

Abstract
Accumulating evidence suggests that microglial cells in the spinal cord play an important role in the development of neuropathic pain. However, it remains largely unknown how glia interact with neurons in the spinal cord after peripheral nerve injury. Recent studies suggest that the chemokine fractalkine may mediate neural/microglial interaction via its sole receptor CX3CR1. We have examined how fractalkine activates microglia in a neuropathic pain condition produced by spinal nerve ligation (SNL). SNL induced an upregulation of CX3CR1 in spinal microglia that began on day 1, peaked on day 3, and maintained on day 10. Intrathecal injection of a neutralizing antibody against CX3CR1 suppressed not only mechanical allodynia but also the activation of p38 MAPK in spinal microglia following SNL. Conversely, intrathecal infusion of fractalkine produced a marked p38 activation and mechanical allodynia. SNL also induced a dramatic reduction of the membrane-bound fractalkine in the dorsal root ganglion, suggesting a cleavage and release of this chemokine after nerve injury. Finally, application of fractalkine to spinal slices did not produce acute facilitation of excitatory synaptic transmission in dorsal horn neurons, arguing against a direct action of fractalkine on spinal neurons. Collectively, our data suggest that (a) fractalkine cleavage (release) after nerve injury may play an important role in neural-glial interaction, and (b) microglial CX3CR1/p38 MAPK pathway is critical for the development of neuropathic pain.
Keywords: Chemokine, MAP kinase, microglia, spinal cord, neural-glial interaction, spinal nerve ligation, intracellular signaling, neuropathic pain
1. Introduction
Chronic pain resulting from nerve damage can be intractable. Analgesic drugs used for treating acute pain are not very effective for neuropathic pain, in part due to our incomplete understanding of the mechanisms underlying the development and maintenance of neuropathic pain. Increasing evidence shows that spinal microglia are activated after nerve injury and contribute to the development of neuropathic pain. Nerve injury induces the expression of microglial markers (e.g., CD11b, TLR4, CD14) within several hours (DeLeo et al., 2004). Microglia are regarded as a major source for the production of proinflammatory cytokines (Hanisch 2002; Koistinaho and Koistinaho, 2002) that are implicated in pain facilitation (DeLeo and Yezierski, 2001; Watkins et al., 2001). Nerve injury upregulates several receptors, such as the chemokine receptors CCR2 and CX3CR1 and the ATP receptor P2X4 in spinal microglia; blocking or deleting these receptors results in decreased neuropathic pain (Abbadie et al., 2003; Tsuda et al., 2003; 2005; Milligan et al., 2004; Verge et al., 2004). A microglial inhibitor minocycline has been shown to prevent/delay neuropathic pain (Raghavendra et al., 2003; Ledeboer et al., 2005) and to attenuate a central neuropathic pain after spinal cord injury (Hains and Waxman, 2006).
Nerve injury also induces a drastic activation (phosphorylation) of p38 mitogen-activated protein kinase (MAPK) in the spinal cord (Jin et al., 2002, 2003; Kim et al., 2002; Schafers et al., 2003). Surprisingly, p38 is not activated in spinal neurons (Ji et al., 2002b). Instead, p38 is activated in spinal cells labeled with OX-42, a marker for microglia, following nerve injury (Jin et al., 2003; Tsuda et al., 2004). In addition to nerve injury, p38 is rapidly activated in spinal microglia in other pain conditions (Svensson et al., 2003; Sweitzer et al., 2004). Several lines of evidence indicates that p38 activation plays an important role in the development of neuropathic pain. Intrathecal administration of the p38 inhibitors was shown to attenuate neuropathic pain symptoms (Jin et al., 2003; Milligan et al., 2003; Schafers et al., 2003; Obata et al., 2004; Tsuda et al., 2004). Therefore, p38 phosphorylation in microglia could serve as a pain-related marker for microglial activation in the spinal cord. Although several studies began to explore the upstream mechanisms responsible for p38 activation in spinal microglia (Svensson et al., 2005; Sung et al., 2005), these mechanisms are still illusive.
Glial cells are believed to regulate pain sensitivity through interacting with neurons. However, little is known about how neural information is conveyed from periphery to spinal glia after peripheral nerve injury. The chemokine fractalkine (also called CXCL1) appears to be an ideal candidate to mediate neural/glial interaction, because (a) fractalkine is produced in neurons of the dorsal root ganglion (DRG) and the spinal cord (Verge et al., 2004; Lindia et al., 2005), and (b) the sole receptor of fractalkine, CX3CR1 is expressed in microglia and required for neuropathic pain facilitation (Milligan et al., 2004; Verge et al., 2004; Lindia et al., 2005). In this study, we show that nerve injury cleaves fractalkine and that CX3CR1 contributes to the development of neuropathic pain by activating p38 in spinal microglia.
2. Methods
2.1. Animals and surgery
Male adult Sprague-Dawley rats (200–260g) were used under Harvard Medical School Animal Care institutional guidelines. The animal room was artificially illuminated from 7:00 am to 7:00 pm. The rats were anesthetized with sodium pentobarbital (40–50 mg/kg, i.p). To produce spinal nerve ligation (SNL), the L5 transverse process was removed to expose the L4, L5 spinal nerves. The L5 spinal nerve was then isolated and tightly ligated with 6-0 silk thread (Kim and Chung, 1992).
2.2. Reagents and intrathecal infusion
Fractalkine was purchased from R & D Systems (Minneapolis) and infused intrathecally via an osmotic pump. Laminectomy was performed under microscope at the spinal level 2 cm below the lumbar enlargement (Ji et al., 2002a). A polyethylene (PE5) catheter was implanted into the intrathecal space of the spinal cord (L4–L5 spinal level). The catheter was connected to an osmotic pump (Alzet) for spinal infusion of fractalkine (3 ng/μl/h for 48 h). The dose was chosen according to a previous study (Milligan et al., 2004).
The neutralizing antibody against rat CX3CR1 was purchased from Torrey Pines Biolabs (1 mg/ml, Houston) and the control rabbit IgG from Sigma. For a single intrathecal infusion, a spinal cord puncture was made with 27G needle between L5 and L6 level to deliver the drug and vehicle to the cerebral spinal fluid (CSF) space around lumbosacral spinal cord (Zhuang et al., 2005; 2006). Animals were briefly anesthetized with sevofluorane before the puncture and 20 μl of liquid (antiserum or control serum) was injected with a microsyringe. Immediately after the needle entry into subarachnoid space (change in resistance), a brisk tail-flick was observed.
