

Abstract
The vascular effects of arsenic in drinking water are global health concerns contributing to human disease worldwide. Arsenic targets the endothelial cells lining blood vessels and endothelial cell activation or dysfunction may underlie the pathogenesis of both arsenic-induced vascular diseases and arsenic-enhanced tumorigenesis. The purpose of the current studies was to demonstrate that exposing mice to drinking water containing environmentally relevant levels of arsenic promoted endothelial cell dysfunction and pathologic vascular remodeling. Increased angiogenesis, neovascularization, and inflammatory cell infiltration was observed in Matrigel plugs implanted in C57BL/6 mice following 5 week exposures to 5-500 ppb arsenic (Soucy et al., 2005). Therefore, functional in vivo effects of arsenic on endothelial cell function and vessel remodeling in an endogenous vascular bed were investigated in the liver. Liver sinusoidal endothelial cells (LSEC) became progressively defenestrated and underwent capillarization to decrease vessel porosity following exposure to 250 ppb arsenic for 2 weeks. Sinusoidal expression of PECAM-1 and laminin-1 proteins, a hallmark of capillarization, was also increased by 2 weeks of exposure. LSEC caveolin-1 protein and caveolae expression were induced after 2 weeks of exposure indicating a compensatory change. Likewise, CD45/CD68 positive inflammatory cells did not accumulate in the livers until after LSEC porosity was decreased; indicating that inflammation is a consequence and not a cause of the arsenic-induced LSEC phenotype. The data demonstrate that the liver vasculature is an early target of pathogenic arsenic effects and that the mouse liver vasculature is a sensitive model for investigating vascular health effects of arsenic.
Keywords: arsenite, endothelial, caveolin-1, PECAM-1, monocyte, macrophage
Introduction
The vascular effects of arsenic in drinking water pose a global public health concern and contribute to disease in tens of millions of people worldwide (reviewed in (Navas-Acien et al., 2005;Engel et al., 1994). While the roles of environmental contaminants in the etiology of vascular diseases and in the vascular contributions to organ dysfunction remain poorly defined, epidemiological studies have associated arsenic exposures with increased risk of cardiovascular diseases (Navas-Acien et al., 2005;Wu et al., 2006;Tseng et al., 2003;Engel et al., 1994) and vascular contributions to liver disease (Mazumder, 2005). Recent reports indicated that high environmental levels of arsenic (10-100 ppm in drinking water) accelerate atherosclerosis (Bunderson et al., 2004;Simeonova et al., 2003) and promote liver vascular channel formation in rodent models (Mazumder, 2005). However, the number of studies that examine thresholds and mechanisms for the vascular effects of environmentally or human relevant arsenic exposures is limited. This limitation is confounded by the current bias that intact rodent models are insensitive to the health effects of arsenic. The vascular effects of arsenic are an exception since vascular activation and dysfunction occur in mice exposed to low ppb levels of arsenic (Kamat et al., 2005;Liu et al., 2006;Soucy et al., 2003;Soucy et al., 2005). The importance of identifying in vivo endpoints that can be used to test for the vascular effects of low to moderate arsenic exposures is underscored by the multiple dose-dependent mechanisms elicited by arsenic exposures.
Angiogenesis, neovascularization in adult tissues, is a complex process of endothelial cell proliferation, migration, and vessel maturation (reviewed in (Hayden et al., 2004;Carmeliet, 2000)). The majority of pathological angiogenesis is accompanied by recruitment of inflammatory and progenitor cells to elaborate growth factors and complete remodeling of the new vessel wall (Ruiz et al., 2006). Thus endothelial cells generate the angiogenic response, but they cannot complete vessel maturation without recruitment of pericytes and smooth muscle cells in a process called vascular myogenesis (Ricousse-Roussanne et al., 2004;Carmeliet, 2000). Pathogenic endothelial cell activation and angiogenesis in rodent models are sensitive to low, environmentally relevant arsenic exposures (Soucy et al., 2003;Soucy et al., 2005;Kamat et al., 2005). Arsenic stimulates the angiogenic process in cultured cells and neovascularization in vivo (Barchowsky et al., 1996;Kao et al., 2003;Kamat et al., 2005;Liu et al., 2006;Soucy et al., 2003;Soucy et al., 2005). However, this stimulation occurs only at low to moderate concentrations of arsenic (5-500 ppb in vivo, 0.1-5.0 μM in cell culture; (Barchowsky et al., 1996;Kao et al., 2003;Soucy et al., 2003;Soucy et al., 2005)). Concentrations of arsenic above 5 μM are toxic to confluent endothelial cells and limit tube formation in culture (Barchowsky et al., 1996;Roboz et al., 2000). Arsenic is more toxic to sub-confluent endothelial cells (Barchowsky et al., 1996) and the threshold for in vivo approximately 1-5 stimulate vascular smooth muscle cells to proliferate and to increase vascular endothelial cell toxicity in angiogenesis assays is μM (Soucy et al., 2003). In contrast, high levels of arsenic (10-50 μM) growth factor (VEGF) expression and secretion (Soucy et al., 2004). The proliferation of the smooth muscle cells may contribute to the atherogenic effects and vessel dysfunction seen following arsenic exposures in humans (Engel et al., 1994;Penn, 1990). VEGF is a primary factor for stimulating angiogenesis and plays a critical role in pathological angiogenesis in both atherosclerosis and tumors (Aggarwal et al., 2006;Carmeliet, 2000;Dalgleish et al., 2006;Hayden et al., 2004).
Angiogenesis is the rate limiting step in tumor growth. As with general effects on vascular cells, arsenic has the dual effects on tumor angiogenesis of low dose promotion and high dose inhibition (Soucy et al., 2003;Liu et al., 2006;Kamat et al., 2005;Roboz et al., 2000;Lew et al., 1999). Depending on the tumor cell type, higher doses of arsenic either inhibit VEGF expression and release (Roboz et al., 2000) or stimulate stress responses that induce VEGF (Duyndam et al., 2003). Low dose arsenic exposures (5-250 ppb in drinking water, or nM to low μM concentrations) increased neovascularization of chicken chorioallantoic membranes, stimulated inflammatory angiogenesis in vivo in a mouse Matrigel assay, and increased vascular density and vessel size in mouse tumors (Soucy et al., 2003;Kamat et al., 2005;Soucy et al., 2005). These studies indicated that vascular cells are highly sensitive to the effects of arsenic and that these in vivo models are useful for identifying mechanisms for the health effects of low dose arsenic exposures. A deficiency in these models; however, is their inability to reveal pathogenic effects of arsenic on vascular beds that are not developing, inflamed, or transformed.
The unique vasculature of the liver represents an endogenous vascular bed that is a primary target for the pathogenic effects of arsenic. The liver is the major organ for arsenic metabolism and the blood in the liver vessels should have one of the highest levels of inorganic arsenic and its metabolites found in the body following exposure to arsenic in drinking water. Liver effects associated with arsenic in drinking water include non-cirrhotic liver fibrosis and to a lesser extent portal hypertension (Mazumder, 2005;Das et al., 2005;Guha Mazumder, 2003). These pathologies are distinguished by increased vascular channels in the portal regions of the liver. In humans, higher levels of chronic arsenic consumption increase urinary levels of porphyrins, a biomarker for liver injury, that are more pronounced in people under 20 years of age (Ng et al., 2005). In addition, cardiac and liver disorders are the major side effects of therapeutic arsenic regimes used to treat leukemia (Shigeno et al., 2005). Despite epidemiological evidence that the liver vasculature is a pathogenic target of chronic arsenic ingestion (Mazumder, 2005), the direct effects of arsenic on the liver vascular cells remain unknown.
