

Abstract
For more than 20 years, the gut has been hypothesized to be the “motor” of multiple organ dysfunction syndrome (MODS). As critical care research has evolved, there have been multiple mechanisms by which the gastrointestinal tract has been proposed to drive systemic inflammation. Many of these disparate mechanisms have proved to be important in the origin and propagation of critical illness. However, this has led to an unusual situation where investigators describing the gut as a “motor” revving the systemic inflammatory response syndrome (SIRS) are frequently describing wholly different processes to support their claim (i.e. increased apoptosis, altered tight junctions, translocation, cytokine production, crosstalk with commensal bacteria, etc). The purpose of this review is to present a unifying theory as to how the gut drives critical illness. Although the gastrointestinal tract is frequently described simply as “the gut,” it is actually made up of a) an epithelium, b) a diverse and robust immune arm, which contains the majority of immune cells in the body, and c) the commensal bacteria, which contain more cells than are present in the entire host organism. We propose that the intestinal epithelium, the intestinal immune system and the intestine’s endogenous bacteria all play vital roles driving MODS, and the complex crosstalk between these three interrelated portions of the gastrointestinal tract are cumulatively what makes the gut a “motor” of critical illness.
INTRODUCTION
The gut was first described as the “motor” of multiple organ failure by Meakins and Marshall at a 1985 panel discussion of the Surgical Infection Society (1). Their hypothesis was based upon evidence that commensal flora was overgrown and increasingly resistant in the ICU setting, especially after antibiotic usage. Worsening gut barrier function was proposed to allow translocation of gut bacteria into the portal and systemic circulation where host defenses were already compromised, leading to propagation of disease. It is interesting to note that the gut was hypothesized to play a driving role in both sepsis and what was then termed “nonbacteremic clinical sepsis” five years before a consensus conference fully defined the terms sepsis, SIRS and MODS (2).
In the ensuing years, numerous studies have further defined and redefined how the gut plays a role in the origin and propagation of critical illness. For example, Moore and colleagues demonstrated that shock induces gut hypoperfusion which, in turn, leads to intestinal reperfusion with production of proinflammatory mediators that can amplify the SIRS response (3). Fink and colleagues have demonstrated that epithelial tight junctions are compromised in critical illness, leading to increased permeability and persistent activation of systemic inflammation (4–6). Alverdy and colleagues have demonstrated that interactions between host and bacterial pathogens lead to gut-derived sepsis, at least partially independent of the proinflammatory nature of bacteremia or of intestinal permeability changes (7). Teixeira and colleagues have demonstrated that germ free mice that entirely lack commensal bacteria have improved survival following intestinal ischemia/reperfusion (I/R) compared to conventional animals (8). Deitch and colleagues have demonstrated that toxic gut-derived substances enter the mesenteric lymph to cause lung damage. Ligating the lymph duct following hemorrhagic shock in a variety of species prevents distant injury from occurring, in what is now called the gut-lymph hypothesis (9). Our group has demonstrated that gut epithelial apoptosis is elevated following sepsis. Prevention of sepsis-induced intestinal apoptosis by overexpression of the anti-apoptotic protein Bcl-2 improves survival in multiple animal models of sepsis (10;11).
The commonality in all of these studies is that some element of gut pathophysiology contributes to critical illness. However, it is striking that the mechanisms underlying how the gut acts as “a motor” in the origin or maintenance of critical illness vary widely between them. This has led us to conclude that while there are multiple correct ways of understanding the role the intestine plays in propagating both SIRS and sepsis in isolation, each of these is incomplete. We believe that there is currently no unifying hypothesis that encompasses the diverse ways in which the gut influences outcome in critical illness. The purpose of this review is to suggest a new paradigm, by viewing the gut as a three-way partnership between its epithelium, immune tissue and commensal bacteria (Fig. 1). In this partnership, each element modifies the others via crosstalk, leading to a state where all components of the gut interact, and the integrated result of this crosstalk acts as a major determinant for survival in MODS. It should be noted that we recognize that crosstalk occurs between the gut and extra-intestinal tissue. While of clear physiologic significance, intestinal/extra-intestinal crosstalk is beyond the scope of this review.
Figure 1.
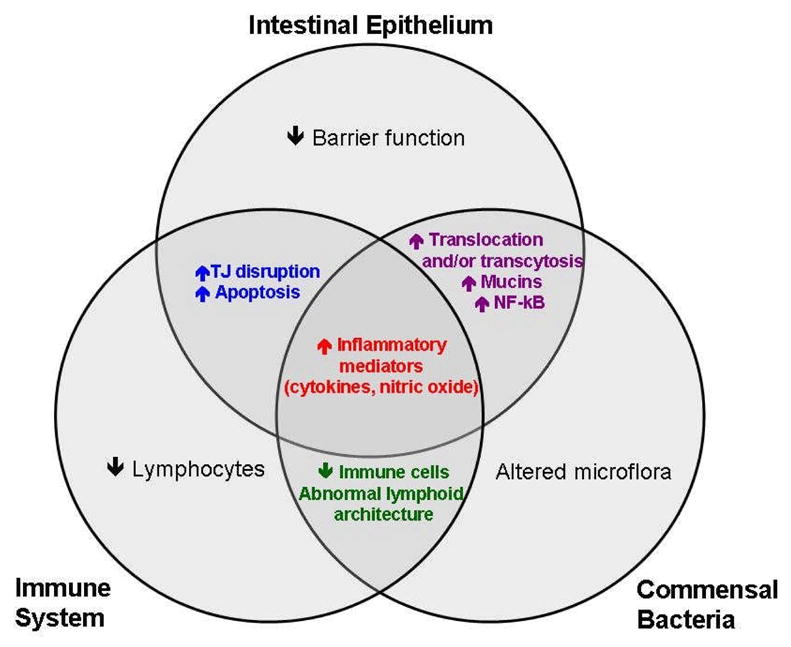
Crosstalk between the intestinal epithelium, immune system, and commensal bacteria is central to initiating the systemic inflammatory response. We hypothesize that the cumulative and integrated result of these multiple interactions is what makes the gut the “motor” of critical illness.
WHAT IS THE GUT?
In broad terms, the gut is comprised of three entities: the epithelium, the mucosal immune system, and the commensal flora (Fig. 2). These are each innervated by the enteric nervous system (described below) and each of these components interact in a complex ecosystem that is under constant surveillance and is tightly regulated (12).
Figure 2.
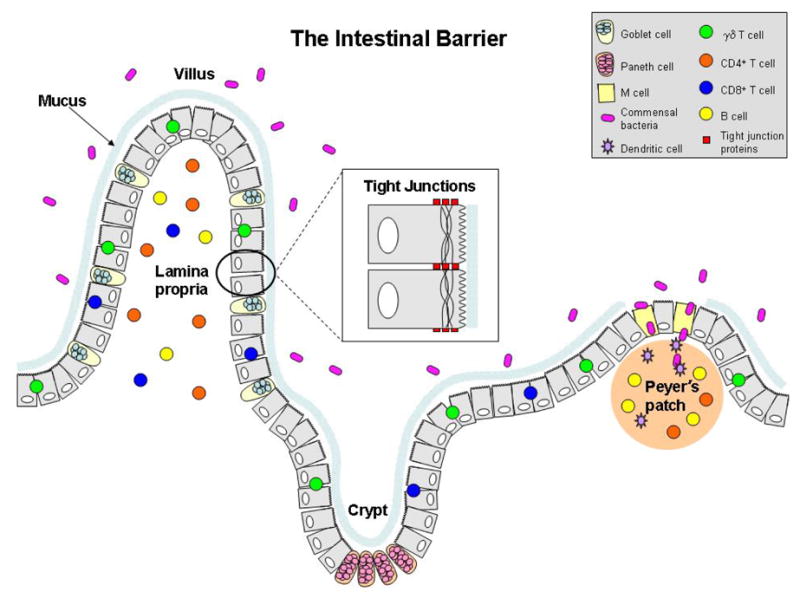
The intestinal barrier. The epithelium, immune cells, and commensal bacteria work together to prevent invasion of potentially harmful substances.
The Epithelium
The epithelium performs the digestive functions of the gastrointestinal tract. However, while the gut absorbs food necessary for host well being, providing the gastrointestinal tract with food is simultaneously critical for intestinal integrity. This is highlighted by the observation that provision of enteral nutrition and specific nutrients such as glutamine in critical illness attenuates increased permeability seen in sepsis and SIRS (13). Additionally the epithelium functions as a barrier to protect against invasion of potentially harmful antigens. Enterocytes are also capable of producing cytokines following an inflammatory stimulus in the absence of bacteremia or translocation (14). However, there are no detectable differences in pro-inflammatory cytokine levels in systemic and portal blood after cecal ligation and puncture (CLP) in rats suggesting the gut epithelium is not the major systemic source of pro-inflammatory cytokines during sepsis (15).