2.3. Western blotting
Animals were rapidly sacrificed on post-SNL day 1, 3, and 10 in an isoflurane chamber (3–4 rats per group). The L5 dorsal horns were rapidly removed and frozen on dry ice (Zhuang et al., 2005). The dorsal horn tissues were homogenized with a hand-held pestle in a SDS sample buffer (10 μl per mg tissue) containing a cocktail of proteinase inhibitors and phosphatase inhibitors (Sigma, Zhuang et al., 2005). Protein samples (30 μg) were separated on SDS-PAGE gel (4–15% gradient mini-gel, Bio-Rad) and transferred to PVDF filters (Millipore). The blots were blocked with 5% milk in PBS with 0.1% Tween-20 for 1 h at room temperature (RT) and incubated over night at 4°C with polyclonal anti-CX3CR1 antibody (Torrey Pines Biolabs, anti-rabbit, 1:1000, in 5% BSA) or fractalkine antibody (Torrey Pines Biolabs, anti-rabbit, 1:1000). The blots were then incubated for 1 h at RT with HRP-conjugated secondary antibody (Amersham, 1:5000), developed in ECL solution (NEN) for 1 min, and exposed onto hyperfilms (Amersham) for 1–30 min. Some blots were further stripped in a buffer (67.5 mM Tris, pH 6.8, 2% SDS, 0.7% β-mercaptoethanol) for 30 min at 50°C and re-probed with ERK2 antibody (1:3000, Cell Signaling) as loading control, since ERK2 levels do not change after tissue and nerve injury (Ji et al., 2002a; Zhuang et al., 2005). Specific bands were evaluated by apparent molecular size and positive control. They were also tested in previous studies (Chapman et al., 2000; Meucci et al., 2000).
2.4. Immunohistochemistry
After defined survival times (1, 2, 3, and 10 days after SNL, or 2 days after CX3CR1 antibody or fractalkine infusion), animals were terminally anesthetized with isoflurane and perfused through the ascending aorta with saline followed by 4% paraformaldehyde with 1.5% picric acid in 0.16 M PB (pH 7.2–7.4, 4°C; Ji et al., 1994). After the perfusion fixation, the L5 spinal cord segments were removed and postfixed in the same fixative overnight, then replaced with 15% sucrose overnight. Transverse spinal sections (free-floating, 30 μm) were cut in a cryostat and processed for immunofluorescence (Jin et al., 2003). All the sections were blocked with 2% goat serum in 0.3% Triton for 1 h at RT and incubated over night at 4°C with anti-CX3CR1 antibody (anti-rabbit, 1:1000; Torrey Pines Biolabs) or p-p38 antibody (anti-rabbit, 1:500, Cell Signaling). The sections were then incubated for 1 h at RT with Cy3-conjugated secondary antibody (1:300, Jackson immunolab). For double immunofluorescence, spinal sections were incubated with a mixture of polyclonal CX3CR1 antibody and monoclonal GFAP antibody (1:5000, Chemicon) or OX-42 antibody (Serotec, 1:5000) over night at 4°C, followed by a mixture of FITC- and CY3-congugated secondary antibodies for 1 h at RT. The stained sections were examined with a Nikon fluorescence microscope, and images were captured with a CCD Spot camera. The specificity of the staining or antibodies was tested in previous studies (Jin et al., 2003; Verge et al., 2004; Lindia et al., 2005). Those cells with distinct contrast to the background were scored as positive and counted in a blinded-manner. The number of p-p38-immunoreactive (IR) cell profiles was quantified from six non-adjacent spinal sections (30 μm) randomly selected from the L5 spinal segments. The region of the spinal cord sampled was the medial two thirds of the dorsal horn (laminae I–III), which was captured under 20x object in a square box (450 x 338 μm, Kawasaki et al., 2004).
2.5. Behavioral analysis
Animals were habituated to the testing environment daily for at least two days before baseline testing. The room temperature and humidity remained stable for all experiments. For testing mechanical sensitivity, animals were put in a plastic box (11 x 13 x 24 cm) on an elevated metal mesh floor and allowed 30 min for habituation. Mechanical paw withdrawal thresholds (PWT) were determined using the methods described by Chaplan et al. (1994). The hindpaw was pressed with one of series von Frey hairs with logarithmically incremental stiffness (0.6, 1, 1.4, 2, 4, 6, 8, 10, 15, and 26 gram, Stoelting; see Zhuang et al., 2006), presented perpendicular to the plantar surface for 4–5 seconds for each hair. The 50% withdrawal threshold was determined using the up-down method of Dixon (Dixon, 1980). We only tested the left hindpaws, which are ipsilateral to nerve injury.
2.6. Spinal cord slice preparation and patch clamp recordings
A portion of the lumbar spinal cord (L4–L5) was removed from adult rats (200 g) under urethane anesthesia (1.5 – 2.0 g/kg, i.p.), and kept in pre-oxygenated ice-cold Krebs solution. Spinal segments were placed in a shallow groove formed in an agar block and glued to the bottom of the microslicer stage with cyanoacrylate adhesive. Transverse slices (600 μm) were cut on a vibrating microslicer. The slices were perfused with Kreb’s solution saturated with 95% O2 and 5% CO2 at 36°C for 3 h before drug stimulation (Kawasaki et al., 2004; 2006).
The whole cell patch-clamp recordings were made from lamina II neurons in voltage clamp mode (Baba et al., 2003; Kohno et al., 2005). Under a dissecting microscope with transmitted illumination, the substantia gelatinosa (SG, lamina II) is clearly visible as a relatively translucent band across the dorsal horn. Patch pipettes were fabricated from thin-walled, borosilicate, glass-capillary tubing (1.5 mm o.d., World Precision Instruments). After establishing the whole-cell configuration, voltage clamped neurons were held at −70 mV for recording spontaneous excitatory postsynaptic current (sEPSC). The resistance of a typical patch pipette is 5–10 Mj, when filled with the internal solution. The pipette solution contains (in mM): K-gluconate 135, KCl 5, CaCl2 0.5, MgCl2 2, EGTA 5, HEPES 5, TEA 5, and ATP-Mg salt 5. Membrane currents were amplified with an Axopatch 200A amplifier (Axon Instruments) in voltage-clamp mode. Signals were filtered at 2 kHz and digitized at 5 kHz. Data were stored with a personal computer using pCLAMP 6 software and analyzed with Axograph 4.0 (Axon Instruments).
2.8. Statistics
All the data are expressed as Mean ± SEM. Differences between groups were compared by Student’s t-test or ANOVA followed by Fisher’s PLSD post hoc analysis (SPSS 10.0 for Window). The criterion for statistical significance was P<0.05.
3. Results
3.1. Spinal nerve ligation (SNL) increases CX3CR1 expression in spinal microglia
Western blot analysis revealed a specific band ( ≈40 kDa) in the spinal cord dorsal horn using a CX3CR1 antibody (Fig 1a). This band was also reported in the brain using the same antibody (Meucci et al., 2000). The intensity of this CX3CR1 band significantly increased after ligation of the L5 spinal nerve (F(3,8)=8.807, P=0.0065, ANOVA). This increase began on day 1, peaked on day 3, and maintained on day 10 after SNL (Fig 1b). Immunohistochemistry confirmed an upregulation of CX3CR1 after nerve injury. Many more CX3CR1-immunoreactive (IR) cells were found in the ipsilateral spinal cord after nerve injury. Although most positive cells were found in the superficial (laminae I–III) dorsal horn, many positive cells were also seen in the ventral horn and in the area around the central canal (Fig 1c). Sham surgery did not produce significant change of CX3CR1 expression in the spinal cord (data not shown). Although CX3CR1 upregulation was predominantly found in the ipsilateral spinal cord, a moderate increase in the contralateral superficial dorsal horn (lamina II) was also evident (Fig 1c).
