

Abstract
Phthalate dioxygenase (PDO) is a member of a class of bacterial oxygenases that contain both Rieske [2Fe-2S] and Fe(II) mononuclear centers. Recent crystal structures of several Rieske dioxygenases showed that they exist as α3β3 multimers with subunits arranged head-to-tail in α and β stacked planar consists of only α-subunits, remains to be solved. Although similar to other Rieske dioxygenases in many aspects, PDO was shown to differ in the mechanism of catalysis. Gel filtration and analytical centrifugation experiments, supplemented with mass spectrometric analysis (both ESI-MS and ESI-GEMMA), in this work showed a hexameric arrangement of subunits in the PDO multimer. Our proposed model for the subunit arrangement in PDO postulates two α3 planar rings one on top the other, similar to the α3β3 arrangement in other Rieske dioxygenases. Unlike other Rieske dioxygenases, this arrangement brings two Rieske and two mononuclear centers, all on separate subunits, into proximity, allowing their cooperation for catalysis. Potential reasons necessitating this unusual structural arrangement are discussed.
Keywords: Phthalate Dioxygenase, Rieske center, Rieske dioxygenase, structure, subunit arrangement
Rieske aromatic ring-hydroxylating dioxygenase systems facilitate the degradation of various aromatic hydrocarbons. These enzymes contain Rieske-type redox-active iron-sulfur clusters ([2Fe-2S]) and mononuclear ferrous centers, and catalyze the formation of cis-dihydrodiols by enantiospecific addition of two hydroxyl groups onto aromatic substrates. This is a first step in many aerobic biodegradation pathways that typically lead to breaching of aromatic rings of otherwise recalcitrant substrates.
Phthalate dioxygenase (PDO), an oxygenase from Burkholderia cepacia (DB01), catalyzes the first step in the breakdown of the aromatic compound phthalate to form the dihydrodiol of phthalate, cis-4,5-dihydro-4,5-dihydroxyphthalate (DHD). It is a member of the Rieske class of bacterial oxygenases. Many such dioxygenases from various organisms and biodegradation pathways have been identified. Although the overall sequence identities are often low between the different Rieske oxygenases (including PDO), there is considerable homology in both the Rieske and the mononuclear domains, suggesting that these regions are structurally similar. Residues ligating the Fe in both the Rieske and the mononuclear centers are fully conserved, as are a number of other residues, including the so-called “bridging” aspartate located on the subunit interface and shown to be critically important for catalysis in NDO [1], AntDO [2], and TDO [3]. In PDO too, it was shown that substitutions of this “bridging” aspartate (Asp178) resulted in enzymes with severely diminished catalytic activity, with both reduction and oxidation rates of the Rieske center affected by the mutations [4]. Additionally, it was shown that substitutions of Asp178 in PDO resulted in significant changes in subunit interactions within the PDO multimer, bringing about a looser arrangement with increased solvent access to the Rieske centers [5].
Although similar to other Rieske dioxygenases in many aspects, PDO was shown to differ somewhat in its mechanism of catalysis. For example, after reduced NDO is exposed to oxygen in the presence of the substrate, the mononuclear iron was found to be in the ferric state [6]. One cis-dihydrodiol product was found per Rieske and mononuclear center that was oxidized. In contrast, upon completion of the reaction in PDO, phthe mononuclear center was found to be in the ferrous state, and one product was formed per two Rieske centers oxidized [7; 8]. The reason for this difference in the mechanism of catalysis is apparently related to differences in structures between the PDO and other Rieske dioxygenases such as NDO or BPDO.
Crystal structures have been successfully determined for a growing number of the dioxygenases. The X-ray structures of naphthalene (NDO) [9], biphenyl (BPDO) [10], nitrobenzene (NBDO) [11], and cumene (CumDO) [12; 13] dioxygenases, as well as 2-oxoquinoline-8-monooxygenase (OMO) [14], and the ring-hydroxylating dioxygenase from Sphingomonas CHY-1 (RHD) [15; 16] all indicated that these enzymes are α3β3 multimers, with the α subunits containing both the Rieske and the mononuclear centers. An exception is carbazole 1,9-dioxygenase (CarDO) [12], which is an α3 trimer. In each case, the α subunits of the trimer are arranged with the subunits oriented head-to-tail, with the Rieske center of one subunit being close (∼ 12 Å) to the mononuclear center of the adjacent subunit. By contrast, the Rieske centers of each subunit are > 40 Å from the mononuclear centers of their own subunits. The β subunit does not affect substrate specificity in NDO, but is apparently important for catalysis, possibly stabilizing the α3 trimer [17; 18]. PDO was shown to lack β subunits, and based on earlier gel filtration data, was believed to be an α4 tetramer of 50 kDa monomers (∼ 200 kDa for the multimer) [19; 20]. Although the tetramer model, by placing two Rieske centers and two mononuclear centers close to each other, allowed for a consistent interpretation of experimental data for kinetics and product formation [8], the α4 multimer was clearly dissimilar to the α3β3 multimeric arrangement found in other known Rieske dioxygenases. It seemed surprising that the enzymes from the same family, which perform essentially similar hydroxylation reactions, would differ so much in the multimeric structural arrangements. Thus, we considered more careful studies to investigate whether the arrangement of subunits within the PDO multimer was hexameric, with the units arranged head-to-tail in two planar rings [4; 5]. Such an arrangement can be formally expressed as α3α3. This arrangement still allowed for two Rieske and two mononuclear centers to be in proximity to each other, and account for our earlier findings on the stoichiometry of DHD formation during catalysis [8]. Because efforts to obtain the crystal structure of PDO so far have not been successful, we have attempted to clarify the question of the multimeric structure of PDO by other methods.
Electrospray ionization (ESI) has demonstrated capabilities with mass spectrometry (MS) to measure the mass of non-covalent multimeric proteins [21]. We therefore used ESI in conjunction with both MS and gas-phase ion mobility analysis [22] to measure the molecular size of the PDO assembly. We complemented this study with gel filtration analysis and differential ultracentrifugation. We also attempted to show how PDO subunits might be arranged within the enzyme multimer to account for existing experimental data on single turnover experiments.