The filtering function of the liver sinusoids is facilitated by the specialized, highly fenestrated sinusoidal endothelial cells (LSEC) (Braet et al., 2002). These cells are unique in the extensive heterogeneity of vascular endothelium throughout the body due to their role in providing low pressure transendothelial cell transport of nutrients and wastes into and out of the liver parenchyma (Braet et al., 2002). Early in development, the LSEC differentiate to lose markers of a continuous endothelium, such as junctional expression of platelet endothelial cell adhesion molecule 1 (PECAM-1/CD31) and a basement membrane containing the matrix protein laminin-1 (Couvelard et al., 1996). In the differentiation process, the SEC gain fenestrae and gaps that allow sieving of circulating nutrients, lipids, and lipoproteins for normal liver metabolism (Braet et al., 2002). The SEC angiogenic process is manifested differently than angiogenesis in endothelial cells of systemic vessels, since there is no increase in sinusoidal vessel number or density. Instead, SEC angiogenesis is a dedifferentiation and maturation process called capillarization with diagnostic hallmarks of SEC defenestration and increased surface expression of PECAM-1 and laminin-1 protein (Braet et al., 2002;DeLeve et al., 2004;Couvelard et al., 1993;Guyot et al., 2006;Tsuneyama et al., 2003). The normally discontinuous SEC become a continuous endothelium with limited transendothelial cell transport due to loss of fenestrae and formation of tight intercellular endothelial junctions (Dubuisson et al., 1995;Braet et al., 2002;Xu et al., 2003;Couvelard et al., 1993). Capillarization precedes vascular remodeling of other liver vessels, such as the hepatic arterioles and the peribiliary vascular plexus causing the shunting of blood flow, vascular channel formation, and eventually liver fibrosis (Couvelard et al., 1993;DeLeve et al., 2004;Li et al., 2005). Liver angiogenesis in general is recognized as an important factor in the pathogenesis not only for portal fibrosis, but also for portal hypertension and progression of hepatocellular carcinomas (Tsuneyama et al., 2003;Ward et al., 2004;Semela et al., 2004;Moreau, 2005;Fernandez et al., 2005). Finally, liver capillarization impacts the systemic vasculature by decreasing liver metabolism of lipids, lipoproteins, and glucose to promote atherogenesis in response to environmental stresses and aging (Braet et al., 2002;Cogger et al., 2004;Hilmer et al., 2005).
Significant gaps remain in the mechanistic understanding of arsenic-induced endothelial cell dysfunction and pathogenesis of vascular diseases caused by low to moderate levels of arsenic in drinking water. However, filling these knowledge gaps has been complicated by a lack of sensitive animal models for in vivo investigation of the molecular pathology of arsenic effects on the cells of endogenous vascular beds. The objective of the following studies was to demonstrate that low to moderate levels of arsenic in drinking water promote inflammatory angiogenesis and vascular remodeling in mouse models. The data demonstrate endothelial cell activation and inflammatory cell infiltration in response to as little as 50 ppb in the mouse Matrigel model. In addition, LSEC undergo capillarization following sub-chronic exposure of mice to 250 ppb in drinking water. The data are the first demonstration of pathogenic effects of environmentally relevant arsenic levels on an endogenous vascular bed and suggest that the mouse liver vasculature is a sensitive model for examining the vascular health effects of low dose arsenic exposures.
Methods
Animal Exposure
Mouse exposures were performed in agreement with institutional guidelines for animal safety and welfare at Dartmouth College and the University of Pittsburgh. The conditions and results for the Dartmouth study have been published, with the exception of data in Fig. 1 (Soucy et al., 2005) .C57BL/6NTac male mice, ages 6-8 weeks weighing approximately 20g were obtained from Taconic Farms (Hudson, NY). Standard mouse chow and drinking water solutions were fed ad libitum to mice housed in boxes of three. Fresh drinking water solutions of sodium arsenite (Fisher Scientific, Pittsburgh, PA) were prepared triweekly using commercially bottled drinking water (Giant Eagle Spring Water, Pittsburgh, PA). Individual mouse consumption of arsenite was not measured, but there were no differences in water consumption, body weights, or liver weights between the groups (data not shown).
Fig. 1.

Arsenic-stimulated inflammatory cell infiltration in mouse Matrigel assays for neovascularization. Matrigel plugs were implanted subcutaneously into control mice and mice exposed for 3 wk to the indicated amount of arsenite in their drinking water. Exposure was continued for an additional 2 wk and the plugs were harvested at time of euthanizing the mice. The plugs were fixed, imbedded in paraffin, and sectioned. Overlapping sections were stained with hematoxylin and eosin (H&E) or immunostained for CD45 positive leukocytes (green channel). Immunostained sections were also stained with DRAQ 5 to visualize nuclei (blue channel). Arrows indicate blood vessels that are defined as luminal structures containing red blood cells (M = abdominal muscle). The images are representative of images from 5 animals in each group.
Matrigel neovascularization assay
In vivo Matrigel neovascularization assays were performed, as previously described (Soucy et al., 2003;Soucy et al., 2005). Briefly, a Matrigel plug containing 50 ng/ml recombinant FGF-2 (PeproTech, Rocky Hill, NJ) was implanted after the mice were exposed to arsenic for 3 wk and exposures were continued for an additional 2 wk. At the end of the exposures, the mice were euthanized and plugs were excised with a portion of adjacent skin and muscle for orientation. The plugs were then fixed in 10% neutral buffered formalin, and embedded in paraffin. One hundred micron thick cross sections were stained with hematoxylin and eosin for counting vessels (identified as cell-lined luminal structures containing red blood cells). The results of arsenic-effects on vessel number in these experiments have been published elsewhere (Soucy et al., 2005). Additional thin slices were immunostained for CD45 positive leukocyte infiltration, essentially as described for PECAM-1 staining (Soucy et al., 2003).
Scanning and Transmission Electron Microscopy
SEM and TEM were used to compare liver sinusoid ultrastructure between control C57BL/6 mice and mice exposed for 5 wk to 250 ppb of arsenite in their drinking water. At the end of the exposure period, three mice in each group were euthanized by IP injection of sodium pentobarbital (Nembutal) and opening the thoracic cavity. The livers were perfusion fixed by flushing with 10 ml of PBS and then perfused with 10 ml 2.5% gluteraldehyde in PBS. Livers were then removed and immersed in 2.5% gluteraldehyde overnight at 4°C. Samples for TEM were processed as described previously.(Ross et al., 2001;Stolz et al., 1999;Wack et al., 2001) Ultrathin, 70 nm sections were imaged on a JEM 1210 TEM (JEOL, Peabody, MA) at 80 kV. For SEM, perfused fixed livers were sliced into approximately 3-mm-thick sections, prepared for imaging as described,(Ross et al., 2001;Stolz et al., 1999;Wack et al., 2001) and imaged with a JSM-6330F scanning electron microscope.