Figure 1.
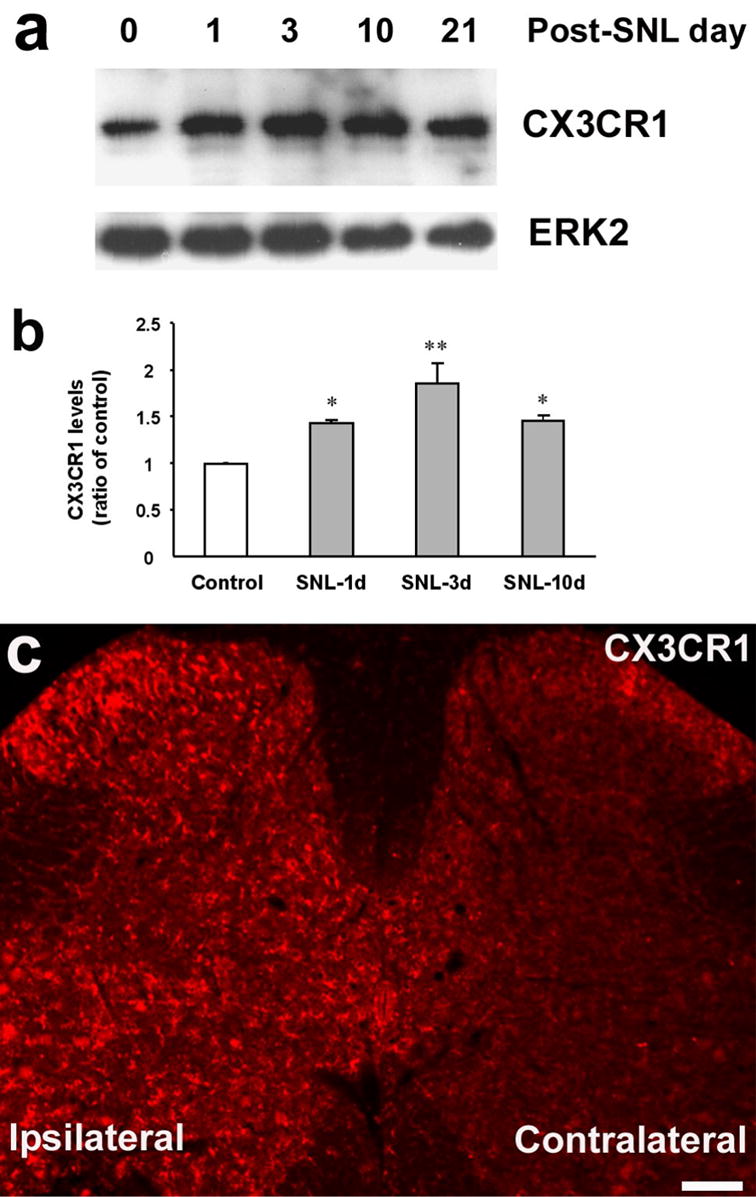
Spinal nerve ligation (SNL) induces CX3CR1 expression in the spinal cord. (a) Western blot analysis shows a sustained increase in the levels of CX3CR1 in the L5 spinal dorsal horn, compared with the control without injury. ERK2 was used as the loading control. (b) Quantification of CX3CR1 levels in the L5-spinal cord from Western blotting. CX3CR1 levels were normalized against corresponding loading control. *, P<0.05, **, P<0.01; compared with control, ANOVA followed by Fisher’s PLSD post-hoc comparison. (c) Immunofluorescence indicates that SNL increases CX3CR1 levels in the ipsilateral spinal cord (L5) 2 days after the injury. Scale, 100 μm.
Double labeling indicated that CX3CR1-IR cells were exclusively microglia, since they also co-expressed the microglial marker OX-42 on both day 2 (Fig 2a-c) and day 10 (Fig 2g). In contrast, CX3CR1 was not expressed in astrocytes, because this receptor did not colocalize with the atroglial marker GFAP (Fig 2f).
Figure 2.
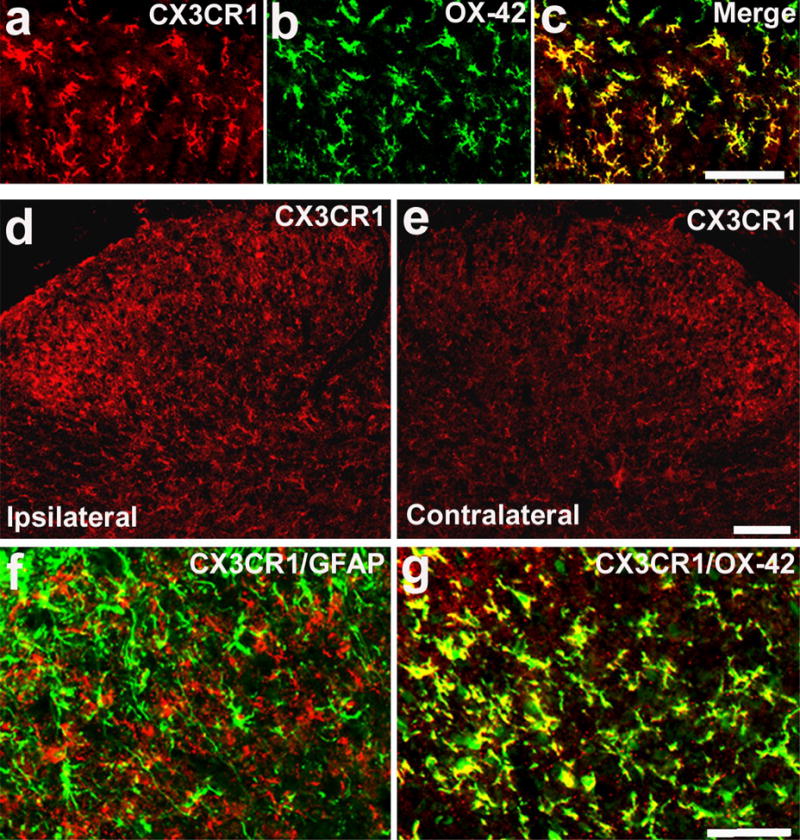
SNL induces CX3CR1 expression in microglia of the spinal cord. (a–c) Double immunofluorescence shows that CX3CR1 (a, red) is colocalized with OX-42 (b, green), a microglial marker, in the medial superficial dorsal horn (laminae II–III), 2 days after SNL. c is the merge of a and b. Yellow fluorescence indicates a double labeling. Scale, 50 μm. (d, e) Immunofluorescence indicates an increase of CX3CR1 expression in the dorsal horn on the ipsilateral (injured) side 10 days after SNL. Scale, 50 μm. (f, g) Double staining indicates that CX3CR1 (red) is not colocalized with astrocytic marker GFAP (f, green), but colocalized with OX-42 (g, green) in the medial ipsilateral dorsal horn (laminae II–IV) on post-SNL day 10. Two singly stained images were merged. Scale, 50 μm.