The mucosal surface of the gut represents the largest body surface in contact with the outside world (approximately 300 m2, roughly the area of a tennis court). The intestinal epithelium consists of a single layer of columnar epithelial cells that are constantly renewed from multipotent stem cells originating in the crypts of Lieberkühn. These stem cells give rise to four major epithelial lineages (16). Three of these --absorptive enterocytes, goblet cells, enteroendocrine cells -- migrate up the villus where they differentiate and ultimately die of apoptosis or are exfoliated whole into the lumen in a process that results in replacement of all cells every 3–5 days (17). In contrast, defensinproducing Paneth cells migrate downward over the course of five to eight days to the crypt base where they reside for approximately three weeks. Specialized M cells are also present in the follicle-associated epithelia, which sample luminal antigens and are responsible for the induction of mucosal immunity (18). Each epithelial cell is in intimate contact with its neighbors, and the integrity of the layer is maintained by apical junctional complexes (19). Tight junctions are the most apical components of the complex and create a dynamic barrier to the paracellular movement of water, solutes, and immune cells (5;20).
Both cellular proliferation and death are altered in the gut epithelium in critical illness. Proliferation is decreased in murine models of Pseudomonas aeruginosa pneumonia, acute lung injury (ALI) and endotoxemia induced by lipopolysaccharide (LPS) injection when measured by 5-bromo-2’deoxyuridine labeling of S-phase cells (21–23). In contrast, proliferation is increased following cecal ligation and puncture (CLP) when measured by 3H thymidine incorporation (24). Both sepsis and noninfectious inflammation cause increased apoptosis in the gut epithelium in numerous animal models as well as in patient autopsy studies (outlined in detail below), and preventing epithelial apoptosis partially abrogates sepsis-induced decreases in proliferation (21).
The Mucosal Immune System
The mucosal immune system functions to a) prevent pathogens from penetrating the epithelium, b) prevent uptake of foreign antigens from a variety of sources including commensal bacteria, food, airborne or particulate matter, and c) prevent pathologic immune responses against luminal antigens if they are successful in crossing mucosal barriers (25).
The gut-associated lymphoid tissue (GALT) is the largest lymphatic organ in the body and is composed of four distinct compartments: Peyer’s patches, mesenteric lymph nodes, the lamina propria, and intraepithelial lymphocytes (IELs). Peyer’s patches are lymphoid follicles that come into contact with antigens from the gut lumen, acting as the inductive arm of gut immunity. Dendritic cells (DC) and macrophages can then travel to the mesenteric lymph nodes and present antigen to T cells as well as induce B cell differentiation. Cells then hone to the lamina propria, which is the main effector portion of gut immunity via B cells and T cells (mostly CD4+). Additionally dendritic cells, macrophages, mast cells and polymorphonuclear leukocytes assist in the lamina propria immune response. In the proximal small intestine, IELs are made up predominantly of gamma-delta T cells, while in the ileum, lymphocytes on the basolateral side of enterocytes are predominantly CD8+ T cells (26;27). The role of IELs in host defense is less well understood than other portions of gut immunity, but gamma-delta T cell knockout animals have increased early mortality after sepsis which is felt to be due, in part, to alterations in IELs (28).
Critical illness has a profound effect upon the number of cells in the mucosal immune system. After I/R, recoverable lymphocytes from the lamina propria and intraepithelial space are present at only 20–30% of levels found in sham animals (29). Additionally, lymphocyte numbers decrease to 60% of sham levels in Peyer’s patches. While this implies that critical illness affects gut effector sites more prominently than inductive sites, it is important to note that lymphocyte numbers recover more quickly in the lamina propria than the Peyer’s patches. Although the mechanism underlying this loss of lymphocytes is not fully understood, sepsis increases apoptosis in lamina propria lymphocytes, IELs and Peyer’s patches (30–32). Additionally, cyclooxygenase inhibitors increase lymphocyte apoptosis while simultaneously attenuating an increase in macrophages in mesenteric lymph nodes following CLP (33).
Commensal Bacteria
While the gut’s endogenous microflora has been known to play some role in host homeostasis for years, its profound influence in both healthy and diseased states has recently been increasingly recognized (34–36). The adult human intestine is home to more than 100 trillion bacteria, which dwarves the number of somatic and germ cells in the entire human organism (37). There are estimated to be between 500–1000 species of bacteria present in our intestines (38). Aerobic, facultative, and anaerobic bacteria are all part of the enteric microflora; however, their distribution changes throughout the length of the gut with anaerobes not present in the stomach but making up greater than 99 percent of the flora in the distal colon.
Although it has been demonstrated that patients with severe SIRS have lower anaerobic bacteria and higher pathogenic bacteria (such as Staphylococcus and Pseudomonas) in their gut, less is known about how critical illness affects the commensal bacteria than how it affects the gut epithelium and immune system (39). In the setting of antibiotic usage, increasingly resistant strains of bacteria are frequently selected out in patients. Selective decontamination of the digestive tract (SDD) is a clinical strategy commonly used in many parts of the world that decreases commensal flora. SDD is associated with a slight mortality decrease in selected patient populations but at the price of an increase in the development of resistant microorganisms (40;41). Importantly, SDD decreases commensal bacteria but does not eliminate them, and successful SDD protocols use a combination of enteral and systemic antibiotics. Similarly, germ free animals have 100% survival when subjected to an I/R insult that is lethal in conventional animals (8). This implies that downregulating or eliminating commensal bacteria may be beneficial in critical illness. In contrast, since commensal bacteria are believed to be a crucial part of host homeostasis, multiple recent studies have looked at the effect of probiotics, in an attempt to recreate the beneficial effects seen with normal gut flora (42). Although this field is in its infancy, a recent randomized trial comparing probiotics to standard care in 65 critically ill trauma patients demonstrated decreased sepsis, length of stay and duration of mechanical ventilation in those treated with exogenous bacteria (43). Additionally, a trial of probiotics in 103 critically ill patients demonstrated a late attenuation of the systemic inflammatory response, although this was not associated with changes in morbidity or mortality (44).
How does the gut initiate or propagate a physiologic state that leads to increased mortality in critical illness?
Although simple on the surface, this question is actually quite challenging to answer because it implies we know why people die when they are critically ill. It is true that there have been enormous advances in the understanding of the pathophysiology of sepsis and SIRS (45;46). However, it is equally true that the vast preponderance of clinical trials for sepsis have yielded negative results despite promising pre-clinical results (46). While clinicians can often support even the most severe organ failure (putting patients on dialysis, pressors, ventilators, etc), huge numbers of patients with septic shock die each year despite maximal medical support. However, when autopsies are performed on patients immediately post-mortem, they are remarkably bland except for increased apoptosis in the gut epithelium (discussed below) and lymphocytes (47).
Despite a fundamental lack of understanding why patients die when they are critically ill, ultimately, there are only two ways in which the gut can initiate or propagate a physiologic state that leads to increased mortality. The first is to allow something that already exists within the intestine (bacteria, endotoxin, other preformed toxins) access to the remainder of the body where it can then have secondary effects. The “leaky gut” hypothesis proposes that a disrupted intestinal epithelium allows translocation of bacteria or other damaging luminal contents. There is strong evidence that loss of barrier function is an early event in critical illness although conclusive data that this leads to translocation of bacteria or other luminal contents in patients has not been firmly established (48). The other mechanism through which the gut can initiate or propagate a state that increases mortality is to produce and then release something (cytokines, toxins) that might have direct or indirect harmful extra-intestinal effects.
There is compelling evidence that elements normally residing within the intestine and elements newly produced within the intestine find their way out of the gut in critical illness. Bacteria and antigens that live within the gut lumen exit the intestine because of increased permeability seen in critical illness. Additionally, both the gut epithelium and the gut immune system can generate an inflammatory response in response to a number of diverse, aberrant stimuli. Whatever substance leaves the intestine (either from its lumen or from its epithelium or immune system) can then gain access to the remainder of the body via the bloodstream or via the lymphatics where it can a) primarily injure distant tissue, b) secondarily interact with the immune system to alter the host inflammatory milieu, or c) cause both primary and secondary damage. In this manner, the gut can act as the “motor” of critical illness in multiple ways, regardless of the origin of the offending agent (epithelial, immune or commensal bacteria), regardless of how it gains access to the remainder of the body (via the bloodstream or lymphatics) and regardless of whether its toxic effects are primary or secondary. The common denominator of the multiple ways the intestine can initiate or propagate critical illness is simply that there is a gut origin or element to the whole body pathophysiology. There is increasing evidence, however, that none of the three portions of the gut act in isolation. Instead, communication within the gut appears to set the stage for how the gut interacts with the remainder of the host.