Morphometric quantitation of fenestrae
Representative 14,000x SEM images of sinusoids were captured in the livers sections from 3 control mice or 3 mice exposed to arsenic. In three separate sinusoids in each mouse, MetaMorph software was used to calculate porosity in an average of area of 2855 ± 1231 μm2 in the vessel walls. Within this area, the area of individual fenestrae (20-500 nm diameter) and gaps (>500 nm diameter) were quantified in μm2. The areas of fenestrae and gaps were summed, divided by the total area of the vessel examined, and multiplied by 100 to give the porosity as percent of open area. The porosities of the three separate vessels per mouse were averaged for a single value used in group comparisons.
Immunofluorescence detection of proteins
At the end of the exposure periods, the livers were excised, and then snap frozen in liquid nitrogen until sectioning. Cryostat sections (8 micron) of excised liver were placed on charged glass slides and fixed for 1 minute in cold methanol. After washing three times with phosphate buffered saline (PBS), the slides were incubated for 1 hour at room temperature (RT) with primary antibodies for: PECAM-1 (1:100; BD Biosciences, San Jose, CA), laminin-1 (1:750; Sigma-Aldrich, St. Louis, MO), CD45 (1:500; BD Biosciences), CD68 (1:500; Serotec, Raleigh, NC), smooth muscle actin (1:500; Sigma), caveolin-1 (1:300; BD Biosciences) diluted in PBS. Slides were then washed three times with PBS and then incubated for 1 hour at RT in the dark with secondary antibodies (Alexa Fluor® 488 Goat anti-rat (H+L) (Invitrogen), Alexa Fluor® 594 Goat anti-mouse (H+L) (Invitrogen), Alexa Fluor® 594 Goat anti-rabbit (H+L) diluted 1:500 (Invitrogen), and nuclear stain DRAQ 5 (1:2000; Biostatus, Leicestershire, UK). After three rinses with PBS, coverslips were mounted with Flouromount G (Southern Biotech, Birmingham, AL) and fluorescent images were captured with an Olympus Fluoview 500 confocal microscope or a Nikon microphot-FXL microscope, fitted with an Olympus CCD digital camera.
Quantitative immunofluorescence of sinusoidal protein levels
Five random 400x immunofluorescent images (40x objective with an additional 10x image magnification) were captured using the same exposure time from in liver sections from five control and five arsenic exposed mice. Using MetaMorph software (Universal Imaging), the images were color separated, changed to monochrome format and the threshold pixel values were set to equal levels. The number of pixels per 400x field was then quantified and the average percentage of positively fluorescent pixels per field in the five fields was calculated to give a single value per mouse.
Statistical analysis
Dose and time dependent changes in mice exposed to arsenic were compared by two-way analysis of variance (ANOVA) followed by Bonferroni's posttest for differences between treatments. All statistical analysis was performed using Prizm 4.0 software (GraphPad, San Diego, CA).
Results
Arsenic exposure enhances inflammatory angiogenesis in vivo
Arsenic causes time- and dose-dependent enhancement of angiogenesis in vivo in chicken chorioallantoic membrane and mouse Matrigel assays (Soucy et al., 2003;Soucy et al., 2005). The threshold for arsenic enhancement of FGF primed angiogenesis in the Matrigel assay was 5 ppb and this enhancement of angiogenic capacity was seen following 5, 10, and 20 wk exposures to 50 and 250 ppb arsenic (Soucy et al., 2005). H&E staining of the Matrigel sections in this earlier study suggested that arsenic induced inflammatory cell infiltration in addition to increasing vascularization of the plugs (Soucy et al., 2005). To confirm this observation, the infiltrating cells were characterized by immunostaining thin sections of the plugs for the leukocyte marker CD45. As shown in Fig. 1, CD45 positive staining was only observed in Matrigel plugs from arsenic exposed animals and was associated with increased vascularization of the plugs.
Sub-chronic As(III) exposure induces defenestration and capillarization of liver sinusoids
The effects of arsenic exposure on functional morphologic change in an endogenous vascular bed were investigated using SEM and TEM to examine the ultrastructure changes in mouse liver sinusoidal endothelium over 5 wks of exposure to 250 ppb of arsenic. As seen in Fig. 2, the SEC in normal mice contain numerous sieve plates with open fenestrae (20-500 nM in diameter) and gaps (>500 nM in diameter). In contrast, the sinusoids in arsenic-exposed mice were defenestrated and the endothelium was continuous indicating capillarization (Fig. 2). Quantitative morphometric analysis revealed that by 2-5 wk Arsenic increased the number of fenestrae per unit surface area of the sinusoid, but decreased the average size of the fenestrae and eliminated gaps to decrease overall sinusoid porosity (i.e. open space per unit area; Fig. 2 graph). The surface of the arsenic exposed sinusoids also showed an increase in associated detritus and projections, some of which were microvilli from the underlying hepatocytes protruding through the SEC fenestrae (Fig. 2). There were no zonal differences along the sinusoids for the effect of arsenic on porosity (data not shown). The TEM images in Fig. 3 confirm that the quantitative decrease in porosity of the sinusoids in arsenic-exposed mice was paralleled by an increased filling of the space of Disse. The images indicate an increased formation of hepatocyte microvilli and similar increases in microvilli have been attributed to a compensatory mechanism to recover lost nutrient uptake (Braet et al., 2002;Wack et al., 2001). The magnified portions of the TEM images showing the space of Disse demonstrate arsenic-stimulated loss of fenestrations and gaps and gain of a rudimentary basement membrane (Fig. 3B).
Fig 2.
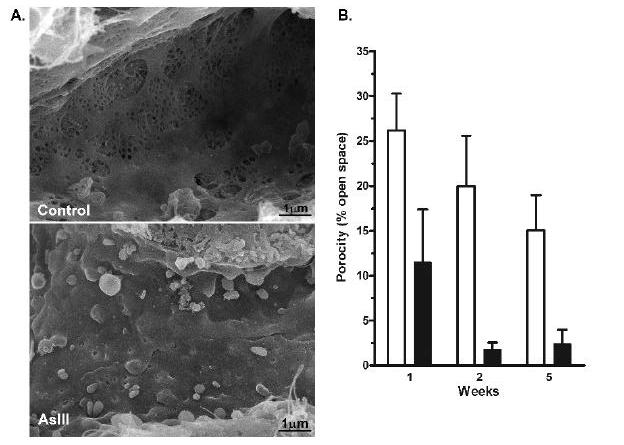
Time-dependent defenestration and capillarization of the liver sinusoidal endothelium after Arsenic exposures. A. SEM images of sinusoidal vessels were captured from thick sections of livers excised from control mice or mice exposed to 250 ppb arsenite in drinking water for 2 wk. The images were captured at a magnification of 14,000x. B. Sinusoid porosity (e.g. percent open space in the sinusoid wall) was determined in control mice and mice exposed to arsenite for 1, 2, or 5 wk, as described in methods. The data in the graph present the mean ± SEM of sinusoid porosity of 3 mice in each group. Statistical analysis using two-way analysis of variance demonstrated a highly significant time effect (p< 0.01) and a significant difference in arsenic-treated animals from control by 2 wk (p<0.05).