3.2. CX3CR1 upregulation contributes to the development of neuropathic pain
To test whether CX3CR1 upregulation has any role in pain hypersensitivity after SNL, we applied a single injection of a neutralizing antibody against CX3CR1 (10 μg, Milligan et al., 2004) into the spinal CSF space by lumbar puncture before surgery and tested mechanical allodynia on day 1 and 2 post-SNL. L5 SNL induced a rapid and marked mechanical allodynia; mechanical thresholds decreased by 80% on day 1 and 2 (Fig 3). Intrathecal injection of the vehicle (control serum) did not affect mechanical allodynia. However, CX3CR1 neutralizing antibody, injected 30 min before the nerve injury, significantly reduced mechanical allodynia on day 1 and 2 (P<0.01, t-test, Fig 3).
Figure 3.
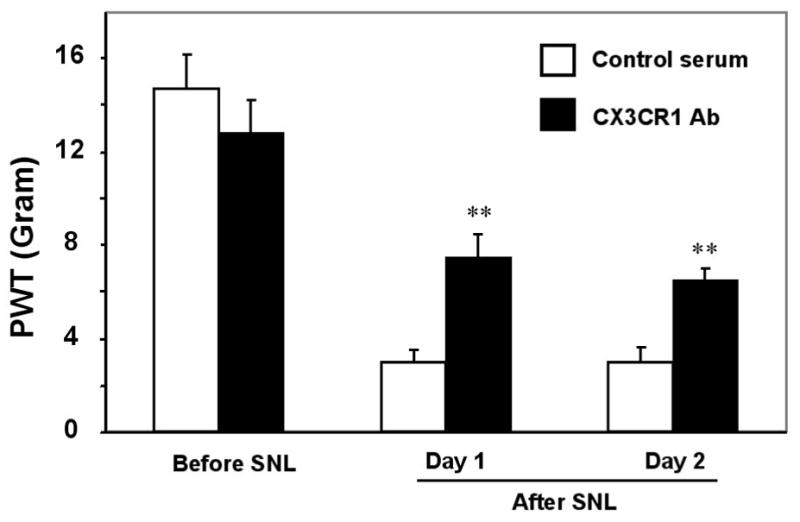
Suppression of SNL-induced neuropathic pain by a neutralizing antibody against CX3CR1. A single injection of a neutralizing CX3CR1 antibody (10 μg, 20 μl) into lumbar CSF space, 30 min before surgery, decreases SNL-induced mechanical allodynia on post-surgery day 1 and day 2. Control serum (10 μg) has no effect. **, P<0.01, compared to corresponding serum control, unpaired t-test, n=6.
3.3. CX3CR1 is required for p38 activation in the spinal cord after SNL
Although sham surgery produced a slight increase in p-p38 levels compared to naïve control, SNL induced a very dramatic increase in p-p38 levels. Numerous p-p38-IR cells were found in the spinal cord, especially in the superficial laminae (I–III) (Fig 4a, b). As we have previously shown (Jin et al., 2003), p-p38 was only expressed in OX-42-IR spinal microglia. Furthermore, SNL-induced p-p38 increase was significantly suppressed by pre-treatment of the neutralizing CX3CR1 antibody (Fig 4d, P<0.05, t-test). The degree of inhibition of p-p38 was correlated well with that of mechanical allodynia on post-SNL day 2 following the same treatment (Fig 3, 4d).
Figure 4.
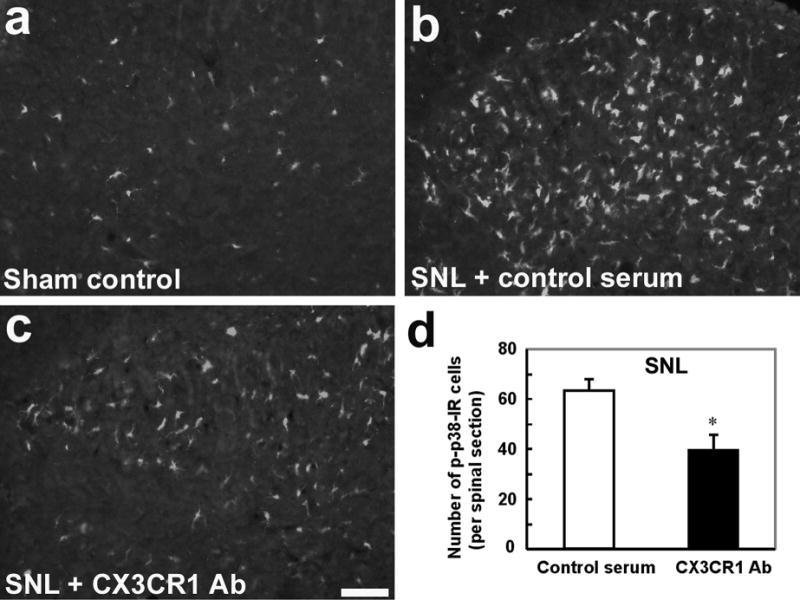
Suppression of SNL-induced p38 activation by a neutralizing antibody against CX3CR1. (a, b) Compared to sham control (a), SNL induces robust p38 activation in the dorsal horn on day 2, as indicated by an increase in the number of p-p38-IR cells (b). (c) A single intrathecal injection of a neutralizing CX3CR1 antibody (10 μg, 20 μl), 30 min before surgery, inhibits SNL-induced p38 activation 2 days after nerve injury. Images in a-c were taken from the medial superficial dorsal horn (laminae I–III). Scale, 50 μm. (d) Number of p-p38-IR cells in the medial superficial dorsal horn (laminae I–III) of SNL rats receiving CX3CR1 antibody or control serum. Animals were perfused for p-p38 staining 2 days after SNL. *, P<0.05, compared to corresponding control serum, n=4, unpaired t-test.
3.4. Spinal infusion of fractalkine activates p38 in microglia and induces mechanical allodynia
To determine whether the CX3CR1 ligand fractalkine can activate p38 in the spinal cord, we continuously infused fractalkine intrathecally for 2 days via an osmotic pump, at the flow rate of 1 μl/h. This infusion (72 ng fractalkine per day) induced a 4-fold increase in spinal p-p38 levels (Fig 5a-d, P<0.01, compared with saline control, t-test). p38 was still activated in spinal microglia after fractalkine infusion (Fig 5c). In parallel, this infusion also induced marked mechanical allodynia on both day 1 and 2 (Fig 6).
Figure 5.