CROSSTALK WITHIN THE GUT
The gut epithelium, mucosal immune system and commensal bacteria all communicate with each other and, in turn, communicate with extraintestinal tissues. While it represents an oversimplification to look at any of these “conversations” in isolation, a reductionist approach can illuminate how crosstalk exists between each of the three components of the gut under basal conditions and how this can be altered in critical illness.
Interactions between the epithelium and mucosal immune system
Epithelial cells serve as immunoeffector cells and are capable of up-regulating surface molecules and secreting cytokines and chemokines to facilitate antigen presentation to immune cells (49). Additionally, enterocytes act as antigen presenting cells themselves and regulate lymphocyte responses in the intestine (50). Enterocytes are also in constant communication with IELs and T cells within the lamina propria and have been shown to exhibit increased MHC class II molecules on the basolateral membrane in states of inflammation (51). The polarized expression of MHC class II molecules on enterocytes suggests that antigen exposure on the luminal side as opposed to the basolateral side elicits different immunologic responses (50).
Permeability
The immune response in critical illness is complex, with an early hyperimmune state and a late hypoimmune state, although both pro- and anti-inflammatory mediators can be found throughout (45;52). Inflammatory mediators can, in turn, modulate epithelial permeability (although it should be noted that some of these mediators originate within immune cells of the intestine, while others originate in an extra-intestinal fashion but have secondary effects on the intestine).
Pro-inflammatory cytokines have been demonstrated to be key modulators of tight junction expression and localization (53). Since the tight junction barrier acts as a physical and functional barrier against paracellular penetration of harmful substances present in the lumen, disruption of the tight junction barrier can result in increased intestinal permeability and subsequent permeation of luminal antigens that promote intestinal inflammation (54). Inflammatory cytokines tumor necrosis factor alpha (TNF-a), interferon gamma (IFN-γ), Interleukin (IL)-4, and IL-13 have been shown to increase permeability in vitro using intestinal epithelial monolayers (55;56). High-mobility group B1 (HMGB1) also increases permeability in hemorrhagic shock, and anti-HMGB1 antibody ameliorates gut barrier function in mice and improves survival (57). Similarly, the receptor for advanced glycation end products (RAGE) induces hyperpermeability and translocation following hemorrhagic shock which is prevented in RAGE knockout mice or by giving soluble RAGE, the extracellular ligand-binding domain of the molecule (58). In contrast to the effect of pro-inflammatory cytokines on gut barrier function, the anti-inflammatory cytokine IL-10 prevents pro-inflammatory-induced increases in intestinal epithelial permeability (59).
The mechanisms of cytokine-induced alterations in tight junction permeability have not been completely elucidated. However, TNF-α and IFN-γ-induced changes are associated with marked increases in myosin light chain phosphorylation and increased permeability can be reversed with an inhibitor of myosin light chain kinase (MLCK). Altered MLCK can disrupt tight junction morphology by reorganizing the actin cytoskeleton and redistributing tight junction proteins (60). Additionally, TNF-α has been shown to increase tight junction permeability by downregulating ZO-1 protein expression and altering junctional protein localization in intestinal epithelial cells (61). These cytokines have also been shown to induce expression of inducible nitric oxide synthase (iNOS) leading to increased production of nitric oxide (NO) in intestinal monolayers (62). NO is a molecule produced by all three components of the gut (enterocytes, immune cells and commensal bacteria) that has been demonstrated to play a major role in gut homeostasis and disease. While endogenous NO production is required for maintaining normal mucosal permeability, derangements in NO production are deleterious to intestinal integrity. Increased production of NO leads to derangements in ATP production and cytoskeletal organization (63). Additionally, incubation of gut epithelial cells with an NO donor causes decreased expression of ZO-1, ZO-3, and occludin, increased expression of claudin-1, and incorrect subcellular localization of ZO-1, occludin, and claudin-1 (64). These cytokine-induced changes are reversed by co-incubation with an NO scavenger. Of note, similar findings have also been demonstrated in vivo. LPS injection causes increased intestinal permeability in mice, with derangements in both tight junction protein levels and subcellular localization. This response can be blocked pharmacologically by blocking iNOS, and this abnormality does not develop in iNOS−/− mice after LPS injection.
Although alterations in permeability are frequently thought to result from mucosal damage, this is not required for bacterial translocation in mice following hemorrhage (65). It has therefore been proposed that although the paracellular pathway is a significant route for bacterial translocation, disruption of tight junctions requires higher levels of cytokines than may be present with a relatively minor inflammatory insult, where transcellular movement of bacteria may occur initially instead (66). In vitro data demonstrates that IFN-γ-mediated translocation of Escherichia coli across epithelial monolayers takes place through a process exploiting lipid raft-mediated transcytosis pathways in times of inflammatory stress (66). It is therefore possible that pro-inflammatory mediators may increase the size and/or number of lipid raft structures on the apical surface of the epithelium, leading to altered translocation.
Apoptosis
It has been estimated that without apoptosis, an 80 year old person would have a 16 kilometer long intestine (67). Our group at Washington University has had a longstanding interest in the role of apoptosis and mortality in sepsis. Initial animal studies examining septic mice following CLP demonstrated increased levels of apoptosis, predominantly in the gut epithelium, Peyer’s patches and extraintestinal lymphocytes in the thymus and spleen (32;68). Additional studies by Ayala and colleagues demonstrated increased intestinal immune apoptosis in lamina propria B cells, lamina propria mononuclear cells, and IELs (30;31;69). These correlate well to an autopsy study performed in our surgical intensive care unit which demonstrated that septic patients have a selective increase in gut epithelial and lymphocyte apoptosis (although additional immune cells have subsequently been found to also have elevated sepsis-induced apoptosis) (47). Of note, gut epithelial and gut immune apoptosis are also elevated in animal models of I/R, trauma/hemorrhage, thermal injury, LPS and necrotizing pancreatitis (70–74). Apoptosis can be initiated by a receptor-mediated pathway or a mitochondrial-mediated pathway, and evidence exists that both play a role in mediating gut epithelial and immune apoptosis (30;31;75).
The functional significance of gut epithelial apoptosis in sepsis has been demonstrated in multiple animal models. Both CLP and Pseudomonas aeruginosa cause sepsis-induced increases in intestinal apoptosis. Importantly, overexpression of the anti-apoptotic protein Bcl-2 in transgenic mice confers a 2–10 fold increase in survival compared to wild type animals subjected to these same septic insults (10;11). The mechanism underlying this survival advantage is not entirely clear. Preventing apoptosis is not associated with changes in systemic cytokines or degree of bacteremia. There is preliminary evidence that altering gut epithelial apoptosis is associated with altered lamina propria apoptosis (unpublished data). There is published precedent for crosstalk where overexpression of Bcl-2 prevents apoptosis in a tissue that does not express the transgene – septic animals that overexpress Bcl-2 in their myeloid cells have decreased gut epithelial apoptosis and overexpression of Bcl-2 in T lymphocytes prevents B lymphocyte apoptosis (76;77). It is possible that decreasing gut apoptosis leads to improved survival by altering the host inflammatory response since apoptotic cells are immunosuppressive, leading to the release of anti-inflammatory cytokines and suppression of pro-inflammatory cytokines (78). This is supported by the fact that adoptive transfer of apoptotic cells worsens survival in CLP, associated with alterations in the host immune response (79). Another possible mechanism through which preventing gut epithelial apoptosis could lead to improved survival is by alterations in intestinal permeability. The rationale behind this potential link primarily comes from in vitro data. Addition of Fas to intestinal monolayers induces apoptosis, decreases transepithelial resistance, and increases permeability to small molecules. However, addition of the pan-caspase inhibitor z-VAD prevents both epithelial apoptosis and barrier dysfunction, suggesting a causal relationship between them (80). Additionally, rats subjected to I/R have both increased intestinal permeability and apoptosis (81), although to date, descriptions of the relationship between barrier and cell death in vivo are associative in nature. It should be noted that gut overexpression of Bcl-2 does not improve survival in a murine model of acute lung injury (82), so as with multiple other ways in which the gut is the “motor” of critical illness, the effect is dependent on the model studied.