Fig. 3.

Arsenic-stimulated capillarization, basement membrane formation, and increased hepatocyte microvilli. A. TEM images of sinusoidal vessels were captured from ∼70 nm thick sections of livers from control mice and mice exposed to arsenite for 2 wk (SD, space of Disse; L, sinusoid lumen). The images are representative of images from 3 mice in each group and do not differ from images of mice exposed to arsenite for 5 wk. B. Portions of images captured at 30,000x are presented to illustrate closure of fenestrations and gain in hepatocyte microvilli in the arsenite -exposed mice, relative to control.
Arsenic induces sinusoidal PECAM-1 and laminin-1 protein expression
SEC junctional expression of PECAM-1 and development of a laminin-1 containing basement membrane are hallmarks of capillarization and vessel maturation.(Davis et al., 2005;DeLeve et al., 2004;Braet et al., 2002) Confocal microscopic image capture and quantitative immunofluorescence analysis for these two proteins were performed on frozen fixed sections to determine whether the arsenic-induced ultrastructural changes observed in Fig. 2 and 3 were accompanied by localized increases in protein expression. The images of tissue from control mice and mice exposed to arsenic for 2 wk in Fig. 4A indicated that PECAM-1 expression was selectively increased in the sinusoids. Consistent with the TEM images in Fig. 3B, basement membrane laminin-1 expression also increased in the sinusoids of arsenic-exposed mice (Fig. 4A). Merging the red and green channels in Fig. 4A revealed punctuate laminin-1 staining (focal red staining) that is adjacent to the sinusoidal PECAM-1. This may indicate that stellate cells, a primary source of laminin-1 (Couvelard et al., 1993), were also activated by arsenic. Consistent with the time course for loss of porosity, quantitative analysis of immunostained sections from mice in each exposure period demonstrated that arsenic exposure caused time-dependent increased expression of PECAM-1, relative to controls (Fig. 4B). As previously reported (Neubauer et al., 2000), the increase in PECAM-1 protein expression at the cell junctions did not correlate with changes in PECAM-1 mRNA levels (data not shown).
Fig. 4.
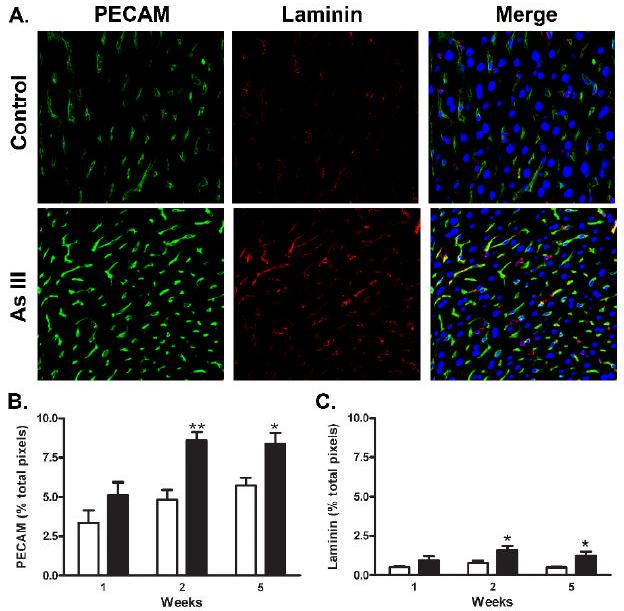
Arsenic induced expression of sinusoidal PECAM-1 and laminin protein. A. Thin sections were prepared from the livers of control mice or mice exposed to arsenite for 2 wk and immunostained for PECAM-1 (green channel) or laminin-1 (red channel). In the merged image, the blue channel was added to show DRAQ 5-stained nuclei. The representative confocal images were captured at 40x with a final magnification 400x. B., C. Morphometric analysis of confocal immunofluorescent images was used to quantify PECAM-1 and laminin-1 protein expression in groups of 6 mice at 1, 2, and 5 wk. Data are expressed as the mean ± SEM percentage of total pixels that stain positive for the respective protein per 400x microscopic field. The data were analyzed by two way analysis of variance and showed both significant time and treatment differences (** = p<0.01 and * = p<0.05).
Arsenic increases caveolin-1 and caveolae in LSEC membranes
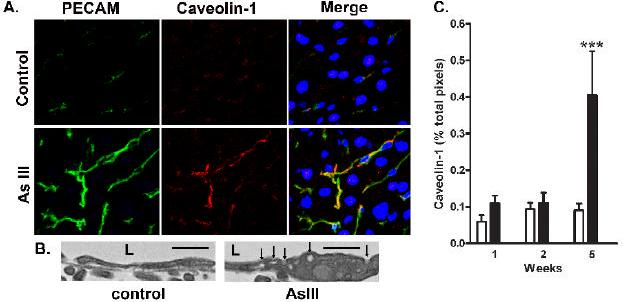
Fenestration of the endothelium is associated with a decreased expression of caveolin-1 and loss or fusion of caveolae (Braet, 2004;Esser et al., 1998). To determine whether arsenic reversed suppressed caveolin-1 expression in liver SEC, thin sections were co-immunostained for caveolin-1 and PECAM-1. The images and graph in Fig. 5 confirm that caveolin-1 expression in normal sinusoids is low. There was no effect of arsenic on caveolin-1 expression at early time points of exposure (Fig 5C). However, by wk 5 of arsenic exposure caveolin-1 protein was greatly increased. The pattern of expression was highly correlated with the increase in LSEC PECAM-1 expression (Fig. 5A) and with an increase in caveolae structures in the LSEC membranes (Fig. 5B). These data suggest that after the LSEC capilliarize, their phenotype continues to mature to convert from transporting bulk constituents through fenestrations to facilitated transendothelial transport through caveolae.
Fig. 5.
Co-localization of Arsenic-stimulated caveolin-1 and PECAM-1 protein expression. A. Sections of livers from mice exposed to arsenite for 5 wk or their time matched controls were immunostained for PECAM-1 (green channel) or caveolin-1 (red channel). In the merged image, the blue channel was added to show DRAQ 5-stained nuclei. The representative confocal images were captured at 40x magnification and magnified 10x. B. Portions of TEM images captured at 30,000x are shown to present the increase in caveolae structures in the LSEC membrane from Arsenic exposed mice relative to controls (L, lumen; arrows point to caveolae; bars = 500 nm). C. The graph presents quantitative morphometric analysis of the immunofluorescent labeled caveolin-1 in the images from 6 mice in each group. Data are expressed as the mean ± SEM percentage of total pixels that stain positive for caveolin-1 protein per 400x microscopic field (*** = p<0.001).