MATERIALS AND METHODS
Plasmids containing WT, D178A, and D178N PDO genes were constructed, and the proteins were expressed in Escherichia coli and purified as described previously [4]. Concentrations of enzymes were determined spectrophotometrically using Δε575 = 2.38 mM-1 cm-1 for the extinction difference between oxidized and reduced PDO.
ESI-MS
A nanoESI-QqTOF analyzer (QSTAR Pulsar XL, m/z range 40,000; Applied Biosystems/MDS Sciex, Concord, ON, Canada) was used for the ESI-MS experiments. Borosilicate glass nanospray emitters that are coated with Au/Pd to allow for 10-50 nL/min spray operation were obtained from Proxeon (Odense, Denmark). Prior to ESI-MS, samples were desalted and concentrated using Millipore Microcon Centrifugal Filter Devices in 20 mM ammonium acetate pH 6.8.
ESI-GEMMA
The details of the GEMMA have been described elsewhere [22; 23; 24]. Briefly, the GEMMA instrument (TSI Inc., St. Paul, MN) consists of an ESI unit with a neutralizing chamber, a differential mobility analyzer (DMA), and a condensation particle counter (CPC). The DMA voltage was scanned and data were recorded using Aerosol Instrument Manager Software. An average density of approximately 0.6 g/cc was used to calculate the molecular mass of the protein complexes. The software corrects for the efficiency of the condensation particle counter as well as for a small fraction of multiply-charged particles, assuming a bipolar charge distribution. The electrophoretic mobility diameter (EMD) maxima for peaks of interest were calculated from this corrected data. Igor Pro 4.08 software (Wavemetrics, Portland, OR) was used for further processing, including smoothing of spectra. From the GEMMA measurements for a variety of proteins and protein complexes ranging in size from small protein dimers to complexes as large as the 700 kDa 20S proteasome and MDa-range viral particles [25,26], an average density of approximately 0.6 g/cc can be used to calculate their molecular masses. The molecular masses calculated from the EMDs have an estimated accuracy of ± 1-5%.
Analytical ultracentrifugation
Sedimentation velocity experiments were performed in an Optima XL-I ultracentrifuge (Beckman-Coulter, Fullerton, CA). A double-sector charcoal-filled Epon centerpiece with sapphire windows and 1.2-mm path-length was used. The reference sector was filled with 420 μL of dialysis buffer and the sample sector with an equivalent volume of sample containing 0.1 - 1 mg/mL protein. Solvent densities and viscosities were calculated with SEDNTERP [27] or measured with a DMA5000 density meter (Anton-Parr, Ashland, VA). Data were processed with the programs SEDFIT 9.3b [28] and SEDPHAT 4.1b [29]. Following thermal equilibration in the centrifuge at 20 °C for at least 1 h at 0 rpm, the velocity experiment was initiated by accelerating the rotor to 30,000 rpm. Boundary positions were determined using both the Rayleigh interference and the absorbance (280 nm) optics with scans taken every 3-5 min. Sedimentation coefficient values derived from velocity data were fitted with SEDPHAT 4.1 using the hybrid local continuous/global discrete model with 2 discrete species. The values for s20,w for different detection methods and protein concentrations were corrected for solvent density and viscosity based on buffer composition, but were not corrected for protein concentration. The mass values were calculated by modeling the diffusional spreading of the boundary as a function of time.
Gel filtration
Experiments were conducted using Sephadex 200 and Sepharose 6 columns from Amersham Biosciences. BioRad protein gel filtration standards were used for calibration. PDO WT or PDO D178A and D178N variants (0.2 mL of 120 μM) were loaded on a column that was equilibrated with 0.1 M HEPES, pH 7.8, 3 mM phthalate, and 0.2 M NaCl. Columns were run at 4 °C, with a flow rate of 0.5 mL/min. Protein was detected spectrophotometrically at 280 nm. For statistical analysis, the traces were approximated by a normal distribution function that was constructed in Excel, using equation (1), where μ is the mean and σ is the standard deviation.
| (1) |
The mean, the standard deviation, and the scaling factor for the amplitude of the signal were used as variables for the fit and were calculated using Solver, a built-in Excel function. These values were optimized to achieve the minimum possible squared deviation(s) between the normal distribution and the experimental data.
All chemicals were of analytical grade and used without further purification.
RESULTS AND DISCUSSION
Gel filtration
Both WT PDO and the D178N variant eluted with the main peak at a position correlating to about 250 kDa. Using a normal function to approximate the signal distribution observed in gel filtration experiments, we calculated for both WT and D178N PDO a value of the mean corresponding to a Stokes radius of ∼ 253 kDa with a σ = 55.6 kDa for the WT and σ = 59.6 for the D178N sample. Statistical analysis shows that the probability (P) that the protein in the sample had a molecular weight of a tetramer or less (P(MW ≤200 kDa)) is about 0.15 for the WT and about 0.14 for D167N variant. Similar probability analysis for a hexamer PDO structure (P(MW) ≥ 300 kDa) give a P = 0.28 for the WT and 0.27 for D178N variant. Although the probability function appears to slightly favor the hexamer, the difference between tetramer, pentamer, or hexamer forms is not statistically significant. However, the data does exclude the possibility that either WT or D178N PDO can be an octomer (∼ 400 kDa), as the probability of such is very low (below 1.4 percent).
Gel filtration is a widely accepted tool to determine molecular weights of protein complexes and aggregates. It is often used to establish binding stoichiometries and multimeric organization states of proteins. However, statistical analysis of the results is rarely presented in published data. As shown above, consideration of mean elution time may not be sufficient for unequivocal allocation of the results to specific protein structures. Statistical analysis using probability functions is necessary to establish distribution profile of gel filtration results and to verify whether the correlation between the expected and experimentally obtained molecular weights has statistical significance.