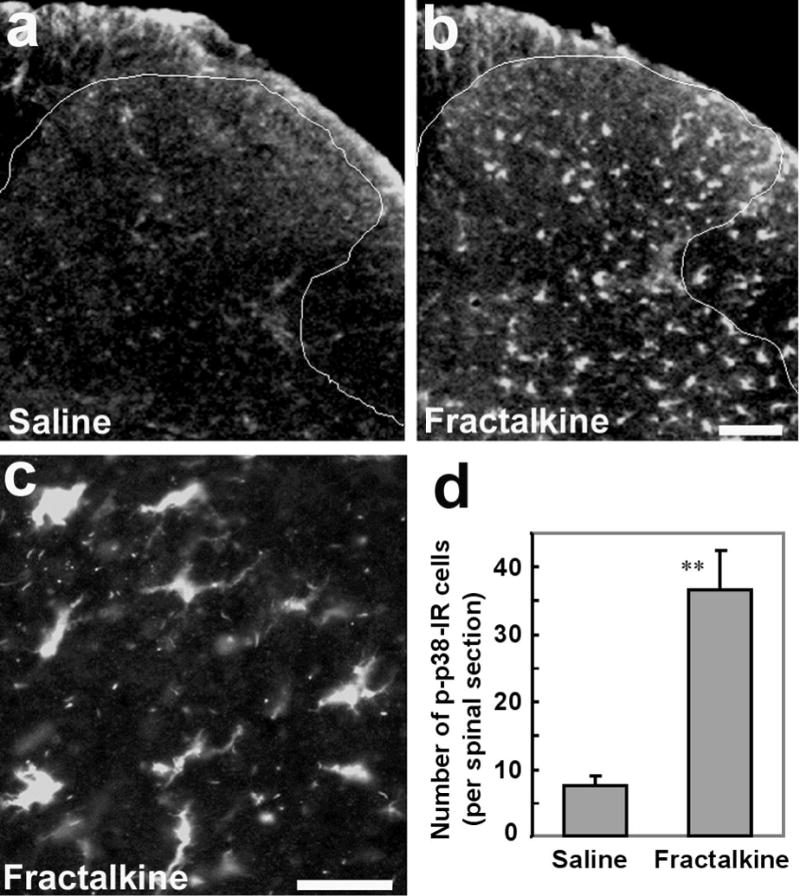
Activation of p38 in the spinal cord following intrathecal infusion of fractalkine. (a, b) Immunofluorescence indicates an increase in p-p38-IR cells in the dorsal horn 2 days after fractalkine infusion via an osmotic pump (3 ng/h). Scale, 50 μm. (c) High magnification image shows p-p38-IR cells in the dorsal horn (lamina III), with morphology of microglia. Scale, 25 μm. (d) Number of p-p38-IR cells in the medial laminae I–III of each spinal section following infusion of saline (1μl/h for 48 h) and fractalkine (3ng/μl/h for 48 h). **, P<0.01, compared to saline control, n=3, unpaired t-test.
Figure 6.
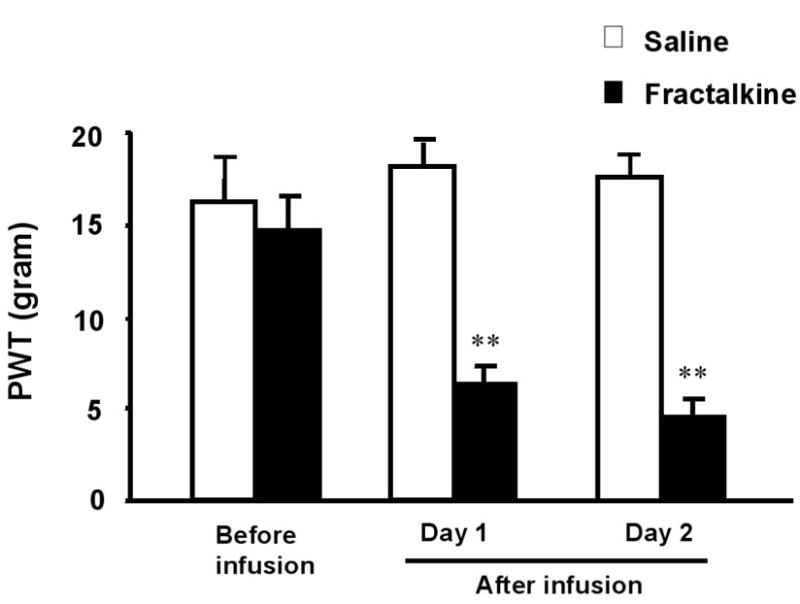
Spinal infusion of fractalkines induces mechanical allodynia. Fractalkine or saline was infused for 2 days as in Fig 5. **, P<0.01, compared to saline control, n=5, unpaired t-test.
3.5. SNL induces cleavage of fractalkine in the injured dorsal root ganglion
It is believed that membrane-bound fractalkine must be cleaved before its release (Chapman et al., 2000; Hundhausen et al., 2003). To determine whether nerve injury induces a cleavage of this chemokine, we examined the expression of membrane-bound and soluble (secreted) forms of fractalkine using Western analysis. In the non-injured conditions, fractalkine in the DRG had two forms, a large form (≈100 kDa) and a small form (≈80 kDa), corresponding to membrane-bound and soluble form, respectively, as shown in other tissues (Chapman et al., 2000; Hundhausen et al., 2003). One day after nerve injury, the large form was almost lost in the injured L5 DRG (Fig 7a). The average intensity of the Western band one day after SNL was only 24 ± 3% of that of the sham control DRG (P<0.01, t-test, n=3), indicating a marked cleavage of fractalkine by SNL (Fig 7b). There appeared to an increase of the soluble fractalkine (band 2, Fig 7a) in the DRG after nerve injury. This 80 kDa band was weaker, because it could be released to the CSF and barely remained in the DRG.
Figure 7.