Leukocyte infiltration into the liver increased following 5 wk of Arsenic exposure
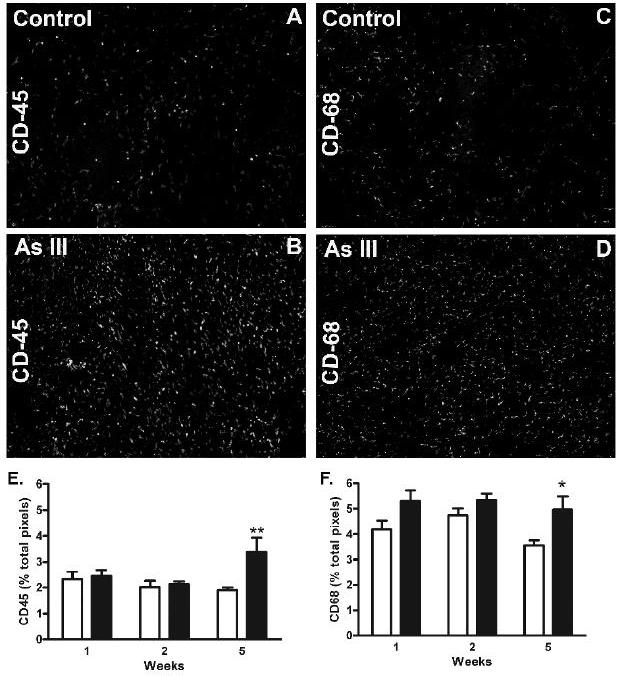
In models of alcohol-induced liver injury, angiogenesis and hepatic vascular remodeling are associated with an influx of monocytes and macrophages that deliver vascular growth and stabilizing factors (Tsuneyama et al., 2003). Since arsenic increased myeloid cell infiltration in the mouse Matrigel assay (Fig. 1), the time dependent effects of arsenic on the liver content of CD45 and CD68 positive cells was examined. Consistent with the data in Fig 1, there were significant increases in both CD45 and CD68 staining in mice exposed to arsenic for 5 wk (Fig. 6). The amount of increase in CD68 positive macrophages is much less striking that the increase in CD45 positive cells indicting that the CD68 positive population represents only a portion of the total CD45 positive infiltrate. There were no differences in CD45 positive cells between control and arsenic-exposed mice at the earlier time points suggesting that the leukocytes are recruited after loss of porosity and capillarization are initiated (Fig. 2). Thus, inflammation appears to be a result and not a cause of arsenic-induced defenestration and capillarization.
Fig. 6.
Arsenic-stimulated liver accumulation of CD45 and CD68 positive cells. Thin sections of liver from mice exposed to arsenite for the indicated time or their time matched controls and immunostained stained for either CD45 (leukocytes/monocytes) or CD68 (macrophages). A-D. Representative images of CD45 or CD68 positive staining in livers of 5 wk control or arsenic-exposed mice were captured at 20x and magnified by an additional 10x. E,F. Quantitative morphometric analysis of the immunofluorescent images from each treatment group was used to determine the percentage of total pixels that stained positive for CD45 or CD68. The graphs present mean ± SEM of values for 6 mice in each group. Two way analysis of variance determined that there was a highly significant time dependence for both CD45 and CD68 staining, as well as significant stimulation by arsenic at 5 wk (* = p<0.05; ** = p<0.01).
Discussion
Neovascularization, angiogenesis, and vessel remodeling in the adult are fundamental to the pathogenesis of a number of diseases caused by environmental arsenic exposures, including cardiovascular ischemic diseases, atherosclerosis, tumorigenesis, and liver fibrosis. Our previous studies were the first to demonstrate that exposure to arsenic in drinking water at or even below the current United States drinking water standard of 10 ppb enhances angiogenesis in an intact mouse model (Soucy et al., 2005). The data in Fig. 1 extend these earlier observations by demonstrating that angiogenesis enhanced by 5 wk arsenic exposures is associated with recruitment of inflammatory cells. This is in keeping with the pathogenesis of angiogenesis in adult tissues (Carmeliet, 2000;Cursiefen et al., 2004;Tsuneyama et al., 2003). However, these data were not unexpected, since the Matrigel plug is an inflammatory model primed with a threshold amount of FGF (Soucy et al., 2005). This model is relevant to tumorigenesis and other studies confirm that low dose arsenic enhances the vascularization of solid tumors (Kamat et al., 2005;Liu et al., 2006;Soucy et al., 2003). The data in the current study go beyond these earlier studies to address the question of whether arsenic initiates or merely enhances pathogenic responses. This study, therefore, is the first to demonstrate functional vascular changes in an endogenous vascular bed that is exposed to arsenic in vivo. The data not only indicate that arsenic initiates defenestration and capillarization of the LSEC, but also demonstrate that arsenic-induced loss of porosity (Fig. 2) precedes inflammatory (Fig. 6) and compensatory changes (Fig.5) in the sinusoidal endothelium.
The data in the current study differ significantly from previous rodent studies of arsenic effects on the liver vasculature (Bashir et al., 2006;Das et al., 2005;Flora et al., 1997;Mazumder, 2005). The major contrast is that in the current study, vascular remodeling was observed following sub-chronic exposure to a moderate arsenic exposure. One previous study in mice demonstrated that prolonged (9 month) exposure to high dose arsenic (50-500 μg/mouse/day by gavage) resulted in liver lipid peroxidation and cytokine release (Das et al., 2005). Assuming equal bioavailability between species, these exposures would be the equivalent of a human drinking 2-20 mg of arsenic/day for approximately 26 years before inflammatory toxicity occurred. This does not fit the demographic of arsenic-induced liver disease in humans, since significant increases in urinary porphyrins, a biomarker for liver injury, are more readily observed in arsenic-exposed humans who are under 20 years of age (Ng et al., 2005). In vivo, high doses of arsenic affect all cells in the liver and promote a significant amount of apoptosis in hepatocytes (Bashir et al., 2006). High doses of arsenic (>5 μM) are known to be toxic to endothelial cells (Barchowsky et al., 1996;Barchowsky et al., 1999;Kao et al., 2003;Roboz et al., 2000) and it is possible that high dose exposures elicit multiple mechanisms for toxicity that mask the pathogenic mechanisms mediating liver diseases in response to environmentally relevant arsenic exposures. In contrast, the current study investigated effects of a relevant ad libitum ingestion of drinking water containing an arsenic level that was near the threshold for observing significant liver disease in humans (250 ppb = ∼ 0.7-.9 μg/mouse/day for 5 weeks; human equivalent ∼ 32 μg/day for 3.75 years). Thus, the data in this study reflect the effects of arsenic on cell phenotype rather than cell death.
Liver injury in response to arsenic has been proposed to be mediated by oxidative stress and increased levels of inflammatory cytokines (Das et al., 2005;Mazumder, 2005). Das et al. showed that nine months was required before overt liver injury was observed in response to high levels of arsenic and that this injury was associated with increased oxidative damage and cytokine release (Das et al., 2005). Capillarization has been shown to occur with aging, an effect that may be caused by progressive oxidative injury to LSEC (Hilmer et al., 2005;Cogger et al., 2004). The data in Fig. 2 demonstrated that capillarization developed over 1-2 weeks of arsenic exposure and was greatly accelerated by arsenic compared to the natural decline in porosity seen in age matched controls. Since arsenite stimulates oxidant production by endothelial cell NAD(P)H oxidase (Smith et al., 2001), it is possible that chronic oxidant stress was responsible for arsenic-induced loss of porosity and phenotypic change in LSEC. However, if this was true, the oxidative stress would have had to be at a low level since total liver hemoxygenase-1 mRNA levels increased by less than 3-fold in following exposure to 250 ppb of arsenite for 5 wks (data not shown). The data in Fig. 6 argue that an inflammatory response resulted from arsenic-initiated capillarization instead of causing it. Leukocytes were not recruited until after the maximal loss in LSEC porosity occurred. Thus, leukocyte oxidant generation was unlikely to have contributed to arsenite-induced LSEC phenotypic change. More studies are needed to determine the threshold for arsenite stimulation of LSEC NAD(P)H oxidases and whether oxidant generation accounts for the mechanism of arsenite induced pathogenic change in LSEC phenotype.