Mass spectrometry
The application of electrospray ionization mass spectrometry (ESI-MS), has demonstrated its utility in structural biology for measuring non-covalently-bound protein-protein, protein-ligand, and protein-oligonucleotide complexes [21; 22; 30; 31; 32]. This form of MS has been termed “native” protein mass spectrometry, because the measurements usually provide the same stoichiometric assembly size as that expected for the solution phase. For the measurement of phthalate dioxygenase, two different forms of ESI-based analyses were performed: ESI with lower resolution ion mobility analysis (gas phase electrophoretic mobility molecular analyzer, or GEMMA) of singly-charged molecules [22; 23; 24; 33] and ESI with a higher resolution quadrupole time-of-flight mass spectrometer for the measurement of multiply-charged molecules [22]. Both methods indicate that the wild-type PDO complex exists predominantly in the hexameric form. The ESI-GEMMA spectra of the PDO proteins at concentrations ranging from 1 μM to 1 nM, in pH 6.8 solutions, consistently yielded an abundant peak near 11.3 nm for the mean electrophoretic mobility diameter (EMD). Based on extensive GEMMA measurements from our laboratory [22; 24; 25; 33] as well as from those of others [23] for a wide size range of protein complexes, an average particle density of 0.6 g/cc was used to estimate a protein molecular weight of 250-280 kDa for PDO (Figure 1). GEMMA spectra of the protein solution in a more acid pH environment (pH 3), which denatured the protein complex, yields an EMD of 6.4 nm, or approximately 45 kDa for the PDO monomer.
Figure 1.

ESI-GEMMA spectrum of WT PDO acquired at pH 6.8. The measured electrophoretic mobility diameter of 11.3 nm corresponds to a molecular weight of approximately 280 kDa.
In agreement with these values, ESI-MS of PDO at pH 3 generated a spectrum for the multiply-charged protein molecules that is consistent with a 48.8 kDa protein monomer (Figure 2A). This correlates well with the molecular weight of 49.78 kDa predicted from the PDO sequence and with the molecular weight of about 50 kDa determined by denaturing SDS-PAGE. Raising the pH to 6.8 shifts the peaks in the mass spectrum (Figure 2B) to much higher mass-to-charge (m/z) values (m/z 7000-8000) compared to the mass spectrum from denaturing solutions (m/z 800-1800). The observation of larger m/z peaks is consistent with previous reports of native protein MS, and this phenomenon has been attributed to lower absolute multiple charging in ESI mass spectrometer due to more compact tertiary-quaternary structures [21; 30]. These m/z 7000-8000 peaks, representing a complex with 38+ through 42+ charges, correspond to a molecular weight of the hexameric complex of 294.8 kDa (Figure 2B). Similar results (not shown) were obtained for both D178A and D178N variants of PDO.
Figure 2.

ESI mass spectra of WT PDO acquired at pH 3 (A) and pH 6.8 (B). The corresponding mass spectra converted to the mass domain are shown in the insets. A denaturing pH 3 solution yields a MW for the monomer of 48,838 ± 3 Da. The protein complex measured under near native pH conditions yields 294,779 ± 53 Da.
The presence of Fe and [2Fe-2S] centers in PDO is also suggested by the ESI-MS measurements. The molecular masses of other metalloproteins that bind [2Fe-2S] and Fe centers have been measured previously using ESI-MS [34]. A hexamer complex from the measured mass of 48.8 kDa for the denatured monomer (without metal-binding) would yield a calculated mass of 293.0 kDa, whereas the measured hexamer complex was measured to be 294.8 kDa. The addition of one 2Fe-2S and one Fe to each subunit would increase the total calculated mass to 294.4 kDa, which is much closer to the measured value.
Similar mass spectrometer experiments were carried out for the Rieske oxygenases, CarDO and NDO. CarDO is an α3 oxygenase with the mass of each α monomer predicted to be 43.8 kDa based on protein sequence. A mass for the denatured monomeric protein was measured to be 44.7 kDa, and the ESI mass spectrum of the native CarDO complex yielded a mass of 134.8 kDa (Figure 3A). From the sum of the measured monomer mass and the addition of one [2Fe-2S] and one Fe per subunit, the calculated mass of the trimer CarDO protein complex is 134.8 kDa, exactly as found for undenatured CarDO. For α3β3 NDO, the predicted masses are 49.6 and 22.9 kDa for the α and β subunits respectively; the masses of the denatured α- and β-subunits were measured to be 49.6 kDa and 22.8 kDa, respectively. The intact α3β3 NDO complex yielded a measured molecular mass of 218.3 kDa (Figure 3B), and a calculated hexamer protein mass of 218.6 kDa (with one [2Fe-2S] and one Fe per subunit). From the ESI-MS data, one cannot definitively conclude that [2Fe-2S] and Fe are stoichiometrically bound to the proteins; however the data are consistent with the hypothesis. Clearly, the MS ESI data resulted in excellent correlations of the determined molecular masses of native multimers of both CarDO and NDO with the expected values. Combined with the molecular masses of denatured protein, this allowed for reasonable predictions of the multimeric structure of theses enzymes.
Figure 3.

ESI mass spectra of CarDO (A) and NDO (B) in 20 mM ammonium acetate, pH 6.8. The corresponding mass spectra converted to the mass domain are shown in the insets. For CarDO, a MW for the denatured monomer and the native α3 trimer was measured to be 44,711 ± 2 Da and 134,853 ± 9 Da, respectively. For NDO, the measured MWs for the denatured α- and β-subunits were 49,607 ± 2 Da and 22,802 ± 0.2 Da, respectively. The MW of the intact α3β3 native NDO complex was measured to be 218,283 ± 58 Da.
Analytical ultracentrifugation
Multiwavelength analytical ultracentrifugation was used for determining the oligomeric size of PDO at both low and high salt concentrations (Figure 4). Table 1 shows the results obtained using the hybrid local continuous/global discrete model in SEDPHAT. Values of s20,w did not exhibit any obvious trend that correlated with protein concentrations; however, the values observed in low salt conditions are consistently larger than those at high salt concentrations (Table 1). The change in shape of a boundary as a function of time can be used to calculate the mass of the particle as long as the boundary shape is not perturbed by phenomena such as electrostatic effects or convection. At pH 7, PDO appears to exhibit a negative surface charge that gives rise to electrostatic effects that may significantly affect the boundary. High salt concentration in the sample would ameliorate these effects. Thus, the difference between the masses determined at low and high salt conditions could be due to differences in electrostatic interactions between the PDO molecules. Alternatively, a rapid hexamer—dodecamer equilibrium process can be invoked to explain low/high salt differences; however, this seems unlikely because of the large net charge on the molecule and the lack of sensitivity of the results to the protein concentration. Overall, the results obtained under the high salt conditions should provide a better estimate for the PDO multimer mass. At high salt the average oligomer size is 6.1 PDO subunits, corresponding to a mass of about 300 kDa, and like the mass spectral results, strongly suggesting a hexameric subunit organization in PDO.