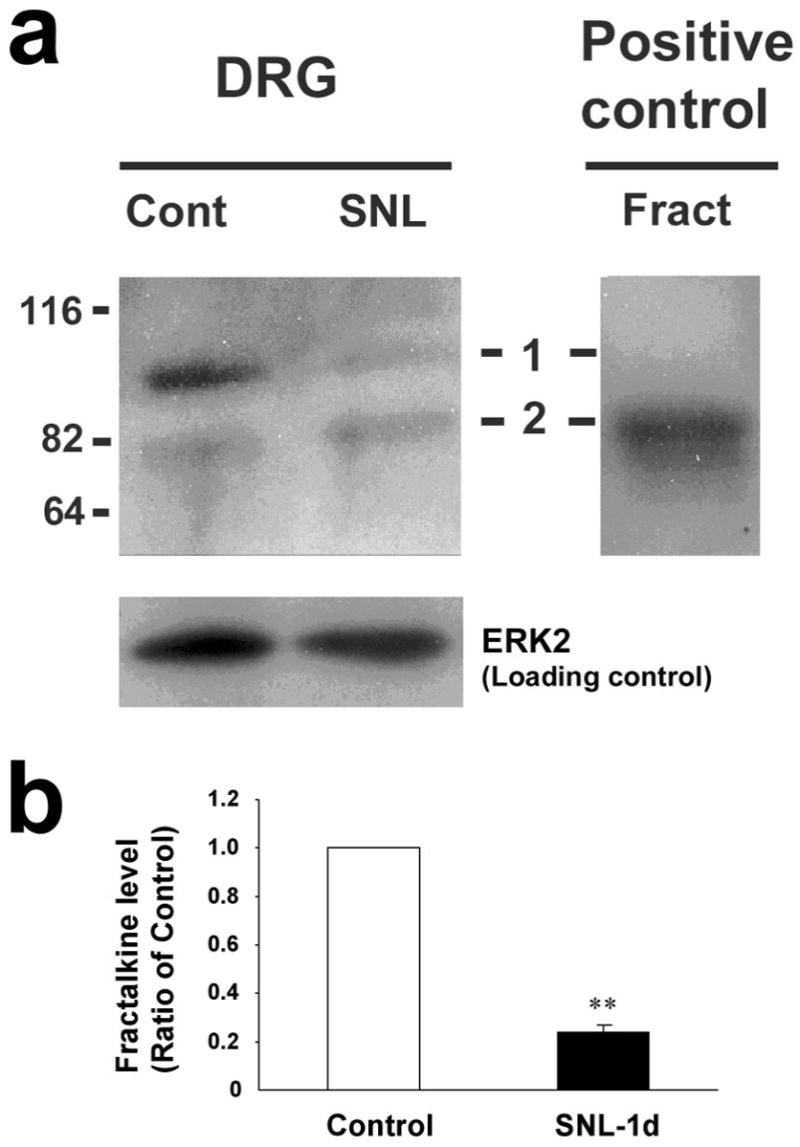
SNL produces cleavage of fractalkine in the injured dorsal root ganglion. (a) Western blot with a fractalkine antibody reveals two bands of fractalkine. The top band (band 1, ≈100 kDa, a membrane-bound form) almost disappears one day after nerve ligation. Band 2 (≈ 80 kDa) is equivalent to the positive control (20 ng of recombinant full-length fractalkine). Molecular weight is indicated on the left edge. (b) Quantification of the large fractalkine band (100 kDa) in the L5 DRG of SNL-injured rats after 1 day and time-matched sham control rats. Fractalkine levels were normalized against corresponding loading control.
3.6. Fractalkine does not acutely change excitatory synaptic transmission in dorsal horn neurons in isolated spinal slices
To determine whether fractalkine has any acute effect on synaptic transmission in the spinal cord, we conducted patch clamp recording in lamina II neurons of the isolated spinal cord slices and examined whether fractalkine could affect spontaneous excitatory synaptic currents (sEPSC). Bath application of fractalkine to spinal slices for 5 min had no effect on sEPSCs (Fig 8a–c). Neither frequency nor amplitude of sEPSC was altered during and after fractalkine perfusion (Fig 8a–e) at concentrations (2 and 20 ng/ml) that are effective to activate ERK/MAPK in spinal cells (data not shown).
Figure 8.
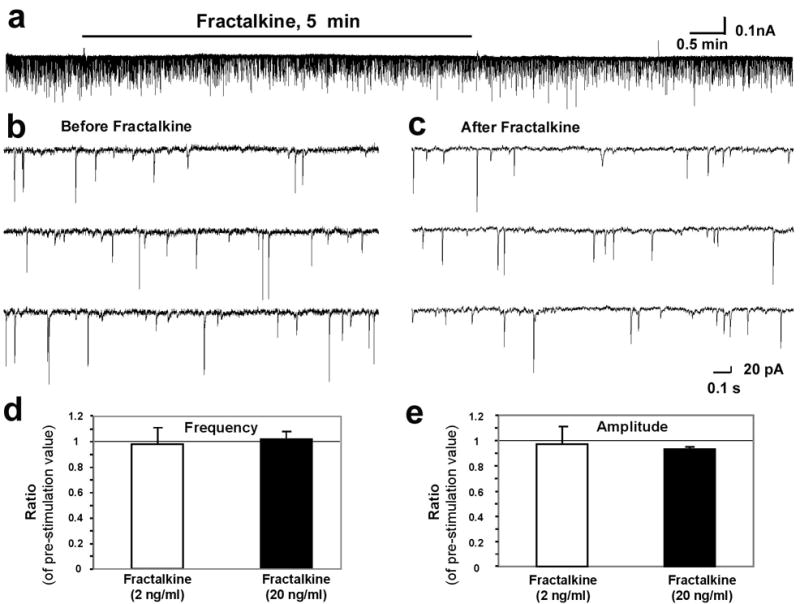
Fractalkine does not produce acute effect on synaptic transmission in the spinal cord. (a) Patch clamp recording in lamina II neurons of the isolated spinal cord slices shows spontaneous excitatory synaptic current (sEPSC) before, during, and after bath application of fractalkine (20 ng/ml, 5 min). (b, c) High magnification shows sEPSC before (a) and after (b) fractalkine (20 ng/ml) application. (d, e) Effects of fractalkine on the frequency (d) and amplitude (e) of mEPSC. Data are presented as the ratio of pre-treatment value. P>0.05, paired t-test, n=3.
4. Discussion
There are several novel findings of this study. First, we show that fractalkine/CX3CR1 is essential for p38 activation after spinal nerve ligation (SNL). Second, we demonstrate that fractalkine in the injured DRG is cleaved after SNL. Third, we show that fractalkine does not produce rapid facilitation of excitatory synaptic transmission in spinal lamina II neurons. We also show in the SNL model that CX3CR1 is induced in spinal microlgia and that this receptor is required for neuropathic pain development. Although CX3CR1 upregulation in spinal microglia has been reported by Watkins’ group in the chronic constriction injury (CCI) model and sciatic inflammatory neuropathy model (SIN) model of neuropathic pain (Verge et al., 2005), it is important to characterize this upregulation in the SNL model, which has provided a base for the following studies.
4.1. p38 activation in spinal microglia and neuropathic pain
Accumulating evidence suggests that microglial cells in the spinal cord play a crucial role in facilitating pain states. However, studies on microglial regulation of pain are hampered by the lack of functional marker for microglial activation. In last several years, different laboratories have shown that p38 is activated in spinal microglia under different pain conditions. Importantly, p38 activation also contributes to the development of pain hypersensitivity (Ji et al., 2002b; Kim et al., 2002; Jin et al., 2003; Svensson et al., 2003, 2005, Tsuda et al., 2004; Cui et al., 2006; Piao et al., 2006). Therefore, p38 activation (phosphorylation) could serve as a marker for microglial activation in the spinal cord under different pain conditions. This marker is also an essential signaling molecule in microglia for pain regulation (Ji and Strichartz, 2004). Minocycline, a microglial inhibitor, is believed to suppress inflammatory and neuropathic pain by inhibiting p38 activation (Hua et al., 2005; Hains and Waxman, 2006). Several studies began to explore the upstream mechanisms causing p38 activation in spinal microglia. For example, p38 was activated in spinal microglia by spinal substance P (Svensson et al., 2003), TNF-αo (Svensson et al., 2005), and IL-1β (Sung et al., 2005). Compared with other pain conditions, nerve injury induces much stronger p38 activation in spinal microglia. We propose that fractalkine is likely to convey signal from neurons to glia by activating p38 in spinal microglia.