Capillarization is the angiogenic process of the liver sinusoidal endothelium and is fundamentally different from angiogenesis in other vascular beds. The main distinction is that there is no increase in vessel number due to the anatomic constraints of the liver sieve plates. Capillarization of LSEC results in ultrastructural phenotypic conversion to endothelial cells with tight intercellular junctions and loss of fenestrations (Couvelard et al., 1993;Dubuisson et al., 1995;Xu et al., 2003). In multiple human and animal studies, capillarization has been demonstrated to precede alcohol-induced liver disease, portal hypertension, cirrhosis, and chronic hepatitis (DeLeve et al., 2004;Dubuisson et al., 1995;Tsuneyama et al., 2003;Xu et al., 2003). Increased SEC membrane PECAM-1 protein expression and deposition of a laminin-1-containing basement membrane are hallmarks of capillarization in injured livers (Couvelard et al., 1993;DeLeve et al., 2004). As seen in Fig. 4, arsenic induces these hallmarks as it promotes defenestration and capillarization of the SEC (Fig. 2 and 3). The time course for gain of PECAM-1 and laminin-1 is identical to the time course for loss of porosity (Fig. 2), indicating that these are reciprocal functions. Thus, as the fenestrations close the cells increase intracellular contacts and transport through the fenestrations is limited by gain of a basement membrane. Infiltration of leukocytes, especially pro-angiogenic myeloid cells, is necessary for supporting pathological angiogenesis, vessel maturation, and remodeling (Brasier et al., 2002;Carmeliet, 2000;Cursiefen et al., 2004;Ruiz et al., 2006). It is possible that the delayed increase in CD45 positive cell infiltration was the result of vessel maturation in the capillarization process rather than a direct effect of arsenic on leukocyte activity. Further indication that consequential or compensatory processes continue to occur in the arsenite exposed mice is the gain in caveolin-1 and caveolae (Fig.5) for transendothelial cell transport.
In summary, there is growing evidence that low level arsenic exposures have functional pathogenic consequences in endothelial cells. Arsenic-enhanced angiogenesis occurs in mouse models and mouse tumors at very (Liu et al., 2006;Soucy et al., 2005;Kamat et al., 2005) making this endpoint one of the most sensitive reported as a health effect of arsenic in rodents. More importantly, angiogenesis is induced in mice by low to moderate human relevant exposures and occurs in response to arsenic at the current United States MCL of 10 ppb (Soucy et al., 2005;Kamat et al., 2005). The data in the current study are the first to demonstrate that moderate environmental exposure to arsenite functionally affects an endogenous vascular bed. While the exposures were not long enough to observe portal fibrosis and there was no measure of portal blood flow, the changes observed were consistent with initial pathogenesis of intrahepatic vascular disease and development of arteriovenous shunts in arsenic-induced human liver diseases (Mazumder, 2005). The data indicate that the intact mouse and liver vasculature are suitable models for investigating arsenic-induced inflammatory and vascular changes that promote both pathogenic vascular cell responses and liver disease. Further studies using this model will be needed to identify the molecular switches and mechanisms through which arsenic stimulates phenotypic change in the SEC without promoting hepatocyte injury. Since liver sinusoidal capillarization can contribute to atherogenic metabolic imbalance, the studies may have great impact on the understanding of the mechanisms for both human liver and vascular diseases associated with chronic environmental exposures to inorganic arsenic.
Acknowledgements
This work was supported by NIEHS grant ES07373 (AB, NVS), NCI grant CA76541 (DBS), and EPA STAR Fellowship FP-91654201 (ACS). Harina Vin was supported by a MaryAnne Stock Student Research Award from the Allegheny-Erie Regional Chapter of the Society of Toxicology. The authors would like to thank Dr. Simon Watkins for the use of the confocal and electron microscopes and processing of tissue at the University of Pittsburgh Center for Biological Imaging. There are no conflicts of interest for any of the authors of this work.
Abbreviations
- LSEC
liver sinusoidal endothelial cell
- PECAM-1
platelet endothelial cell adhesion molecule (CD31)
- SEM
scanning electron microscopy
- TEM
transmission electron microscopy
- PBS
phosphate-buffered saline
References
- Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: How hot is the link? Biochem.Pharmacol. 2006 doi: 10.1016/j.bcp.2006.06.029. Epub. [DOI] [PubMed] [Google Scholar]
- Barchowsky A, Dudek EJ, Treadwell MD, Wetterhahn KE. Arsenic Induces Oxidant Stress And NF-KappaB Activation In Cultured Aortic Endothelial Cells. Free Radic.Biol.Med. 1996;21:783–790. doi: 10.1016/0891-5849(96)00174-8. [DOI] [PubMed] [Google Scholar]
- Barchowsky A, Roussel RR, Klei LR, James PE, Ganju N, Smith KR, Dudek EJ. Low Levels of Arsenic Trioxide Stimulate Proliferative Signals in Primary Vascular Cells without Activating Stress Effector Pathways. Toxicol Appl Pharmacol. 1999;159:65–75. doi: 10.1006/taap.1999.8723. [DOI] [PubMed] [Google Scholar]
- Bashir S, Sharma Y, Irshad M, Nag TC, Tiwari M, Kabra M, Dogra TD. Arsenic induced apoptosis in rat liver following repeated 60 days exposure. Toxicology. 2006;217:63–70. doi: 10.1016/j.tox.2005.08.023. [DOI] [PubMed] [Google Scholar]
- Braet F. How molecular microscopy revealed new insights into the dynamics of hepatic endothelial fenestrae in the past decade. Liver Int. 2004;24:532–539. doi: 10.1111/j.1478-3231.2004.0974.x. [DOI] [PubMed] [Google Scholar]
- Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol. 2002;1:1–17. doi: 10.1186/1476-5926-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasier AR, Recinos A, III, Eledrisi MS. Vascular inflammation and the reninangiotensin system. Arterioscler.Thromb.Vasc.Biol. 2002;22:1257–1266. doi: 10.1161/01.atv.0000021412.56621.a2. [DOI] [PubMed] [Google Scholar]