Figure 4.

Interference optics detection of PDO sedimentation in low salt conditions (see Table 2). Concentration of PDO and the fitted results in fringe units against radial position are shown at 27 minute intervals (9 minutes for the last interval) during the velocity experiment (A); the differences between the observed and calculated fringes are offset by -.5 units to aid in visualization (B) and the distribution of sedimenting species with a given s value against the sedimentation coefficient corrected to 20°C and water are in (C).
Table 1.
Sedimentation coefficients and molecular mass of PDO as determined by Analytical ultracentrifugation.
| Conditionsa | Detection method | Loading concentration (signal units) | s20,W, S (± 0.2 S) | Mass, kDa (± 10 kDa) | PDO oligomer sizeb |
|---|---|---|---|---|---|
| Low salt | Interference | 3.40 | 11.78 | 363 | 7.3 |
| Low salt | Absorbance at 405 nm | 0.12 | 11.99 | 372 | 7.6 |
| Low salt | Absorbance at 455 nm | 0.16 | 11.99 | 387 | 7.9 |
| High salt | Interference | 0.39 | 11.50 | 318 | 6.5 |
| High salt | Absorbance at 280 nm | 0.34 | 11.81 | 283 | 5.8 |
| High salt | Interference | 0.92 | 11.37 | 301 | 6.1 |
| High salt | Absorbance at 280 nm | 0.72 | 11.66 | 284 | 5.8 |
Buffer for low salt was 20 mM KPO4, pH 6.9; for high salt, 50 mM KPO4, 0.5 MKCl, pH 7. Buffers contained 5 mM phthalate.
Based on monomer mass of 49 kDa.
Numerical Modeling
The dioxygenation of phthalate requires two electrons and both atoms of an oxygen molecule being incorporated into the product. When reduced, fully active PDO reacts with phthalate and oxygen. Both electrons necessary for DHD formation are ultimately provided by the Rieske centers of the enzyme. At the end of the reaction, the mononuclear iron center of PDO is found in the ferrous state [7; 8]; it is possible that iron acts as a transient electron donor in catalysis and then becomes reduced by a nearby Rieske center. Thus, in single turnover experiments only one equivalent of DHD is formed per every two Rieske centers oxidized; furthermore, only one active iron mononuclear center per two Rieske centers is necessary to generate the entire product possible. When only some iron mononuclear centers are active (e.g., in the case of APO-PDO partially reconstituted with iron), the amount of product produced would depend on the distribution of the Fe(II) among all available mononuclear centers. Depending on the arrangement of the subunits in the multimer, each Rieske center may be able to transfer electrons to one or more Fe-mononuclear centers. With each mononuclear center being in contact with at least two Rieske centers, full product could result from only 50% reconstituted PDO. The particular arrangement present will also affect the estimated amount of DHD produced in the course of oxidation of reduced PDO. In our model the PDO hexamer consists of two PDO trimers, each arranged in a head-to-tail α subunit arrangement as observed in many other Rieske dioxygenases [9; 10; 11; 12; 13; 14]. The arrangement, presented in Figure 5A, has two mononuclear iron centers and two Rieske centers from four different subunits near to each other (Figure 5B), and this motif occurs at three positions in the hexamer.
Figure 5.
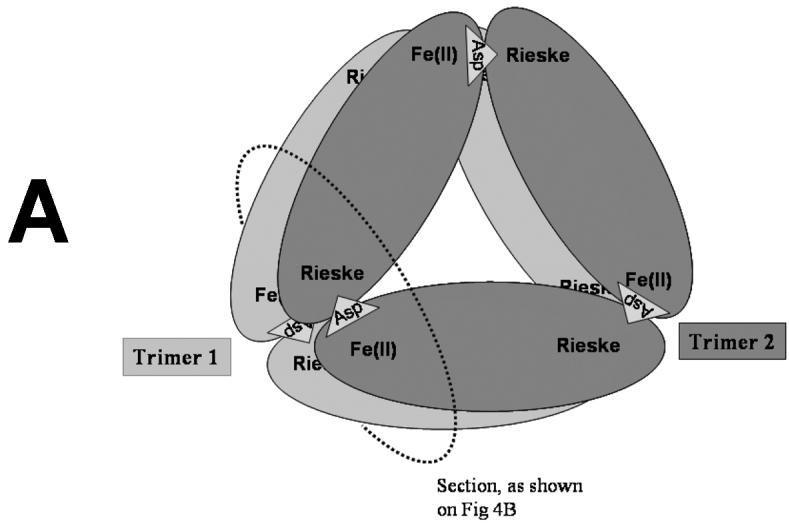
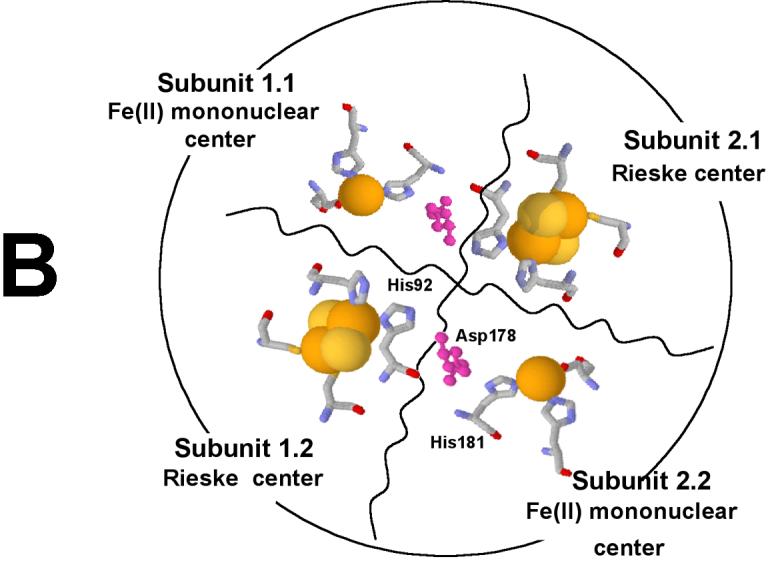
Proposed α6 model of PDO (A). The PDO monomers are arranged in two α3 rings stacked one on top of the other. Rieske and Fe-mononuclear sites of four different subunits are located near each other (B) as indicated by previously reported kinetic and product analysis results [7; 8]. The model was constructed on the basis of structural data from studies of NDO [9].