It remains to be investigated how p38 activation in spinal microglia contributes to pain sensitization. Upon activation, microglia produce proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 (Hanisch 2002; Koistinaho and Koistinaho, 2002). p38 activation is implicated in the synthesis of theses cytokines (reviewed Ji and Strichartz, 2004). SNL-induced spinal synthesis of IL-1β is suppressed by p38 inhibitor (Zhuang and Ji, unpublished observation). IL-1β may facilitate pain by enhancing excitatory synaptic transmission in dorsal horn neurons (Kawasaki and Ji, 2006) and inducing the expression of COX-2 in the spinal cord (Samad et al., 2001). p38 may also regulate the expression of other genes (e.g., P2X4, CX3CR1) that are upregulated in spinal microglia after nerve injury. Additionally, p38 can acutely induce the synthesis of PGE2 via activation of phospholipase A2 (Yaksh et al., 2006).
4.2. CX3CR1 upregulation in spinal microglia and neuropathic pain
We have shown that nerve injury upregulates CX3CR1 in spinal microglia using the SNL model. Our results are consistent with previous studies in the CCI, SIN, and SNL model (Verge et al., 2004; Lindia et al., 2005). However, our study has included additional characterization of CX3CR1 upregulation. First, Western blotting analysis indicated a time course of the upregulation. This upregulation was rapid and was already significant after one day. Although declined from the peak, the upregulation was still evident after 10 days. Second, we have shown that CX3CR1 is exclusively expressed in microglia at different times (2 and 10 days) of nerve injury. CX3CR1 upregulation was widespread and could be found all over the dorsal and ventral horn, but still restricted to the ipsilateral spinal cord. The same distribution pattern after SNL has also been observed for other microglial molecules, such as OX-42 and p-p38 (Jin et al., 2003). This is in part due to the fact that L5-SNL produces more severe nerve damage in the L5 segment than other neuropathic pain models with peripheral nerve injury. However, the site that shows most CX3CR1 upregulation is the superficial dorsal horn. It may not be necessary for microglia to be anatomically close to nociceptive neurons. The mediators produced by microglia could be diffused to affect nociceptive neurons.
In support of a previous study by Milligan et al. (2004), we have shown that CX3CR1 upregulation is required for neuropathic pain development in the SNL model. Since we only tested pain behavior in the first 2 days after SNL, this study focused on the induction mechanism of neuropathic pain. However, CX3CR1 neutralizing antibody was shown to reverse neuropathic pain when given several days after injury (Milligan et al., 2004). Therefore, CX3CR1 is also involved in the maintenance of neuropathic pain. However, whether CX3CR1 is still important for late maintenance (e.g., >3 weeks) of neuropathic pain remains unclear.
In particular, we have shown that CX3CR1 upregulation is essential for the activation of p38 in spinal microglia after SNL. We have also shown that intrathecal fractalkine infusion activates p38 in spinal microglia and induces mechanical allodynia. Therefore, it is likely that activation of p38 is an underlying mechanism for CX3CR1 to regulate neuropathic pain. In addition, activation of CX3CR1 receptor may also facilitate pain via inducing the release of IL-1β (Johnston et al., 2004).
4.3. Fractalkine’s expression, cleavage, and spinal action
Our Western analysis shows that fractalkine is heavily expressed in the DRG. Unlike its receptor, fractalkine is expressed in neurons including primary sensory neurons in the DRG and spinal neurons (Verge et al., 2004; Lindia et al., 2005). Fractalkine is also expressed in spinal astrocytes after nerve injury (Lindia et al., 2005).
Fractalkine has 2 forms, a membrane-bound form (large size) and a soluble, secreted form (small size). These two forms mediate cell adhesion and chemotaxis, respectively (Hundhausen et al., 2003). Chapman et al. (2000) have shown that membrane-bound fractalkine in cortical neurons is cleaved and liberated in response to glutamate-induced neuronal hyperactivity and toxicity. Consistently, out data show that fractalkine in the DRG also has two forms, a membrane-bound form and a secreted from. Importantly, we have shown for the first time that membrane-bound fractalkine is rapidly lost after nerve injury in the injured DRG, indicating a cleavage of this chemokine. The cleaved fractalkine could be rapidly released into CSF, thus, difficult to be detected in the DRG samples. Metalloproteinases, such as disintegrin-like metalloproteinase ADAM10, are implicated in the cleavage of membrane fractalkine (Chapman et al., 2000; Ludwig et al., 2002; Hundhausen et al., 2003).
Our electrophysiological data show that fractalkine does not produce acute effect on excitatory synaptic transmission in lamina II dorsal horn neurons, arguing against a direct effect of this chemokine on dorsal horn neurons. This funding supports the notion that fractalkine receptor (CX3CR1) is not expressed in neurons in the spinal cord. However, caution must be taken, since we only tested lamina II neurons. We cannot exclude the possibility that fractalkine may directly act on other dorsal horn neurons such as lamina V neurons that receive input from A-beta fiber responsible for signaling light touch. Although fractalkine may not produce direct activation in dorsal horn neurons, it might produce delayed activation of dorsal horn neurons via glial-neural interaction, which is supported by a recent electrophysiological study (Owolabi and Saab, 2006). It is also consistent with the finding that intrathecal fractalkine induces delayed pain behavior that manifests after 40 min (Milligan et al., 2004). Fractalkine was shown to induce IL-1β release from isolated spinal cord (Johnston et al., 2004). It is likely that fractalkine acts on CX3CR1 receptor to release IL-1β from spinal microglia, which then produces delayed activation/sensitization of dorsal horn neurons, leading to pain hypersensitivity. Interestingly, a recent study shows that IL-1β release in the spinal cord requires p38 activation (Clark et al., 2006).
4.4. Concluding remarks
It is becoming generally accepted that glial cells in the spinal cord play important roles in the generation of pathological pain states, such as neuropathic pain. Glial cells must interact with neurons in order to regulate pain sensitivity (Ji and Wen, 2006). Fractalkine is a unique chemokine that plays an important role in mediating neural-glial interaction. We find that fractalkine produces no acute facilitation of excitatory synaptic transmission of dorsal horn neurons. We also find that SNL induces a marked cleavage of fractalkine in the DRG. Upon release, this chemokine can bind to its upregulated CX1CR1 receptor in spinal microglia, leading to the activation of p38 MAPK. We suggest that the CX3CR1/p38 cascade contributes to the development of neuropathic pain. Inhibitors that can block this pathway in spinal microglia may be useful for the management of neuropathic pain.