- Bunderson M, Brooks DM, Walker DL, Rosenfeld ME, Coffin JD, Beall HD. Arsenic exposure exacerbates atherosclerotic plaque formation and increases nitrotyrosine and leukotriene biosynthesis. Toxicol Appl.Pharmacol. 2004;201:32–39. doi: 10.1016/j.taap.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat.Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- Cogger VC, Muller M, Fraser R, McLean AJ, Khan J, Le Couteur DG. The effects of oxidative stress on the liver sieve. J.Hepatol. 2004;41:370–376. doi: 10.1016/j.jhep.2004.04.034. [DOI] [PubMed] [Google Scholar]
- Couvelard A, Scoazec JY, Dauge MC, Bringuier AF, Potet F, Feldmann G. Structural and functional differentiation of sinusoidal endothelial cells during liver organogenesis in humans. Blood. 1996;87:4568–4580. [PubMed] [Google Scholar]
- Couvelard A, Scoazec JY, Feldmann G. Expression of cell-cell and cell-matrix adhesion proteins by sinusoidal endothelial cells in the normal and cirrhotic human liver. Am.J.Pathol. 1993;143:738–752. [PMC free article] [PubMed] [Google Scholar]
- Cursiefen C, Chen L, Borges LP, Jackson D, Cao J, Radziejewski C, D'Amore PA, Dana MR, Wiegand SJ, Streilein JW. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J.Clin.Invest. 2004;113:1040–1050. doi: 10.1172/JCI20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgleish AG, O'Byrne K. Inflammation and cancer: the role of the immune response and angiogenesis. Cancer Treat.Res. 2006;130:1–38. [PubMed] [Google Scholar]
- Das S, Santra A, Lahiri S, Guha Mazumder DN. Implications of oxidative stress and hepatic cytokine (TNF-alpha and IL-6) response in the pathogenesis of hepatic collagenesis in chronic arsenic toxicity. Toxicol Appl.Pharmacol. 2005;204:18–26. doi: 10.1016/j.taap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ.Res. 2005;97:1093–1107. doi: 10.1161/01.RES.0000191547.64391.e3. [DOI] [PubMed] [Google Scholar]
- DeLeve LD, Wang X, Hu L, McCuskey MK, McCuskey RS. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am.J.Physiol Gastrointest.Liver Physiol. 2004;287:G757–G763. doi: 10.1152/ajpgi.00017.2004. [DOI] [PubMed] [Google Scholar]
- Dubuisson L, Boussarie L, Bedin CA, Balabaud C, Bioulac-Sage P. Transformation of sinusoids into capillaries in a rat model of selenium-induced nodular regenerative hyperplasia: an immunolight and immunoelectron microscopic study. Hepatology. 1995;21:805–814. [PubMed] [Google Scholar]
- Duyndam MC, Hulscher ST, van der WE, Pinedo HM, Boven E. Evidence for a role of p38 kinase in hypoxia-inducible factor 1-independent induction of vascular endothelial growth factor expression by sodium arsenite. J Biol Chem. 2003;278:6885–6895. doi: 10.1074/jbc.M206320200. [DOI] [PubMed] [Google Scholar]
- Engel RR, Hopenhayn-Rich C, Receveur O, Smith AH. Vascular effects of chronic arsenic exposure: a review. Epidemiologic Review. 1994;16:184–208. doi: 10.1093/oxfordjournals.epirev.a036150. [DOI] [PubMed] [Google Scholar]
- Esser S, Wolburg K, Wolburg H, Breier G, Kurzchalia T, Risau W. Vascular endothelial growth factor induces endothelial fenestrations in vitro. J Cell Biol. 1998;140:947–959. doi: 10.1083/jcb.140.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez M, Mejias M, Angermayr B, Garcia-Pagan JC, Rodes J, Bosch J. Inhibition of VEGF receptor-2 decreases the development of hyperdynamic splanchnic circulation and portal-systemic collateral vessels in portal hypertensive rats. J.Hepatol. 2005;43:98–103. doi: 10.1016/j.jhep.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Flora SJ, Pant SC, Malhotra PR, Kannan GM. Biochemical and histopathological changes in arsenic-intoxicated rats coexposed to ethanol. Alcohol. 1997;14:563–568. doi: 10.1016/s0741-8329(97)00048-7. [DOI] [PubMed] [Google Scholar]
- Guha Mazumder DN. Chronic arsenic toxicity: clinical features, epidemiology, and treatment: experience in West Bengal. J.Environ.Sci Health Part A Tox.Hazard.Subst.Environ.Eng. 2003;38:141–163. doi: 10.1081/ese-120016886. [DOI] [PubMed] [Google Scholar]
- Guyot C, Lepreux S, Combe C, Doudnikoff E, Bioulac-Sage P, Balabaud C, Desmouliere A. Hepatic fibrosis and cirrhosis: The (myo)fibroblastic cell subpopulations involved. Int.J.Biochem.Cell Biol. 2006;38:135–151. doi: 10.1016/j.biocel.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Hayden MR, Tyagi SC. Vasa vasorum in plaque angiogenesis, metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: a malignant transformation. Cardiovasc.Diabetol. 2004;3:1–16. doi: 10.1186/1475-2840-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilmer SN, Cogger VC, Fraser R, McLean AJ, Sullivan D, Le Couteur DG. Age-related changes in the hepatic sinusoidal endothelium impede lipoprotein transfer in the rat. Hepatology. 2005;42:1349–1354. doi: 10.1002/hep.20937. [DOI] [PubMed] [Google Scholar]
- Kamat CD, Green DE, Curilla S, Warnke L, Hamilton JW, Sturup S, Clark C, Ihnat MA. Role of HIF signaling on tumorigenesis in response to chronic low-dose arsenic administration. Toxicol Sci. 2005;86:248–257. doi: 10.1093/toxsci/kfi190. [DOI] [PubMed] [Google Scholar]
- Kao YH, Yu CL, Chang LW, Yu HS. Low Concentrations of Arsenic Induce Vascular Endothelial Growth Factor and Nitric Oxide Release and Stimulate Angiogenesis In Vitro. Chem.Res Toxicol. 2003;16:460–468. doi: 10.1021/tx025652a. [DOI] [PubMed] [Google Scholar]
- Lew YS, Brown SL, Griffin RJ, Song CW, Kim JH. Arsenic trioxide causes selective necrosis in solid murine tumors by vascular shutdown. Cancer Res. 1999;59:6033–6037. [PubMed] [Google Scholar]
- Li J, Niu JZ, Wang JF, Li Y, Tao XH. Pathological mechanisms of alcohol-induced hepatic portal hypertension in early stage fibrosis rat model. World J Gastroenterol. 2005;11:6483–6488. doi: 10.3748/wjg.v11.i41.6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Pan SG, Dong XS, Qiao HQ, Jiang HC, Krissansen GW, Sun XY. Opposing effects of arsenic trioxide on hepatocellular carcinomas in mice. Cancer Science. 2006;97:675–681. doi: 10.1111/j.1349-7006.2006.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder DN. Effect of chronic intake of arsenic-contaminated water on liver. Toxicol Appl.Pharmacol. 2005;206:169–175. doi: 10.1016/j.taap.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Moreau R. VEGF-induced angiogenesis drives collateral circulation in portal hypertension. J.Hepatol. 2005;43:6–8. doi: 10.1016/j.jhep.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Navas-Acien A, Sharrett AR, Silbergeld EK, Schwartz BS, Nachman KE, Burke TA, Guallar E. Arsenic Exposure and Cardiovascular Disease: A Systematic Review of the Epidemiologic Evidence. Am.J.Epidemiol. 2005;162:1037–1049. doi: 10.1093/aje/kwi330. [DOI] [PubMed] [Google Scholar]