We have previously tested what levels of product will be formed with partially iron-depleted PDO [8]. Here we have calculated how these levels of product formation can be accounted for based on the hexameric structure of the enzyme. The Berkeley Madonna Version 8.0.1 program was used to model the fractional reconstitution of the mononuclear centers of APO-PDO with different levels of added Fe(II). The main assumptions used in the model are: i) Fe(II) preferentially binds at the mononuclear centers rather than adventitiously at other regions of PDO; ii) All mononuclear centers can bind Fe(II); and iii) There is no significant cooperativity in iron binding at the mononuclear centers (random Fe(II) binding at the mononuclear centers) and no preferential binding of oxygen to a given mononuclear iron. This seems reasonable because for WT PDO in the presence of phthalate, Fe(II) does bind tightly to the WT PDO (Kd about 1 μM) with no observed cooperativity [4]. At 1:1 stoichiometry between free Fe(II) and PDO monomers, each monomer would be fully populated; at lower relative concentrations of free Fe(II) only some of the mononuclear sites would contain iron, thus creating species containing different amounts of iron bound at different subunits of PDO multimer. Concentrations of each particular species relative to the total PDO monomer concentration would then be a function of amount of free iron in the solution. The fractional concentration of mononuclear centers of the hexamer that has iron incorporated, can be calculated as a function of different levels of added iron. Figure 6 shows possible PDO multimeric species for various distributions and amounts of Fe(II) bound at the mononuclear centers for the model presented in figure 5. The relative amounts of each species at different levels of exogenously added free Fe(II) is presented in Table 2.
Figure 6.
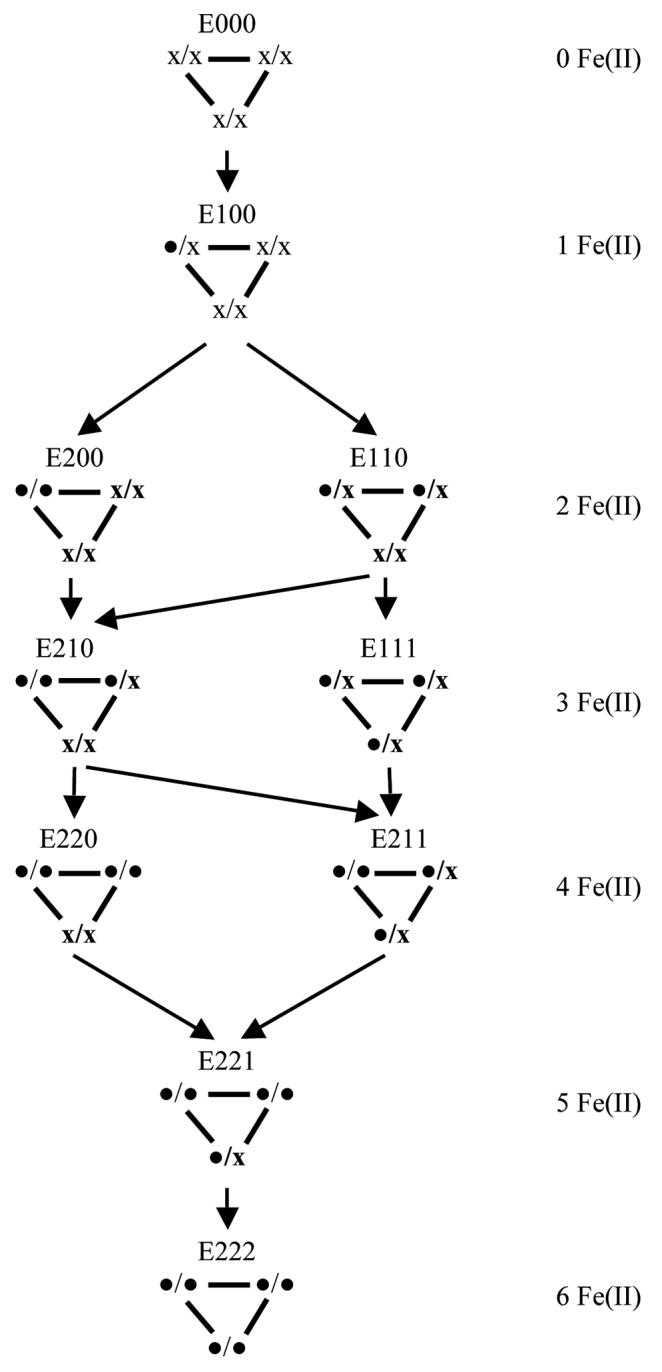
Binding of Fe(II) to the mononuclear centers of the PDO hexamer. The PDO hexamer has three assemblages each containing two Rieske and two mononuclear centers. Each mononuclear Fe(II) center may either contain ( •) or lack ( x ) iron. The model does not differentiate which of the two mononuclear centers within a given assemblage binds Fe(II). The model has a 120° rotational symmetry; thus, structures that can be reduced to one another by rotation are not differentiated.
Table 2.
Distribution of variously populated Fe-mononuclear centers in PDO and DHD production at different relative concentrations of exogenous iron.
| E000 | E100 | E200 | E110 | E210 | E111 | E220 | E211 | E221 | E222 | Product, % | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Products per hexamer | 0 | 1 | 1 | 2 | 2 | 3 | 2 | 3 | 3 | 3 | Modelb | Experimentc |
| F(II) a, % | % of the form (Eijk) in solution | |||||||||||
| ≥100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 0.50 | 0.50 |
| 50 | 10.0 | 9.0 | 14.9 | 6.7 | 11.4 | 7.3 | 8.4 | 13.6 | 11.9 | 7.7 | 0.38 | 0.32 |
| 25 | 31.3 | 21.5 | 14.9 | 10.1 | 8.3 | 4.7 | 4.3 | 2.7 | 1.8 | 0.4 | 0.18 | 0.18 |
“Fe(II), %” is the amount of Fe(II) added to the solution for PDO reconstitution expressed as a fraction of available Fe-mononuclear centers.