Acknowledgments
The work was supported by NIH RO1DE 17794 to RRJ.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba H, Ji RR, Kohno T, Moore KA, Ataka T, Wakai A, Okamoto M, Woolf CJ. Removal of GABAergic inhibition facilitates polysynaptic A fiber-mediated excitatory transmission to the superficial spinal dorsal horn. Mol Cell Neurosci. 2003;24:818–830. doi: 10.1016/s1044-7431(03)00236-7. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci. 2000;20:RC87. doi: 10.1523/JNEUROSCI.20-15-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, D’Aquisto F, Gentry C, Marchand F, McMahon SB, Malcangio M. Rapid co-release of interleukin 1beta and caspase 1 in spinal cord inflammation. J Neurochem. 2006 Aug 29; doi: 10.1111/j.1471-4159.2006.04126.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Cui Y, Chen Y, Zhi JL, Guo RX, Feng JQ, Chen PX. Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res. 2006;1069:235–243. doi: 10.1016/j.brainres.2005.11.066. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist. 2004;10:40–52. doi: 10.1177/1073858403259950. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Hua XY, Svensson CI, Matsui T, Fitzsimmons B, Yaksh TL, Webb M. Intrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eur J Neurosci. 2005;22:2431–2440. doi: 10.1111/j.1460-9568.2005.04451.x. [DOI] [PubMed] [Google Scholar]
- Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102:1186–1195. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002a;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002b;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Ji RR, Wen YR. Neural-glial interaction in the spinal cord for the development and maintenance of nerve injury-induced neuropathic pain. Drug Dev Res. 2006;67:331–338. [Google Scholar]
- Ji RR, Zhang X, Wiesenfeld-Hallin Z, Hokfelt T. Expression of neuropeptide Y and neuropeptide Y (Y1) receptor mRNA in rat spinal cord and dorsal root ganglia following peripheral tissue inflammation. J Neurosci. 1994;14:6423–6434. doi: 10.1523/JNEUROSCI.14-11-06423.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SX, Woolf CJ, Ji RR. Activation of p38 MAP kinase in the dorsal root ganglion and spinal cord after spinal nerve ligation. Soc Neurosci Abstr. 2002;28:655.13. [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, Langer S, Martin D, Green P, Fleshner M, Leinwand L, Maier SF, Watkins LR. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci. 2004;24:7353–7365. doi: 10.1523/JNEUROSCI.1850-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Ji RR. Effects of proinflammatory cytokines on synaptic transmission and phosphorylation of the transcription factor CREB in dorsal horn neurons in spinal cord slices. Soc Neurosci Abstr. 2006:643.15. [Google Scholar]
- Kawasaki Y, Kohno T, Ji RR. Different effects of opioid and cannabinoid on C-fiber-induced ERK activation in dorsal Horn neurons in normal and spinal nerve-ligated Rats. J Pharmacol Exp Ther. 2006;316:601–607. doi: 10.1124/jpet.105.093583. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der Meer C, Befort K, Woolf CJ, Ji RR. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24:8310–8321. doi: 10.1523/JNEUROSCI.2396-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Kim SY, Bae JC, Kim JY, Lee HL, Lee KM, Kim DS, Cho HJ. Activation of p38 MAP kinase in the rat dorsal root ganglia and spinal cord following peripheral inflammation and nerve injury. Neuroreport. 2002;13:2483–2486. doi: 10.1097/00001756-200212200-00021. [DOI] [PubMed] [Google Scholar]
- Kohno T, Ji RR, Ito N, Allchorne AJ, Befort K, Karchewski LA, Woolf CJ. Peripheral axonal injury results in reduced mu opioid receptor pre- and post-synaptic action in the spinal cord. Pain. 2005;117:77–87. doi: 10.1016/j.pain.2005.05.035. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J. Role of p38 and p44/42 mitogen-activated protein kinases in microglia. Glia. 2002;40:175–183. doi: 10.1002/glia.10151. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Lindia JA, McGowan E, Jochnowitz N, Abbadie C. Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J Pain. 2005;6:434–438. doi: 10.1016/j.jpain.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Miller RJ. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci U S A. 2000;97:8075–8080. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Twining C, Chacur M, Biedenkapp J, O’Connor K, Poole S, Tracey K, Martin D, Maier SF, Watkins LR. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci. 2003;23:1026–1040. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, O’Connor KA, Verge GM, Chapman G, Green P, Foster AC, Naeve GS, Maier SF, Watkins LR. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci. 2004;20:2294–2302. doi: 10.1111/j.1460-9568.2004.03709.x. [DOI] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24:10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owolabi SA, Saab CY. Fractalkine and minocycline alter neuronal activity in the spinal cord dorsal horn. FEBS Lett. 2006;580:4306–4310. doi: 10.1016/j.febslet.2006.06.087. [DOI] [PubMed] [Google Scholar]
- Piao ZG, Cho IH, Park CK, Hong JP, Choi SY, Lee SJ, Lee S, Park K, Kim JS, Oh SB. Activation of glia and microglial p38 MAPK in medullary dorsal horn contributes to tactile hypersensitivity following trigeminal sensory nerve injury. Pain. 2006;121:219–231. doi: 10.1016/j.pain.2005.12.023. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Schafers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003;23:2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CS, Wen ZH, Chang WK, Chan KH, Ho ST, Tsai SK, Chang YC, Wong CS. Inhibition of p38 mitogen-activated protein kinase attenuates interleukin-1beta-induced thermal hyperalgesia and inducible nitric oxide synthase expression in the spinal cord. J Neurochem. 2005;94:742–752. doi: 10.1111/j.1471-4159.2005.03226.x. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, Catalano R, Feng Y, Protter AA, Scott B, Yaksh TL. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. 2003;86:1534–1544. doi: 10.1046/j.1471-4159.2003.01969.x. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Schafers M, Jones TL, Powell H, Sorkin LS. Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett. 2005;379:209–213. doi: 10.1016/j.neulet.2004.12.064. [DOI] [PubMed] [Google Scholar]
- Sweitzer SM, Peters MC, Ma JY, Kerr I, Mangadu R, Chakravarty S, Dugar S, Medicherla S, Protter AA, Yeomans DC. Peripheral and central p38 MAPK mediates capsaicin-induced hyperalgesia. Pain. 2004;111:278–285. doi: 10.1016/j.pain.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45:89–95. doi: 10.1002/glia.10308. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci. 2004;20:1150–1160. doi: 10.1111/j.1460-9568.2004.03593.x. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci 2001. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Kokotos G, Svensson CI, Stephens D, Kokotos CG, Fitzsimmons B, Hadjipavlou-Litina D, Hua XY, Dennis EA. Systemic and intrathecal effects of a novel series of phospholipase A2 inhibitors on hyperalgesia and spinal prostaglandin E2 release. J Pharmacol Exp Ther. 2006;316:466–475. doi: 10.1124/jpet.105.091686. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide JNK inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]