Gut-lymph hypothesis
In hemorrhagic shock, toxic gut-derived factors are carried in the mesenteric lymph (not the portal vein) where they secondarily induce distant organ injury (9;83). Importantly, lung injury induced by trauma-hemorrhagic shock can be completely abrogated by ligation of the lymph duct in rats as well as in porcine and primate models (84;85). Similar findings have been noted in rat models of thermal injury, cardiac injury, and I/R. Mesenteric lymph from shocked rats also induces endothelial injury and activates neutrophils although a gender dimorphism exists wherein males are more susceptible to injury than females, likely due to sex hormone protection. Recent data suggests that Amiloride reduces the capacity of post-shock lymph to prime neutrophils without having an effect on endothelial cells. The effect of this inhibitor of Na+/H+ exchangers and Na+ channels on mesenteric lymph is also associated with decreased gut permeability (86). While the exact components in gut-derived lymph responsible for distant organ injury have not been identified, evidence indicates that the factors are larger than 100 kD and potentially located in the lipid fraction of lymph (87;88).
Interactions between the epithelium and commensal bacteria
The gut epithelium directly senses commensal bacteria by means of pattern recognition receptors (PPR), which include the membrane-bound Toll-like receptors (TLRs) and the intracellular nucleotide-binding oligomerization domain proteins (89). PPRs recognize conserved structures on bacteria and viruses and activate pro-inflammatory pathways to alert the host to infection via activation of the transcription factor nuclear factor kappa B (NFkB) (90).
In vitro data suggests that, in isolation, the gut epithelium can mount a pro-inflammatory response against commensal bacteria (91). However, commensal bacteria do not typically cause disease in healthy patients, and there is growing evidence that crosstalk between the gut epithelium and commensal bacteria helps maintain epithelial integrity in an intact host. When the epithelium is damaged, it must be repaired quickly in order to prevent bacterial penetration and subsequent systemic inflammation. An example of what happens when this relationship is altered is recent data that suggests that HIV-1 infection leads to breakdown of the follicle-associated epithelium. In turn, this leads to bacterial translocation, increased levels of plasma LPS and chronic activation of the immune system (92).
Activation of TLRs by commensal bacteria play a critical role in initiating mucosal repair. Mice given broad-spectrum antibiotics are more susceptible to dextran sulfate sodium (DSS)-induced colitis than fully colonized mice. Importantly, recolonization of antibiotic-treated animals with commensal bacteria restores their ability to initiate mucosal repair (93). Further, functional TLR signaling is required for protection against DSS-induced colitis, even in the presence of commensal bacteria since myeloid differentiation factor (MyD)88-deficient mice, which lack an adaptor molecule essential for TLR signaling, exhibit profound mucosal damage when subjected to colitis. One mechanism through which this occurs is that commensal bacteria interact with host TLRs, which in turn leads to IL-6 and cytokine-induced neutrophil chemoattractant (KC)-1 production. Removing commensal bacteria eliminates this MyD88-dependent epithelial cytokine production. Additionally, commensal flora direct expression of cytoprotective heat shock proteins (HSP) 25 and 72 in gut epithelial cells via soluble factors that activate mitogen activated protein kinases (94–96). TLR signaling also appears to be important in mediating how commensal flora induces HSP 25 and 72 in epithelial cells, since this is not seen in MyD88 knockout animals (93).
Commensal bacteria also interact with the gut epithelium by affecting mucus production. Although it was long thought that the sole function of mucus was to protect and lubricate the epithelial surface, it is now known that mucins are essential for growth, epithelial renewal, differentiation and intestinal integrity (97). Commensal bacteria are able to inhibit pathogenic bacterial adherence to the intestinal epithelium by inducing increased production of mucins (98). Alterations in normal commensal flora secondarily lead to changes in goblet cell function and in the chemical composition of intestinal mucus (99). When germ free mice are conventionalized with commensal bacteria, they have a decrease in the number of goblet cells and alterations in mucin composition in a time-dependent manner (100). In contrast, LPS (which can be derived from commensal gram-negative bacteria) has been shown to increase production and secretion of mucins (101).
Commensal bacteria also induce the epithelium to produce agents which, in turn, kill selected bacteria. Specifically, Paneth cells produce a potent bactericidal protein called angiogenin 4 (Ang4) which is released in secretory granules into the lumen in response to bacterial signals and preferentially targets gram-positive bacteria. (102). It has been postulated that Ang4 helps to maintain the predominantly gram-negative microflora in the gut. While Ang4 production is defective in germ free mice, monoassociation with Bacteroides thetaiotaomicron induces full expression of Ang4 in Paneth cells (as well as leading to development of a complete submucosal capillary network in 10 days).
Commensal bacteria can also actively attenuate transcription of epithelial-produced inflammatory cytokines by interfering with the transcription factor NFkB. In its unactivated state, NFkB is bound to an inhibitor, IkB, preventing pro-inflammatory gene transcription. When a stimulus leads to phosphorylation of IkB, ubiquitination occurs. In turn, NFkB is released and translocates into the nucleus to modulate gene transcription. Commensal bacteria can blocking IkB ubiquitination and trigger export of the RelA subunit out of the nucleus upon NFkB translocation (103;104). As such, commensal-epithelial crosstalk may be responsible for the tolerance of the gut to the endogenous bacteria and antigens it is continuously in contact with. It is important to note that induction of gut epithelial NFkB induces a potent systemic pro-inflammatory response. When gut epithelial-specific NFkB activation is prevented by ablation of IkB kinase, mice subjected to intestinal I/R fail to develop MODS or lung edema and have reduced induction of intestinal inflammation-associated genes compared to wild type animals (105). This does come at a price, however, since these animals have a marked upregulation in gut epithelial apoptosis, suggesting that NFkB is both a potent pro-inflammatory and local anti-apoptotic factor.
Less is known about epithelial-commensal crosstalk in SIRS, but there is increasing evidence that this is altered in critical illness. As outlined earlier, in patients with severe SIRS, hyperpermeability, immunosuppression and antibiotic usage can alter the delicate balance of the enteric microflora, leaving the intestinal mucosa with less “beneficial” bacteria such as Bifidobacterium and Lactobacillus and higher "pathogenic" bacteria such Staphylococcus and Pseudomonas compared to healthy volunteers (39). While the precise functional significance of this is unclear, it is known that enteric pathogens worsen barrier function and increase secretion in intestinal epithelial cells in vitro (106). Enteropathogenic Escherichia coli infection induces a marked increase in tight junction permeability, potentially by altering myosin light chain kinase (107). Additionally, intestinal epithelial cells either incubated with Escherichia coli or subjected to hypoxia and reoxygenation have normal amounts of apoptosis, but a combination of the two insults results in a four-fold increase in cell death, suggesting that the endogenous gut flora might have pathogenic effects when the host is subjected to systemic stress (108).
Interactions between the mucosal immune system and commensal bacteria
Commensal bacteria are required for the normal development of mucosal immunity and, in turn, the mucosal immune system continuously samples luminal bacteria allowing tolerance and controlling host inflammation (109). Dendritic cells (DC) extend dendrites into the intestinal lumen to sample antigens and bacteria. Even though these extensions penetrate the epithelium, integrity of the gut barrier is preserved because DC express tight junction proteins and maintain a tight seal with adjacent epithelial cells (110). This process is regulated by the chemokine CX3CL1, which is required for extension of DC protrusions (111). Of note, mucosal DC are predominantly located in the distal small intestine, coinciding with increased bacterial density and CX3CL1 production suggesting that not only is the epithelium required for proper functioning of mucosal DC, but bacteria themselves may be essential as well.
Studies have also shown that DC digest and carry commensals into mesenteric lymph nodes where they induce IgA secretion which, in turn, can protect against penetration by other commensal bacteria (112). This process occurs without help from T cells, thereby preventing a systemic immune response. Additionally, commensal bacteria do not penetrate past the mesenteric lymph nodes, meaning any immune responses are confined to the mucosal level (113).
Comparing mucosal immunity in germ free and wild type mice illustrates the importance of the commensal bacteria on gut immune development. Germ free mice have hypoplastic Peyer’s patches and decreased numbers of IgA-producing plasma cells and lamina propria lymphocytes while colonization of germ free mice restores these deficiencies (114). Further, specific components of individual species of commensal bacteria can have profound effects on mucosal immunity when introduced into animals lacking endogenous bacteria. This is demonstrated by experiments where germ free mice were monoassociated with Bacteroides fragilis with or without bacterial polysaccharide, an immunomodulatory component of the organism (115). Mice given the mutated Bacteroides fragilis without bacterial polysaccharide appeared similar to other germ free mice, with deficiencies in CD4+ T cells, abnormal lymphoid architecture and a skewing towards Th2 cytokine production. However, animals given intact Bacteroides fragilis appeared similar to wild type animals with normal levels of CD4+ T cells, normal lymphoid architecture and a shift toward a Th1 predominant phenotype.