- Neubauer K, Wilfling T, Ritzel A, Ramadori G. Platelet-endothelial cell adhesion molecule-1 gene expression in liver sinusoidal endothelial cells during liver injury and repair. J.Hepatol. 2000;32:921–932. doi: 10.1016/s0168-8278(00)80096-3. [DOI] [PubMed] [Google Scholar]
- Ng JC, Wang JP, Zheng B, Zhai C, Maddalena R, Liu F, Moore MR. Urinary porphyrins as biomarkers for arsenic exposure among susceptible populations in Guizhou province, China. Toxicol Appl.Pharmacol. 2005;206:176–184. doi: 10.1016/j.taap.2004.09.021. [DOI] [PubMed] [Google Scholar]
- Penn A. International Commission for Protection Against Environmental Mutagens and Carcinogens. ICPEMC Working Paper 7/1/1. Mutational events in the etiology of arteriosclerotic plaques. Mutation Research. 1990;239:149–162. doi: 10.1016/0165-1110(90)90003-t. [DOI] [PubMed] [Google Scholar]
- Ricousse-Roussanne S, Barateau V, Contreres JO, Boval B, Kraus-Berthier L, Tobelem G. Ex vivo differentiated endothelial and smooth muscle cells from human cord blood progenitors home to the angiogenic tumor vasculature. Cardiovasc.Res. 2004;62:176–184. doi: 10.1016/j.cardiores.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Roboz GJ, Dias S, Lam G, Lane WJ, Soignet SL, Warrell RP, Jr., Rafii S. Arsenic trioxide induces dose- and time-dependent apoptosis of endothelium and may exert an antileukemic effect via inhibition of angiogenesis. Blood. 2000;96:1525–1530. [PubMed] [Google Scholar]
- Ross MA, Sander CM, Kleeb TB, Watkins SC, Stolz DB. Spatiotemporal expression of angiogenesis growth factor receptors during the revascularization of regenerating rat liver. Hepatology. 2001;34:1135–1148. doi: 10.1053/jhep.2001.29624. [DOI] [PubMed] [Google Scholar]
- Ruiz d. A., Luttun A, Carmeliet P. An SDF-1 trap for myeloid cells stimulates angiogenesis. Cell. 2006;124:18–21. doi: 10.1016/j.cell.2005.12.023. [DOI] [PubMed] [Google Scholar]
- Semela D, Dufour JF. Angiogenesis and hepatocellular carcinoma. J.Hepatol. 2004;41:864–880. doi: 10.1016/j.jhep.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Shigeno K, Naito K, Sahara N, Kobayashi M, Nakamura S, Fujisawa S, Shinjo K, Takeshita A, Ohno R, Ohnishi K. Arsenic Trioxide Therapy in Relapsed or Refractory Japanese Patients with Acute Promyelocytic Leukemia: Updated Outcomes of the Phase II Study and Postremission Therapies. Int.J.Hematol. 2005;82:224–229. doi: 10.1532/IJH97.05044. [DOI] [PubMed] [Google Scholar]
- Simeonova PP, Hulderman T, Harki D, Luster MI. Arsenic exposure accelerates atherogenesis in apolipoprotein E(−/−) mice. Environ.Health Perspect. 2003;111:1744–1748. doi: 10.1289/ehp.6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, Klei LR, Barchowsky A. Arsenite stimulates plasma membrane NADPH oxidase in vascular endothelial cells. Am.J Physiol. 2001;280:L442–L449. doi: 10.1152/ajplung.2001.280.3.L442. [DOI] [PubMed] [Google Scholar]
- Soucy NV, Ihnat MA, Kamat CD, Hess L, Post MJ, Klei LR, Clark C, Barchowsky A. Arsenic stimulates angiogenesis and tumorigenesis in vivo. Toxicol Sci. 2003;76:271–279. doi: 10.1093/toxsci/kfg231. [DOI] [PubMed] [Google Scholar]
- Soucy NV, Klei LR, Mayka DD, Barchowsky A. Signaling pathways for arsenic-stimulated vascular endothelial growth factor-a expression in primary vascular smooth muscle cells. Chem.Res.Toxicol. 2004;17:555–563. doi: 10.1021/tx034193q. [DOI] [PubMed] [Google Scholar]
- Soucy NV, Mayka D, Klei LR, Nemec AA, Bauer JA, Barchowsky A. Neovascularization and angiogenic gene expression following chronic arsenic exposure in mice. Cardiovasc.Toxicol. 2005;5:29–42. doi: 10.1385/ct:5:1:029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz DB, Ross MA, Salem HM, Mars WM, Michalopoulos GK, Enomoto K. Cationic colloidal silica membrane perturbation as a means of examining changes at the sinusoidal surface during liver regeneration. Am.J.Pathol. 1999;155:1487–1498. doi: 10.1016/S0002-9440(10)65464-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng CH, Chong CK, Tseng CP, Hsueh YM, Chiou HY, Tseng CC, Chen CJ. Long-term arsenic exposure and ischemic heart disease in arseniasis-hyperendemic villages in Taiwan. Toxicol Lett. 2003;137:15–21. doi: 10.1016/s0378-4274(02)00377-6. [DOI] [PubMed] [Google Scholar]
- Tsuneyama K, Ohba K, Zen Y, Sato Y, Niwa H, Minato H, Nakanuma Y. A comparative histological and morphometric study of vascular changes in idiopathic portal hypertension and alcoholic fibrosis/cirrhosis. Histopathology. 2003;43:55–61. doi: 10.1046/j.1365-2559.2003.01658.x. [DOI] [PubMed] [Google Scholar]
- Wack KE, Ross MA, Zegarra V, Sysko LR, Watkins SC, Stolz DB. Sinusoidal ultrastructure evaluated during the revascularization of regenerating rat liver. Hepatology. 2001;33:363–378. doi: 10.1053/jhep.2001.21998. [DOI] [PubMed] [Google Scholar]
- Ward NL, Haninec AL, Van Slyke P, Sled JG, Sturk C, Henkelman RM, Wanless IR, Dumont DJ. Angiopoietin-1 causes reversible degradation of the portal microcirculation in mice: implications for treatment of liver disease. Am.J.Pathol. 2004;165:889–899. doi: 10.1016/S0002-9440(10)63351-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MM, Chiou HY, Hsueh YM, Hong CT, Su CL, Chang SF, Huang WL, Wang HT, Wang YH, Hsieh YC, Chen CJ. Effect of plasma homocysteine level and urinary monomethylarsonic acid on the risk of arsenic-associated carotid atherosclerosis. Toxicol.Appl.Pharmacol. 2006 doi: 10.1016/j.taap.2006.05.005. Epub. [DOI] [PubMed] [Google Scholar]
- Xu B, Broome U, Uzunel M, Nava S, Ge X, Kumagai-Braesch M, Hultenby K, Christensson B, Ericzon BG, Holgersson J, Sumitran-Holgersson S. Capillarization of hepatic sinusoid by liver endothelial cell-reactive autoantibodies in patients with cirrhosis and chronic hepatitis. Am.J.Pathol. 2003;163:1275–1289. doi: 10.1016/S0002-9440(10)63487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]