Predicted by the described model from the distribution of Fe(II) between the mononuclear centers of PDO, with the subunits arranged in a hexameric structure as on Figure 5.
Data from [7].
Based on the calculated iron distribution profile using the assumptions stated above, the maximum amount of DHD that could be produced was calculated. This amount depends on the number and distribution of iron-containing mononuclear centers. Similarly to our previous analysis [8], the amount of product produced by each particular species shown on figure 6, was calculated on the basis of the following assumptions: iv) Only one active mononuclear center per two Rieske centers is necessary for DHD formation (no net electrons are provided by the Fe(II) incorporated into the mononuclear center), and if an active site were available, two Rieske centers would cooperate to irreversibly provide one electron each for DHD formation; v) Electron transfer is slow between the Rieske centers of individual hexameric enzyme molecules, as well as between the Rieske and mononuclear centers within a given monomer; vi) All Rieske centers can be oxidized, but only those coupled to a mononuclear Fe(II) center will give rise to a product. These assumptions naturally follow from the reported experimental results and the model proposed previously [4; 7; 8].
The quantities of DHD expected to be produced by each species shown in figure 6 (between zero and 3, depending on the pattern of Fe(II) distribution among the iron mononuclear centers in PDO multimer) is shown in Table 2. For example, E100 species would be able to produce one molecule of product per multimer (per six Rieske centers oxidized), similar to the E200 species. That is because in the E200 species both Fereconstituted mononuclear centers are located near to the same two Rieske centers, and thus the total amount of DHD produced would be limited by the available electron supply from the adjacent Rieske centers (2 electrons). On the other hand, E110, E210, and E220 would produce two molecules of product, and E111, E211, E221, and E222 would produce three molecules of DHD per 6 Rieske centers oxidized. Data presented in the table are based on the additional assumption vii) that all mononuclear centers are active, which is true at least for the WT enzyme used in both this study and previously [8]. Combined, the amounts of DHD produced for each possible distribution of Fe(II) among the iron mononuclear centers and the fractional content of these species determine the amount of DHD PDO hexamer would produce at each level of extrageneous free iron.
Dialysis of PDO in the presence of EDTA results in the loss of iron from the iron mononuclear center with the development of E000 species. For that species, no DHD is formed in the course of reduced PDO reacting with oxygen. Reconstitution of such PDO with measured, sub-stoichiometric amounts of Fe(II) results in partial reconstitution of the mononuclear centers, with the distribution of iron among the centers in PDO multimer determining the amount of DHD produced in the single turnover oxidation of reduced PDO. Both estimates, based on the model shown on figure 5 and the assumptions detailed above, and the experimentally observed amounts of DHD produced [8], are presented in Table 2. There is good correlation between the predicted and experimentally observed amounts of DHD.
Overall, the proposed hexameric structure of PDO (Figure 5) has many similarities to that of the enzymes in NDO family. The main difference is instead of α and β trimeric planar rings, both rings in PDO are composed of α subunits. Thus, the PDO structure can be formally expressed as α3α3, to emphasize the division of the subunits into two trimers. Such an arrangement brings two Rieske centers and two mononuclear centers from four subunits located on two planar rings in proximity to one another, allowing for both Rieske centers to participate in the catalysis at a single Fe-mononuclear site. This process leaves the mononuclear iron in the ferrous state after the oxidation of the Rieske center and the formation of the product. No such effect was observed in NDO and similar enzymes, as they obviously lack the second Rieske center proximal to the mononuclear iron. To the best of our knowledge, this is the first time such cooperativity has been shown for Rieske oxygenases. As detailed above, in many other Rieske dioxygenases the Rieske center of one subunit is positioned for delivering electrons to the Fe-mononuclear center on the other subunit. However, none appear to have the capacity for two Rieske centers to participate in electron transfer to the same Fe-mononuclear center, or alternatively, for any one Rieske center to deliver electrons to two different Fe-mononuclear centers.
CarDO appears to be stable and catalytically active in an α3 form [12; 35], while in both NDO [9] and in TDO [36] the β subunit, although it does not contain any activity centers, appears to be essential for enzyme activity, and plays supposedly a structural role. Cooperative interactions between α subunits in PDO, as described above, implies that in PDO both trimer α3 rings are essential for catalysis. It remains yet unknown whether such an interaction observed in single turnover experiments is a valid model for steady-state turnover, where consecutive electron transfers through the same Rieske center are possible. It also remains to be determined whether the α3 rings in PDO have a structural function, stabilizing the multimer, similar to that in NDO and TDO, or, at least in steady-state catalysis, they can function independently.
The presented evidence is consistent with an α3α3 model of subunit arrangement in PDO.
Although superficially similar to αβ arrangement observed in most Rieske dioxygenases, this structure has probably arisen from different functional requirements placed on the Fe-mononuclear center by the need to accommodate phthalate as the substrate. Further comparison of functional and structural differences and similarities between these structures could further improve our understanding of the specifics of the catalysis mediated by the joint action of Rieske and iron-mononuclear centers.
ACKNOWLEDGEMENT
MT and DPB thank Dr. Ray Trieval for permitting us the use of the Amersham Biosciences system and gel filtration columns. The UCLA Functional Proteomics Center was established and equipped by a grant from the W. M. Keck Foundation. JAL acknowledges support from the UCLA-DOE Institute for Genomics and Proteomics, and the National Institutes of Health (RR 20004). JB would like to express gratitude to Pravinda Kumar (Purdue) for assistance in the ultracentrifugation experiments and to acknowledge the support of the National Institutes of Health (R01-GM52381 to Jeffrey T. Bolin). Authors want to thank Prof. Hideaki Nojiri (Biotechnology Research Center, The University of Tokyo) for his generous gift of the CarDO sample, Dr. Ramaswamy Subramanian (Department of Biochemistry Roy J and Lucille A Carver College of Medicine The University of Iowa) for his gift of the NDO sample, and Dr. Jeffrey Bolin (Purdue) for general assistance and helpful discussions. This work was supported by NIH grant GM20877 to D.P.B.