The enteric nervous system
The enteric nervous system represents a fourth element of the intestine. Nerve fibers from the sympathetic nervous system innervate gut mucosa and lymphoid tissue where they have both pro- and anti-inflammatory functions depending on concentration, receptor affinity, presence of cotransmitters and relationship to whole body inflammatory state (116). Gut-derived norepinephrine has been demonstrated to prime Kupffer cells, leading to pro-inflammatory cytokine release and propagation of SIRS (117). Crosstalk between the enteric nervous system and the intestinal epithelium, immune system and commensal bacteria is not addressed above because, to date, there are minimal data on the specific role of the enteric nervous system in critical illness. It is likely, however, that crosstalk exists and plays an important role in the pathophysiology of critical illness. This statement is based upon multiple studies of crosstalk between the central nervous system and the immune system in critical illness as well as crosstalk between the enteric nervous system and the gut in homeostasis and chronic inflammatory disease. For example, in multiple experimental models of critical illness, an inflammatory reflex has been described where the parasympathetic vagus nerve inhibits cytokine release (118). Although the vagus nerve innervates multiple organs throughout the body, stimulation has been demonstrated to have intestine-specific effects, such as decreasing murine postoperative ileus by activating STAT3 in intestinal macrophages (119). Additionally, increasing evidence suggests that host-derived regulators from the enteric nerve system engage in crosstalk with both the immune system and the epithelium in determining whether the host mounts a chronic inflammatory response to commensal bacteria (120).
CONCLUSIONS
The gut (broadly defined) is made up of an epithelium, a mucosal immune system, an overwhelming number of commensal bacteria, and the enteric nervous system, and each of these interacts with each other, and, in turn, interacts with the remainder of the host. There is overwhelming evidence that the intestine plays a crucial role in the pathophysiology of sepsis. This is demonstrated by multiple seminal manuscripts defining and redefining gut-derived sepsis and how the gut acts as the “motor” of SIRS. We believe the paradigm presented herein takes multiple correct but disparate theories and finds a unifying way to encompass each of them. Under basal conditions, the gut has a remarkably complex interplay that plays a key role in host homeostasis. In critical illness, all elements of the gut are markedly perturbed and alterations in crosstalk yield both beneficial and pathologic responses. A more complete understanding of how the three arms of the intestine communicate with each other and how the cumulative output of this conversation communicates with the host outside of the gut represents both a potent challenge and a tremendous opportunity in critical care research.
Acknowledgments
This study was supported by funding from National Institutes of Health (grant nos. GM072808-01, GM66202-01).
Reference List
- 1.Carrico CJ, Meakins JL, Marshall JC, Fry D, Maier RV. Multiple-organ-failure syndrome. The gastrointestinal tract: the "motor" of MOF. Arch Surg. 1986;121:196–208. doi: 10.1001/archsurg.1986.01400020082010. [DOI] [PubMed] [Google Scholar]
- 2.American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864–874. [PubMed] [Google Scholar]
- 3.Hassoun HT, Kone BC, Mercer DW, Moody FG, Weisbrodt NW, Moore FA. Post-injury multiple organ failure: the role of the gut. Shock. 2001;15:1–10. doi: 10.1097/00024382-200115010-00001. [DOI] [PubMed] [Google Scholar]
- 4.Fink MP. Intestinal epithelial hyperpermeability: update on the pathogenesis of gut mucosal barrier dysfunction in critical illness. Curr Opin Crit Care. 2003;9:143–151. doi: 10.1097/00075198-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Han X, Fink MP, Yang R, Delude RL. Increased iNOS activity is essential for intestinal epithelial tight junction dysfunction in endotoxemic mice. Shock. 2004;21:261–270. doi: 10.1097/01.shk.0000112346.38599.10. [DOI] [PubMed] [Google Scholar]
- 6.Fink MP, Delude RL. Epithelial Barrier Dysfunction: A Unifying Theme to Explain the Pathogenesis of Multiple Organ Dysfunction at the Cellular Level. Crit Care Clin. 2005;21:177–196. doi: 10.1016/j.ccc.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Alverdy JC, Laughlin RS, Wu L. Influence of the critically ill state on host-pathogen interactions within the intestine: gut-derived sepsis redefined. Crit Care Med. 2003;31:598–607. doi: 10.1097/01.CCM.0000045576.55937.67. [DOI] [PubMed] [Google Scholar]
- 8.Souza DG, Vieira AT, Soares AC, Pinho V, Nicoli JR, Vieira LQ, Teixeira MM. The essential role of the intestinal microbiota in facilitating acute inflammatory responses. J Immunol. 2004;173:4137–4146. doi: 10.4049/jimmunol.173.6.4137. [DOI] [PubMed] [Google Scholar]
- 9.Deitch EA, Xu D, Kaise VL. Role of the gut in the development of injury- and shock induced SIRS and MODS: the gut-lymph hypothesis, a review. Front Biosci. 2006;11:520–528. doi: 10.2741/1816. [DOI] [PubMed] [Google Scholar]
- 10.Coopersmith CM, Stromberg PE, Dunne WM, Davis CG, Amiot DM, Buchman TG, Karl IE, Hotchkiss RS. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. JAMA. 2002;287:1716–1721. doi: 10.1001/jama.287.13.1716. [DOI] [PubMed] [Google Scholar]
- 11.Coopersmith CM, Chang KC, Swanson PE, Tinsley KW, Stromberg PE, Buchman TG, Karl IE, Hotchkiss RS. Overexpression of Bcl-2 in the intestinal epithelium improves survival in septic mice. Crit Care Med. 2002;30:195–201. doi: 10.1097/00003246-200201000-00028. [DOI] [PubMed] [Google Scholar]
- 12.McCracken VJ, Lorenz RG. The gastrointestinal ecosystem: a precarious alliance among epithelium, immunity and microbiota. Cell Microbiol. 2001;3:1–11. doi: 10.1046/j.1462-5822.2001.00090.x. [DOI] [PubMed] [Google Scholar]
- 13.Wischmeyer PE. Glutamine: role in gut protection in critical illness. Curr Opin Clin Nutr Metab Care. 2006;9:607–612. doi: 10.1097/01.mco.0000241672.09676.03. [DOI] [PubMed] [Google Scholar]
- 14.Mainous MR, Ertel W, Chaudry IH, Deitch EA. The gut: a cytokine-generating organ in systemic inflammation? Shock. 1995;4:193–199. [PubMed] [Google Scholar]
- 15.Koo DJ, Zhou M, Jackman D, Cioffi WG, Bland KI, Chaudry IH, Wang P. Is gut the major source of proinflammatory cytokine release during polymicrobial sepsis? Biochim Biophys Acta. 1999;1454:289–295. doi: 10.1016/s0925-4439(99)00045-9. [DOI] [PubMed] [Google Scholar]
- 16.Cheng H, Leblond CP. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian Theory of the origin of the four epithelial cell types. Am J Anat. 1974;141:537–561. doi: 10.1002/aja.1001410407. [DOI] [PubMed] [Google Scholar]
- 17.Hall PA, Coates PJ, Ansari B, Hopwood D. Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis. J Cell Sci. 1994;107:3569–3577. doi: 10.1242/jcs.107.12.3569. [DOI] [PubMed] [Google Scholar]
- 18.Mach J, Hshieh T, Hsieh D, Grubbs N, Chervonsky A. Development of intestinal M cells. Immunol Rev. 2005;206:177–189. doi: 10.1111/j.0105-2896.2005.00281.x. [DOI] [PubMed] [Google Scholar]
- 19.Utech M, Bruwer M, Nusrat A. Tight junctions and cell-cell interactions. Methods Mol Biol. 2006;341:185–195. doi: 10.1385/1-59745-113-4:185. [DOI] [PubMed] [Google Scholar]
- 20.Johnson LG. Applications of imaging techniques to studies of epithelial tight junctions. Adv Drug Deliv Rev. 2005;57:111–121. doi: 10.1016/j.addr.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 21.Coopersmith CM, Stromberg PE, Davis CG, Dunne WM, Amiot DM, Karl IE, Hotchkiss RS, Buchman TG. Sepsis from Pseudomonas aeruginosa pneumonia decreases intestinal proliferation and induces gut epithelial cell cycle arrest. Crit Care Med. 2003;31:1630–1637. doi: 10.1097/01.CCM.0000055385.29232.11. [DOI] [PubMed] [Google Scholar]
