bugula neritina. J Nat Prod. 2007;70:67–74. doi: 10.1021/np060361d. [DOI] [PubMed] [Google Scholar]
- 25.Partida-Martinez LP, Hertweck C. A gene cluster encoding rhizoxin biosynthesis in “Burkholderia rhizoxina”, the bacterial endosymbiont of the fungus Rhizopus microsporus. Chembiochem. 2007;8:41–45. doi: 10.1002/cbic.200600393. [DOI] [PubMed] [Google Scholar]
- 26.Jin M, Fischbach MA, Clardy J. A biosynthetic gene cluster for the acetyl-CoA carboxylase inhibitor andrimid. J Am Chem Soc. 2006;128:10660–10661. doi: 10.1021/ja063194c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williamson RT, Boulanger A, Vulpanovici A, Roberts MA, Gerwick WH. Structure and absolute stereochemistry of phormidolide, a new toxic metabolite from the marine cyanobacterium Phormidium sp. J Org Chem. 2002;67:7927–7936. doi: 10.1021/jo020240s. [DOI] [PubMed] [Google Scholar]
- 28.Rahman AS, Hothersall J, Crosby J, Simpson TJ, Thomas CM. Tandemly duplicated acyl carrier proteins, which increase polyketide antibiotic production, can apparently function either in parallel or in series. J Biol Chem. 2005;280:6399–6408. doi: 10.1074/jbc.M409814200. [DOI] [PubMed] [Google Scholar]
- 29.Metz JG, Roessler P, Facciotti D, Levering C, Dittrich F, Lassner M, Valentine R, Lardizabal K, Domergue F, Yamada A, Yazawa K, Knauf V, Browse J. Production of polyunsaturated fatty acids by polyketide synthases in both prokaryotes and eukaryotes. Science. 2001;293:290–293. doi: 10.1126/science.1059593. [DOI] [PubMed] [Google Scholar]
- 30.de Lorenzo V, Eltis L, Kessler B, Timmis KN. Analysis of Pseudomonas gene products using lacIq/Ptrp-lac plasmids and transposons that confer conditional phenotypes. Gene. 1993;123:17–24. doi: 10.1016/0378-1119(93)90533-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.