Abbreviations
- PDO
phthalate dioxygenase
- WT PDO
enzyme with no substitutions in the sequence
- APO-PDO
PDO with the mononuclear center lacking the iron
- NDO
naphthalene dioxygenase
- TDO
toluene dioxygenase
- AntDO
anthranilate dioxygenase
- BPDO
biphenyl dioxygenase
- BZDO
benzoate dioxygenase
- NBDO
nitrobenzene dioxygenase
- CarDO
carbazole 1,9a-dioxygenase
- CumDO
cumene dioxygenase
- OMO
2-Oxoquinoline-8-monooxygenase
- ESI-MS
electrospray ionization mass spectrometry
- GEMMA
gas phase electrophoretic mobility molecular analyzer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCIES
- [1].Parales RE, Parales JV, Gibson DT. Aspartate 205 in the catalytic domain of naphthalene dioxygenase is essential for activity. J Bacteriol. 1999;181:1831–7. doi: 10.1128/jb.181.6.1831-1837.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Beharry ZM, Eby DM, Coulter ED, Viswanathan R, Neidle EL, Phillips RS, Kurtz DM., Jr. Histidine ligand protonation and redox potential in the rieske dioxygenases: role of a conserved aspartate in anthranilate 1,2-dioxygenase. Biochemistry. 2003;42:13625–36. doi: 10.1021/bi035385n. [DOI] [PubMed] [Google Scholar]
- [3].Jiang H, Parales RE, Lynch NA, Gibson DT. Site-directed mutagenesis of conserved amino acids in the alpha subunit of toluene dioxygenase: potential mononuclear non-heme iron coordination sites. J Bacteriol. 1996;178:3133–3139. doi: 10.1128/jb.178.11.3133-3139.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pinto A, Tarasev M, Ballou DP. Substitutions of the “bridging” aspartate 178 result in profound changes in the reactivity of the Rieske center of phthalate dioxygenase. Biochemistry. 2006;45:9032–41. doi: 10.1021/bi060216z. [DOI] [PubMed] [Google Scholar]
- [5].Tarasev M, Pinto A, Kim D, Elliott SJ, Ballou DP. The “bridging” aspartate 178 in phthalate dioxygenase facilitates interactions between the Rieske center and the iron(II)--mononuclear center. Biochemistry. 2006;45:10208–16. doi: 10.1021/bi060219b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wolfe MD, Parales JV, Gibson DT, Lipscomb JD. Single turnover chemistry and regulation of O2 activation by the oxygenase component of naphthalene 1,2-dioxygenase. J Biol Chem. 2001;276:1945–1953. doi: 10.1074/jbc.M007795200. [DOI] [PubMed] [Google Scholar]
- [7].Tarasev M, Rhames F, Ballou DP. Rates of the Phthalate Dioxygenase Reaction with Oxygen Are Dramatically Increased by Interactions with Phthalate and Phthalate Oxygenase Reductase. Biochemistry. 2004;43:12799–12808. doi: 10.1021/bi0490587. [DOI] [PubMed] [Google Scholar]
- [8].Tarasev M, Ballou DP. Chemistry of the catalytic conversion of phthalate into its cis-dihydrodiol during the reaction of oxygen with the reduced form of phthalate dioxygenase. Biochemistry. 2005;44:6197–207. doi: 10.1021/bi047724y. [DOI] [PubMed] [Google Scholar]
- [9].Kauppi B, Lee K, Carredano E, Parales RE, Gibson DT, Eklund H, Ramaswamy S. Structure of an aromatic-ring-hydroxylating dioxygenase-naphthalene 1,2-dioxygenase. Structure. 1998;6:571–586. doi: 10.1016/s0969-2126(98)00059-8. [DOI] [PubMed] [Google Scholar]
- [10].Furusawa Y, Nagarajan V, Tanokura M, Masai E, Fukuda M, Senda T. Crystal structure of the terminal oxygenase component of biphenyl dioxygenase derived from Rhodococcus sp. strain RHA1. Journal of Molecular Biology. 2004;342:1041–52. doi: 10.1016/j.jmb.2004.07.062. [DOI] [PubMed] [Google Scholar]
- [11].Friemann R, Ivkovic-Jensen MM, Lessner DJ, Yu CL, Gibson DT, Parales RE, Eklund H, Ramaswamy S. Structural insight into the dioxygenation of nitroarene compounds: the crystal structure of nitrobenzene dioxygenase. Journal of Molecular Biology. 2005;348:1139–51. doi: 10.1016/j.jmb.2005.03.052. [DOI] [PubMed] [Google Scholar]
- [12].Nojiri H, Ashikawa Y, Noguchi H, Nam JW, Urata M, Fujimoto Z, Uchimura H, Terada T, Nakamura S, Shimizu K, Yoshida T, Habe H, Omori T. Structure of the terminal oxygenase component of angular dioxygenase, carbazole 1,9a-dioxygenase. Journal of Molecular Biology. 2005;351:355–70. doi: 10.1016/j.jmb.2005.05.059. [DOI] [PubMed] [Google Scholar]
- [13].Dong X, Fushinobu S, Fukuda E, Terada T, Nakamura S, Shimizu K, Nojiri H, Omori T, Shoun H, Wakagi T. Crystal structure of the terminal oxygenase component of cumene dioxygenase from Pseudomonas fluorescens IP01. Journal of Bacteriology. 2005;187:2483–90. doi: 10.1128/JB.187.7.2483-2490.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Martins BM, Svetlitchnaia T, Dobbek H. 2-Oxoquinoline 8-monooxygenase oxygenase component: active site modulation by Rieske-[2Fe-2S] center oxidation/reduction. Structure (Camb) 2005;13:817–24. doi: 10.1016/j.str.2005.03.008. [DOI] [PubMed] [Google Scholar]
- [15].Jakoncic J, Jouanneau Y, Meyer C, Stojanoff V. The crystal structure of the ring-hydroxylating dioxygenase from Sphingomonas CHY-1. Febs J. 2007;274:2470–81. doi: 10.1111/j.1742-4658.2007.05783.x. [DOI] [PubMed] [Google Scholar]