- 22.Husain KD, Stromberg PE, Woolsey CA, Turnbull IR, Dunne WM, Javadi P, Buchman TG, Karl IE, Hotchkiss RS, Coopersmith CM. Mechanisms of decreased intestinal epithelial proliferation and increased apoptosis in murine acute lung injury. Crit Care Med. 2005;33:2350–2357. doi: 10.1097/01.ccm.0000182797.89252.a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Potoka DA, Upperman JS, Zhang XR, Kaplan JR, Corey SJ, Grishin A, Zamora R, Ford HR. Peroxynitrite inhibits enterocyte proliferation and modulates Src kinase activity in vitro. Am J Physiol Gastrointest Liver Physiol. 2003;285:G861–G869. doi: 10.1152/ajpgi.00412.2002. [DOI] [PubMed] [Google Scholar]
- 24.Rafferty JF, Noguchi Y, Fischer JE, Hasselgren PO. Sepsis in rats stimulates cellular proliferation in the mucosa of the small intestine. Gastroenterology. 1994;107:121–127. doi: 10.1016/0016-5085(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 25.Haynes BF. Gut microbes out of control in HIV infection. Nat Med. 2006;12:1351–1352. doi: 10.1038/nm1206-1351. [DOI] [PubMed] [Google Scholar]
- 26.Acheson DW, Luccioli S. Microbial-gut interactions in health and disease. Mucosal immune responses. Best Pract Res Clin Gastroenterol. 2004;18:387–404. doi: 10.1016/j.bpg.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Tamura A, Soga H, Yaguchi K, Yamagishi M, Toyota T, Sato J, Oka Y, Itoh T. Distribution of two types of lymphocytes (intraepithelial and lamina-propria-associated) in the murine small intestine. Cell Tissue Res. 2003;313:47–53. doi: 10.1007/s00441-003-0706-4. [DOI] [PubMed] [Google Scholar]
- 28.Chung CS, Watkins L, Funches A, Lomas-Neira J, Cioffi WG, Ayala A. Deficiency of gammadelta T lymphocytes contributes to mortality and immunosuppression in sepsis. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1338–R1343. doi: 10.1152/ajpregu.00283.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukatsu K, Sakamoto S, Hara E, Ueno C, Maeshima Y, Matsumoto I, Mochizuki H, Hiraide H. Gut ischemia-reperfusion affects gut mucosal immunity: a possible mechanism for infectious complications after severe surgical insults. Crit Care Med. 2006;34:182–187. doi: 10.1097/01.ccm.0000196207.86570.16. [DOI] [PubMed] [Google Scholar]
- 30.Chung CS, Wang W, Chaudry IH, Ayala A. Increased apoptosis in lamina propria B cells during polymicrobial sepsis is FasL but not endotoxin mediated. Am J Physiol Gastrointest Liver Physiol. 2001;280:G812–G818. doi: 10.1152/ajpgi.2001.280.5.G812. [DOI] [PubMed] [Google Scholar]
- 31.Chung CS, Xu YX, Wang W, Chaudry IH, Ayala A. Is Fas ligand or endotoxin responsible for mucosal lymphocyte apoptosis in sepsis? Arch Surg. 1998;133:1213–1220. doi: 10.1001/archsurg.133.11.1213. [DOI] [PubMed] [Google Scholar]
- 32.Hiramatsu M, Hotchkiss RS, Karl IE, Buchman TG. Cecal ligation and puncture (CLP) induces apoptosis in thymus, spleen, lung, and gut by an endotoxin and TNF-independent pathway. Shock. 1997;7:247–253. doi: 10.1097/00024382-199704000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Osterberg J, Ljungdahl M, Haglund U. Influence of cyclooxygenase inhibitors on gut immune cell distribution and apoptosis rate in experimental sepsis. Shock. 2006;25:147–154. doi: 10.1097/01.shk.0000189843.78729.e2. [DOI] [PubMed] [Google Scholar]
- 34.Wells C, Hess D, Erlandsen S. Impact of the indigenous flora in animal models of shock and sepsis. Shock. 2004;22:562–568. doi: 10.1097/01.shk.0000145935.24344.2d. [DOI] [PubMed] [Google Scholar]
- 35.Madara J. Building an intestine -- architectural contributions of commensal bacteria. N Engl J Med. 2004;351:1685–1686. doi: 10.1056/NEJMcibr042621. [DOI] [PubMed] [Google Scholar]
- 36.Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 37.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 38.Xu J, Gordon JI. Inaugural Article: Honor thy symbionts. Proc Natl Acad Sci U S A. 2003;100:10452–10459. doi: 10.1073/pnas.1734063100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimizu K, Ogura H, Goto M, Asahara T, Nomoto K, Morotomi M, Yoshiya K, Matsushima A, Sumi Y, Kuwagata Y, Tanaka H, Shimazu T, Sugimoto H. Altered gut flora and environment in patients with severe SIRS. J Trauma. 2006;60:126–133. doi: 10.1097/01.ta.0000197374.99755.fe. [DOI] [PubMed] [Google Scholar]
- 40.Nathens AB, Marshall JC. Selective decontamination of the digestive tract in surgical patients: a systematic review of the evidence. Arch Surg. 1999;134:170–176. doi: 10.1001/archsurg.134.2.170. [DOI] [PubMed] [Google Scholar]
- 41.Leone M, Albanese J, Antonini F, Nguyen-Michel A, Martin C. Long-term (6-year) effect of selective digestive decontamination on antimicrobial resistance in intensive care, multiple-trauma patients. Crit Care Med. 2003;31:2090–2095. doi: 10.1097/01.CCM.0000079606.16776.C5. [DOI] [PubMed] [Google Scholar]
- 42.Floch MH, Montrose DC. Use of probiotics in humans: an analysis of the literature. Gastroenterol Clin North Am. 2005;34:547–570. doi: 10.1016/j.gtc.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Kotzampassi K, Giamarellos-Bourboulis EJ, Voudouris A, Kazamias P, Eleftheriadis E. Benefits of a synbiotic formula (Synbiotic 2000Forte) in critically Ill trauma patients: early results of a randomized controlled trial. World J Surg. 2006;30:1848–1855. doi: 10.1007/s00268-005-0653-1. [DOI] [PubMed] [Google Scholar]
- 44.McNaught CE, Woodcock NP, Anderson AD, MacFie J. A prospective randomised trial of probiotics in critically ill patients. Clin Nutr. 2005;24:211–219. doi: 10.1016/j.clnu.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 45.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 46.Russell JA. Management of sepsis. N Engl J Med. 2006;355:1699–1713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- 47.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 48.Moore FA, Moore EE, Poggetti R, McAnena OJ, Peterson VM, Abernathy CM, Parsons PE. Gut bacterial translocation via the portal vein: a clinical perspective with major torso trauma. J Trauma. 1991;31:629–636. doi: 10.1097/00005373-199105000-00006. [DOI] [PubMed] [Google Scholar]
- 49.Acheson DW, Luccioli S. Microbial-gut interactions in health and disease. Mucosal immune responses. Best Pract Res Clin Gastroenterol. 2004;18:387–404. doi: 10.1016/j.bpg.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 50.Hershberg RM, Mayer LF. Antigen processing and presentation by intestinal epithelial cells - polarity and complexity. Immunol Today. 2000;21:123–128. doi: 10.1016/s0167-5699(99)01575-3. [DOI] [PubMed] [Google Scholar]
- 51.Kaiserlian D, Vidal K, Revillard JP. Murine enterocytes can present soluble antigen to specific class II-restricted CD4+ T cells. Eur J Immunol. 1989;19:1513–1516. doi: 10.1002/eji.1830190827. [DOI] [PubMed] [Google Scholar]
- 52.Osuchowski MF, Welch K, Siddiqui J, Remick DG. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J Immunol. 2006;177:1967–1974. doi: 10.4049/jimmunol.177.3.1967. [DOI] [PubMed] [Google Scholar]
- 53.Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, Nusrat A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164–6172. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- 54.Anderson JM, Van Itallie CM. Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol. 1995;269:G467–G475. doi: 10.1152/ajpgi.1995.269.4.G467. [DOI] [PubMed] [Google Scholar]
- 55.Adams RB, Planchon SM, Roche JK. IFN-gamma modulation of epithelial barrier function. Time course, reversibility, and site of cytokine binding. J Immunol. 1993;150:2356–2363. [PubMed] [Google Scholar]
- 56.Ceponis PJ, Botelho F, Richards CD, McKay DM. Interleukins 4 and 13 increase intestinal epithelial permeability by a phosphatidylinositol 3-kinase pathway. Lack of evidence for STAT 6 involvement. J Biol Chem. 2000;275:29132–2917. doi: 10.1074/jbc.M003516200. [DOI] [PubMed] [Google Scholar]
- 57.Yang R, Harada T, Mollen KP, Prince JM, Levy RM, Englert JA, Gallowitsch-Puerta M, Yang L, Yang H, Tracey KJ, Harbrecht BG, Billiar TR, Fink MP. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med. 2006;12:105–114. doi: 10.2119/2006-00010.Yang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raman KG, Sappington PL, Yang R, Levy RM, Prince JM, Liu S, Watkins SK, Schmidt AM, Billiar TR, Fink MP. The role of RAGE in the pathogenesis of intestinal barrier dysfunction after hemorrhagic shock. Am J Physiol Gastrointest Liver Physiol. 2006;291:G556–G565. doi: 10.1152/ajpgi.00055.2006. [DOI] [PubMed] [Google Scholar]
- 59.Madsen KL, Lewis SA, Tavernini MM, Hibbard J, Fedorak RN. Interleukin 10 prevents cytokine-induced disruption of T84 monolayer barrier integrity and limits chloride secretion. Gastroenterology. 1997;113:151–159. doi: 10.1016/s0016-5085(97)70090-8. [DOI] [PubMed] [Google Scholar]
- 60.Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, Mrsny RJ, Turner JR. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology. 2002;123:163–172. doi: 10.1053/gast.2002.34235. [DOI] [PubMed] [Google Scholar]
- 61.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 62.Chavez AM, Menconi MJ, Hodin RA, Fink MP. Cytokine-induced intestinal epithelial hyperpermeability: role of nitric oxide. Crit Care Med. 1999;27:2246–2251. doi: 10.1097/00003246-199910000-00030. [DOI] [PubMed] [Google Scholar]
- 63.Salzman AL, Menconi MJ, Unno N, Ezzell RM, Casey DM, Gonzalez PK, Fink MP. Nitric oxide dilates tight junctions and depletes ATP in cultured Caco-2BBe intestinal epithelial monolayers. Am J Physiol. 1995;268:G361–G373. doi: 10.1152/ajpgi.1995.268.2.G361. [DOI] [PubMed] [Google Scholar]
- 64.Han X, Fink MP, Delude RL. Proinflammatory cytokines cause NO*-dependent and -independent changes in expression and localization of tight junction proteins in intestinal epithelial cells. Shock. 2003;19:229–237. doi: 10.1097/00024382-200303000-00006. [DOI] [PubMed] [Google Scholar]
- 65.Gunji H, Scarth S, Carlson GL, Warhurst G, Little RA, Hopkins SJ. Variability of bacterial translocation in the absence of intestinal mucosal damage following injury and the influence of interleukin-6. Pathophysiology. 2006;13:39–49. doi: 10.1016/j.pathophys.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 66.Clark E, Hoare C, Tanianis-Hughes J, Carlson GL, Warhurst G. Interferon gamma induces translocation of commensal Escherichia coli across gut epithelial cells via a lipid raft-mediated process. Gastroenterology. 2005;128:1258–1267. doi: 10.1053/j.gastro.2005.01.046. [DOI] [PubMed] [Google Scholar]
- 67.Melino G. The Sirens' song. Nature. 2001;412:23. doi: 10.1038/35083653. [DOI] [PubMed] [Google Scholar]
- 68.Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG, Karl IE. Apoptosis in lymphoid and parenchymal cells during sepsis: findings in normal and T- and B-cell-deficient mice. Crit Care Med. 1997;25:1298–1307. doi: 10.1097/00003246-199708000-00015. [DOI] [PubMed] [Google Scholar]
- 69.Chung CS, Xu YX, Chaudry IH, Ayala A. Sepsis induces increased apoptosis in lamina propria mononuclear cells which is associated with altered cytokine gene expression. J Surg Res. 1998;77:63–70. doi: 10.1006/jsre.1998.5339. [DOI] [PubMed] [Google Scholar]
- 70.Ikeda H, Suzuki Y, Suzuki M, Koike M, Tamura J, Tong J, Nomura M, Itoh G. Apoptosis is a major mode of cell death caused by ischaemia and ischaemia/reperfusion injury to the rat intestinal epithelium. Gut. 1998;42:530–537. doi: 10.1136/gut.42.4.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu YX, Ayala A, Monfils B, Cioffi WG, Chaudry IH. Mechanism of intestinal mucosal immune dysfunction following trauma- hemorrhage: increased apoptosis associated with elevated Fas expression in Peyer's patches. J Surg Res. 1997;70:55–60. doi: 10.1006/jsre.1997.5111. [DOI] [PubMed] [Google Scholar]
- 72.Wolf SE, Ikeda H, Matin S, Debroy MA, Rajaraman S, Herndon DN, Thompson JC. Cutaneous burn increases apoptosis in the gut epithelium of mice. J Am Coll Surg. 1999;188:10–16. doi: 10.1016/s1072-7515(98)00260-9. [DOI] [PubMed] [Google Scholar]
- 73.Cinel I, Buyukafsar K, Cinel L, Polat A, Atici S, Tamer L, Oral U. The role of poly(adp-ribose) synthetase inhibition in preventing endotoxemia-induced intestinal epithelial apoptosis. Pharmacol Res. 2002;46:119–127. doi: 10.1016/s1043-6618(02)00075-0. [DOI] [PubMed] [Google Scholar]
- 74.Wang X, Wang B, Wu K, Xu M, Gong Z. Growth hormone downregulated the excessive apoptosis of ileal intestinal epithelial cells in rats during the early course of acute necrotizing pancreatitis. Pancreas. 2002;25:205–209. doi: 10.1097/00006676-200208000-00016. [DOI] [PubMed] [Google Scholar]
- 75.Coopersmith CM, O'Donnell D, Gordon JI. Bcl-2 inhibits ischemia-reperfusion-induced apoptosis in the intestinal epithelium of transgenic mice. Am J Physiol. 1999;276:G677–G686. doi: 10.1152/ajpgi.1999.276.3.G677. [DOI] [PubMed] [Google Scholar]
- 76.Iwata A, Stevenson VM, Minard A, Tasch M, Tupper J, Lagasse E, Weissman I, Harlan JM, Winn RK. Over-expression of Bcl-2 provides protection in septic mice by a trans effect. J Immunol. 2003;171:3136–3141. doi: 10.4049/jimmunol.171.6.3136. [DOI] [PubMed] [Google Scholar]
- 77.Hotchkiss RS, Swanson PE, Knudson CM, Chang KC, Cobb JP, Osborne DF, Zollner KM, Buchman TG, Korsmeyer SJ, Karl IE. Overexpression of Bcl-2 in transgenic mice decreases apoptosis and improves survival in sepsis. J Immunol. 1999;162:4148–4156. [PubMed] [Google Scholar]
- 78.Chen W, Frank ME, Jin W, Wahl SM. TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity. 2001;14:715–725. doi: 10.1016/s1074-7613(01)00147-9. [DOI] [PubMed] [Google Scholar]
- 79.Hotchkiss RS, Chang KC, Grayson MH, Tinsley KW, Dunne BS, Davis CG, Osborne DF, Karl IE. Adoptive transfer of apoptotic splenocytes worsens survival, whereas adoptive transfer of necrotic splenocytes improves survival in sepsis. Proc Natl Acad Sci U S A. 2003;100:6724–6729. doi: 10.1073/pnas.1031788100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abreu MT, Palladino AA, Arnold ET, Kwon RS, McRoberts JA. Modulation of barrier function during Fas-mediated apoptosis in human intestinal epithelial cells. Gastroenterology. 2000;119:1524–1536. doi: 10.1053/gast.2000.20232. [DOI] [PubMed] [Google Scholar]
- 81.Sun Z, Wang X, Deng X, Lasson A, Wallen R, Hallberg E, Andersson R. The influence of intestinal ischemia and reperfusion on bidirectional intestinal barrier permeability, cellular membrane integrity, proteinase inhibitors, and cell death in rats. Shock. 1998;10:203–212. doi: 10.1097/00024382-199809000-00009. [DOI] [PubMed] [Google Scholar]
- 82.Husain KD, Stromberg PE, Javadi P, Buchman TG, Karl IE, Hotchkiss RS, Coopersmith CM. BCL-2 Inhibits Gut Epithelial Apoptosis Induced by Acute Lung Injury in Mice but Has No Effect On Survival. Shock. 2003;20:437–443. doi: 10.1097/01.shk.0000094559.76615.1c. [DOI] [PubMed] [Google Scholar]
- 83.Magnotti LJ, Upperman JS, Xu DZ, Lu Q, Deitch EA. Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and promotes lung injury after hemorrhagic shock. Ann Surg. 1998;228:518–527. doi: 10.1097/00000658-199810000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Senthil M, Brown M, Xu DZ, Lu Q, Feketeova E, Deitch EA. Gut-lymph hypothesis of systemic inflammatory response syndrome/multiple-organ dysfunction syndrome: validating studies in a porcine model. J Trauma. 2006;60:958–965. doi: 10.1097/01.ta.0000215500.00018.47. [DOI] [PubMed] [Google Scholar]