- [16].Jakoncic J, Jouanneau Y, Meyer C, Stojanoff V. The catalytic pocket of the ring-hydroxylating dioxygenase from Sphingomonas CHY-1. Biochem Biophys Res Commun. 2007;352:861–6. doi: 10.1016/j.bbrc.2006.11.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Parales JV, Parales RE, Resnick SM, Gibson DT. Enzyme specificity of 2-nitrotoluene 2,3-dioxygenase from Pseudomonas sp. strain JS42 is determined by the C-terminal region of the alpha subunit of the oxygenase component. J Bacteriol. 1998;180:1194–9. doi: 10.1128/jb.180.5.1194-1199.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Parales RE, Emig MD, Lynch NA, Gibson DT. Substrate specificities of hybrid naphthalene and 2,4-dinitrotoluene dioxygenase enzyme systems. J Bacteriol. 1998;180:2337–44. doi: 10.1128/jb.180.9.2337-2344.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Batie CJ, LaHaie E, Ballou DP. Purification and characterization of phthalate oxygenase and phthalate oxygenase reductase from Pseudomonas cepacia. J Biol Chem. 1987;262:1510–1518. [PubMed] [Google Scholar]
- [20].Batie CJ, Ballou DP. Phthalate dioxygenase. Methods Enzymol. 1990;188:61–70. doi: 10.1016/0076-6879(90)88013-z. [DOI] [PubMed] [Google Scholar]
- [21].Loo JA. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom Rev. 1997;16:1–23. doi: 10.1002/(SICI)1098-2787(1997)16:1<1::AID-MAS1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- [22].Loo JA, Berhane B, Kaddis CS, Wooding KM, Xie Y, Kaufman SL, Chernushevich IV. Electrospray ionization mass spectrometry and ion mobility analysis of the 20S proteasome complex. J Am Soc Mass Spectrom. 2005;16:998–1008. doi: 10.1016/j.jasms.2005.02.017. [DOI] [PubMed] [Google Scholar]
- [23].Bacher G, Szymanski WW, Kaufman SL, Zollner P, Blaas D, Allmaier G. Charge-reduced nano electrospray ionization combined with differential mobility analysis of peptides, proteins, glycoproteins, noncovalent protein complexes and viruses. J Mass Spectrom. 2001;36:1038–52. doi: 10.1002/jms.208. [DOI] [PubMed] [Google Scholar]
- [24].Poderycki MJ, Kickhoefer VA, Kaddis CS, Raval-Fernandes S, Johansson E, Zink JI, Loo JA, Rome LH. The vault exterior shell is a dynamic structure that allows incorporation of vault-associated proteins into its interior. Biochemistry. 2006;45:12184–93. doi: 10.1021/bi0610552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kaddis CS, Lomeli SH, Yin S, Berhane B, Apostol MI, Kickhoefer VA, Rome LH, Loo JA. Sizing large proteins and protein complexes by electrospray ionization mass spectrometry and ion mobility. J Am Soc Mass Spectrom. 2007;18:1206–1216. doi: 10.1016/j.jasms.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kaddis CS, Loo JA. Native protein MS and ion mobility large flying proteins with ESI. Analytical chemistry. 2007;79:1778–1784. doi: 10.1021/ac071878c. [DOI] [PubMed] [Google Scholar]
- [27].Laue TM, Shah BD, Ridgeway TM, Pelletier SL. In: Analytical Ultracentrifugation in Biochemistry and Polymer Science. Harding SE, Rowe AJ, Horton JC, editors. The Royal Society of Chemistry; Cambridge, UK: 1992. pp. 90–125. [Google Scholar]
- [28].Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Balbo A, Minor KH, Velikovsky CA, Mariuzza RA, Peterson CB, Schuck P. Studying multiprotein complexes by multisignal sedimentation velocity analytical ultracentrifugation. Proc Natl Acad Sci U S A. 2005;102:81–86. doi: 10.1073/pnas.0408399102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Heck AJR, van den Heuvel RHH. Investigation of Intact Protein Complexes by Mass Spectrometry. Mass Spectrom. Rev. 2004;23:368–389. doi: 10.1002/mas.10081. [DOI] [PubMed] [Google Scholar]
- [31].van den Heuvel RH, Heck AJ. Native protein mass spectrometry: from intact oligomers to functional machineries. Curr Opin Chem Biol. 2004;8:519–26. doi: 10.1016/j.cbpa.2004.08.006. [DOI] [PubMed] [Google Scholar]
- [32].Ruotolo BT, Giles K, Campuzano I, Sandercock AM, Bateman RH, Robinson CV. Evidence for macromolecular protein rings in the absence of bulk water. Science. 2005;310:1658–61. doi: 10.1126/science.1120177. [DOI] [PubMed] [Google Scholar]
- [33].Kani K, Warren CM, Kaddis CS, Loo JA, Landgraf R. Oligomers of ERBB3 have two distinct interfaces that differ in their sensitivity to disruption by heregulin. J Biol Chem. 2005;280:8238–47. doi: 10.1074/jbc.M410944200. [DOI] [PubMed] [Google Scholar]
- [34].Johnson KA, Verhagen MF, Brereton PS, Adams MW, Amster IJ. Probing the stoichiometry and oxidation states of metal centers in iron-sulfur proteins using electrospray FTICR mass spectrometry. Analytical chemistry. 2000;72:1410–1418. doi: 10.1021/ac991183e. [DOI] [PubMed] [Google Scholar]
- [35].Nam JW, Nojiri H, Noguchi H, Uchimura H, Yoshida T, Habe H, Yamane H, Omori T. Purification and characterization of carbazole 1,9a-dioxygenase, a three-component dioxygenase system of Pseudomonas resinovorans strain CA10. Appl Environ Microbiol. 2002;68:5882–90. doi: 10.1128/AEM.68.12.5882-5890.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jiang H, Parales RE, Gibson DT. The alpha subunit of toluene dioxygenase from Pseudomonas putida F1 can accept electrons from reduced FerredoxinTOL but is catalytically inactive in the absence of the beta subunit. Appl Environ Microbiol. 1999;65:315–8. doi: 10.1128/aem.65.1.315-318.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]