- 85.Deitch EA, Forsythe R, Anjaria D, Livingston DH, Lu Q, Xu DZ, Redl H. The role of lymph factors in lung injury, bone marrow suppression, and endothelial cell dysfunction in a primate model of trauma-hemorrhagic shock. Shock. 2004;22:221–228. doi: 10.1097/01.shk.0000133592.55400.83. [DOI] [PubMed] [Google Scholar]
- 86.Fujiyoshi N, Feketeova E, Lu Q, Xu DZ, Hasko G, Deitch EA. Amiloride moderates increased gut permeability and diminishes mesenteric lymph-mediated priming of neutrophils in trauma/hemorrhagic shock. Surgery. 2006;140:810–817. doi: 10.1016/j.surg.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 87.Adams CA, Jr, Xu DZ, Lu Q, Deitch EA. Factors larger than 100 kd in post-hemorrhagic shock mesenteric lymph are toxic for endothelial cells. Surgery. 2001;129:351–363. doi: 10.1067/msy.2001.111698. [DOI] [PubMed] [Google Scholar]
- 88.Gonzalez RJ, Moore EE, Biffl WL, Ciesla DJ, Silliman CC. The lipid fraction of post-hemorrhagic shock mesenteric lymph (PHSML) inhibits neutrophil apoptosis and enhances cytotoxic potential. Shock. 2000;14:404–408. doi: 10.1097/00024382-200014030-00028. [DOI] [PubMed] [Google Scholar]
- 89.Philpott DJ, Girardin SE. The role of Toll-like receptors and Nod proteins in bacterial infection. Mol Immunol. 2004;41:1099–1108. doi: 10.1016/j.molimm.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 90.Macdonald TT, Monteleone G. Immunity, inflammation, and allergy in the gut. Science. 2005;307:1920–1925. doi: 10.1126/science.1106442. [DOI] [PubMed] [Google Scholar]
- 91.Akhtar M, Watson JL, Nazli A, McKay DM. Bacterial DNA evokes epithelial IL-8 production by a MAPK-dependent, NF-kappaB-independent pathway. FASEB J. 2003;17:1319–1321. doi: 10.1096/fj.03-0950fje. [DOI] [PubMed] [Google Scholar]
- 92.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 93.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 94.Tao Y, Drabik KA, Waypa TS, Musch MW, Alverdy JC, Schneewind O, Chang EB, Petrof EO. Soluble factors from Lactobacillus GG activate MAPKs and induce cytoprotective heat shock proteins in intestinal epithelial cells. Am J Physiol Cell Physiol. 2006;290:C1018–C1030. doi: 10.1152/ajpcell.00131.2005. [DOI] [PubMed] [Google Scholar]
- 95.Arvans DL, Vavricka SR, Ren H, Musch MW, Kang L, Rocha FG, Lucioni A, Turner JR, Alverdy J, Chang EB. Luminal bacterial flora determines physiological expression of intestinal epithelial cytoprotective heat shock proteins 25 and 72. Am J Physiol Gastrointest Liver Physiol. 2005;288:G696–G704. doi: 10.1152/ajpgi.00206.2004. [DOI] [PubMed] [Google Scholar]
- 96.Kojima K, Musch MW, Ren H, Boone DL, Hendrickson BA, Ma A, Chang EB. Enteric flora and lymphocyte-derived cytokines determine expression of heat shock proteins in mouse colonic epithelial cells. Gastroenterology. 2003;124:1395–1407. doi: 10.1016/s0016-5085(03)00215-4. [DOI] [PubMed] [Google Scholar]
- 97.Corfield AP, Carroll D, Myerscough N, Probert CS. Mucins in the gastrointestinal tract in health and disease. Front Biosci. 2001;6:D1321–D1357. doi: 10.2741/corfield. [DOI] [PubMed] [Google Scholar]
- 98.Mack DR, Ahrne S, Hyde L, Wei S, Hollingsworth MA. Extracellular MUC3 mucin secretion follows adherence of Lactobacillus strains to intestinal epithelial cells in vitro. Gut. 2003;52:827–833. doi: 10.1136/gut.52.6.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Deplancke B, Gaskins HR. Microbial modulation of innate defense: goblet cells and the intestinal mucus layer. Am J Clin Nutr. 2001;73:1131S–41S. doi: 10.1093/ajcn/73.6.1131S. [DOI] [PubMed] [Google Scholar]
- 100.Fukushima K, Sasaki I, Ogawa H, Naito H, Funayama Y, Matsuno S. Colonization of microflora in mice: mucosal defense against luminal bacteria. J Gastroenterol. 1999;34:54–60. doi: 10.1007/s005350050216. [DOI] [PubMed] [Google Scholar]
- 101.Smirnova MG, Guo L, Birchall JP, Pearson JP. LPS up-regulates mucin and cytokine mRNA expression and stimulates mucin and cytokine secretion in goblet cells. Cell Immunol. 2003;221:42–49. doi: 10.1016/s0008-8749(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 102.Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat Immunol. 2003;4:269–273. doi: 10.1038/ni888. [DOI] [PubMed] [Google Scholar]
- 103.Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG, Pettersson S, Conway S. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol. 2004;5:104–112. doi: 10.1038/ni1018. [DOI] [PubMed] [Google Scholar]
- 104.Neish AS, Gewirtz AT, Zeng H, Young AN, Hobert ME, Karmali V, Rao AS, Madara JL. Prokaryotic regulation of epithelial responses by inhibition of IkappaB-alpha ubiquitination. Science. 2000;289:1560–1563. doi: 10.1126/science.289.5484.1560. [DOI] [PubMed] [Google Scholar]
- 105.Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med. 2003;9:575–581. doi: 10.1038/nm849. [DOI] [PubMed] [Google Scholar]
- 106.Resta-Lenert S, Barrett KE. Enteroinvasive bacteria alter barrier and transport properties of human intestinal epithelium: role of iNOS and COX-2. Gastroenterology. 2002;122:1070–1087. doi: 10.1053/gast.2002.32372. [DOI] [PubMed] [Google Scholar]
- 107.Yuhan R, Koutsouris A, Savkovic SD, Hecht G. Enteropathogenic Escherichia coli-induced myosin light chain phosphorylation alters intestinal epithelial permeability. Gastroenterology. 1997;113:1873–1882. doi: 10.1016/s0016-5085(97)70006-4. [DOI] [PubMed] [Google Scholar]
- 108.Diebel LN, Liberati DM, Dulchavsky SA, Diglio CA, Brown WJ. Enterocyte apoptosis and barrier function are modulated by SIgA after exposure to bacteria and hypoxia/reoxygenation. Surgery. 2003;134:574–580. doi: 10.1016/s0039-6060(03)00302-7. [DOI] [PubMed] [Google Scholar]
- 109.Kelly D, Conway S. Bacterial modulation of mucosal innate immunity. Mol Immunol. 2005;42:895–901. doi: 10.1016/j.molimm.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 110.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl JP, Ricciardi-Castagnoli P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 111.Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG, Littman DR, Reinecker HC. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254–258. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 112.Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science. 2004;303:1662–1665. doi: 10.1126/science.1091334. [DOI] [PubMed] [Google Scholar]
- 113.Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H, Zinkernagel RM. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. 2000;288:2222–2226. doi: 10.1126/science.288.5474.2222. [DOI] [PubMed] [Google Scholar]
- 114.Umesaki Y, Setoyama H. Structure of the intestinal flora responsible for development of the gut immune system in a rodent model. Microbes Infect. 2000;2:1343–1351. doi: 10.1016/s1286-4579(00)01288-0. [DOI] [PubMed] [Google Scholar]
- 115.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 116.Straub RH, Wiest R, Strauch UG, Harle P, Scholmerich J. The role of the sympathetic nervous system in intestinal inflammation. Gut. 2006;55:1640–1649. doi: 10.1136/gut.2006.091322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Miksa M, Wu R, Zhou M, Wang P. Sympathetic excitotoxicity in sepsis: pro-inflammatory priming of macrophages by norepinephrine. Front Biosc. 2005;10:2217, 2229. doi: 10.2741/1691. [DOI] [PubMed] [Google Scholar]
- 118.Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117:289–296. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6:844–851. doi: 10.1038/ni1229. [DOI] [PubMed] [Google Scholar]
- 120.Haller D. Intestinal epithelial cell signalling and host-derived negative regulators under chronic inflammation: to be or not to be activated determines the balance towards commensal bacteria. Neurogastroenterol Motil. 2006;18:184–199. doi: 10.1111/j.1365-2982.2006.00762.x. [DOI] [PubMed] [Google Scholar]