

Abstract
Porphyrins that bear one-carbon oxygenic substituents (hydroxymethyl, formyl, ester) directly attached to the macrocycle afford a compact architecture that is attractive for diverse applications. Routes to 9 porphyrins bearing such groups in distinct architectures (A4-, trans-A2-, trans-A2B2-, trans-AB- and trans-AB2C-porphyrins) have been explored (A = hydroxymethyl), including porphyrins bearing two one-carbon units in different oxidation states (hydroxymethyl/ester, formyl/ester). The hydroxymethyl group was introduced via TBDMS-protected dipyrromethane precursors.
Keywords: Porphyrin, Dipyrromethane, Hydroxymethyl, Formyl
1. Introduction
Porphyrins and hydroporphyrins equipped with compact substituents are attractive for diverse applications. The core macrocycle lacking any peripheral substituents other than hydrogen is of modest molecular weight (porphine, C20H14N4, 310.4 u) whereas common substituted porphyrins are substantially larger (β-octaethylporphyrin, C36H46N4, 534.8 u) if not nearly twice that (meso-tetraphenylporphyrin, C44H30N4, 614.7 u). Tailoring porphyrins by modifying the meso-aryl groups typically further increases the size and molecular weight, which is undesirable for a number of applications including the following:
For medical applications wherein molecules passively cross the blood-brain barrier, a molecular weight of less than ∼800 u is considered essential.1
For the development of manganese porphyrins that function as catalytic superoxide dismutase mimics in therapeutic applications, potent electron-withdrawing substituents attached to the porphyrin nucleus are required (to shift the electrochemical potential) while maintaining low molecular weight and tailored hydrophilic or amphipathic character.2,3
For molecular information storage applications wherein redox-active molecules are assembled on an electroactive surface, small size permits a high charge density.4,5
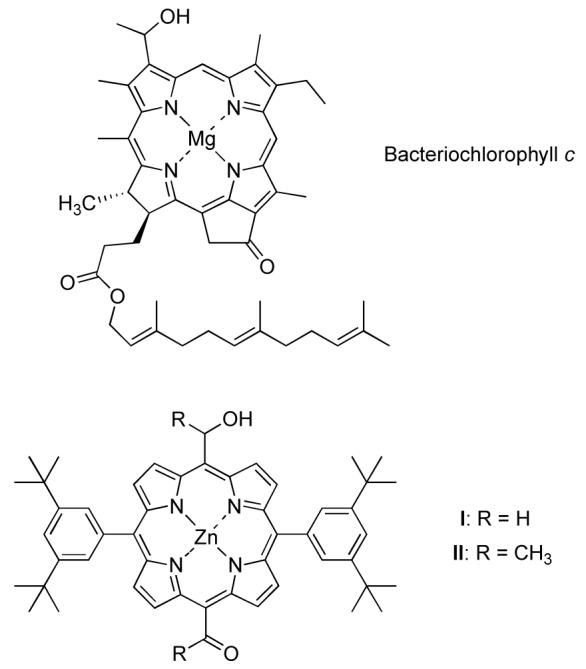
For the self-assembly of porphyrins in a manner analogous to that of bacteriochlorophyll c,6,7 attachment of coordinative groups directly to the porphyrin meso positions provided a viable architecture8-10 whereas the same groups at the p-positions of meso-aryl groups failed.11
To create porphyrinic architectures suitable for these and many other types of fundamental studies, we have begun developing motifs and synthetic routes to access substituted macrocycles that generally contain no β-substituents and few if any meso-aryl substituents. As one example, a symmetrically branched alkyl moiety (“swallowtail”) bearing phosphonate or phosphate groups at the termini has been introduced and found to impart high water solubility to porphyrins.12 As a second example, a new route to the fully unsubstituted porphine13 enables this core macrocycle to be used in substitution chemistry. In chlorin chemistry, we have developed routes to sparsely substituted chlorins14 so that the effects of auxochromic groups (formyl, acetyl, vinyl, ethynyl, aryl)15,16 can be clearly delineated. Senge has developed novel routes to porphyrins that are sparsely substituted and where the substituents are one-carbon entities.17,18
Porphyrins bearing diverse one-carbon substituents and derivatives (e.g., formyl,17-32 carboxy,2,20,33 alkoxycarbonyl,2,29,34,35 amide,2 hydroxymethyl,4,20,23,24,27,29,30 alkoxymethyl,28 N-alkylaminomethyl,28,36 imino,20,21,28,37 trifluoromethyl,2,3,38 cyano,20,31,37,39-41 nitrile-oxide,42 amidino31) have been examined and serve a broad range of applications. A common procedure to access porphyrins bearing many types of one-carbon oxygenic substituents is to selectively formylate one meso position followed by conversion of the formyl group to other one-carbon units such as hydroxymethyl,8,23,24 carboxy,33 oxime,20 etc. Although selective Vilsmeier formylation at one porphyrin meso position proceeds in high yield,17,21,22,25,27,29 the method is ineffective when the starting porphyrin is unsymmetrical and has multiple reactive positions.
Statistical approaches have often been employed to prepare meso-substituted porphyrins that bear two different one-carbon oxygenic substituents. Examples are provided by the intriguing model porphyrins I and II prepared to mimic the self-assembling features of bacteriochlorophyll c; the porphyrins contain α-hydroxyalkyl and acyl groups disposed at two trans meso positions and solubilizing 3,5-di-tert-butylphenyl groups at the other two meso positions (Chart 1). The choice of meso-substituents in the model compound (versus β-substituents in the biological target) stems from the ostensibly more facile synthetic methodology for introducing meso versus β-substituents in the porphyrin macrocycle. However, the synthesis of I entailed preparation of 5,15-bis(3,5-di-tert-butylphenyl)porphyrin followed by copper metalation, diformylation, copper demetalation, and statistical reduction to achieve the hydroxymethyl and formyl substituent pattern.8 The synthesis of II entailed treatment of 5,15-bis(3,5-di-tert-butylphenyl)porphyrin to meso-bromination, palladium-mediated ethynylation, hydration to form the diacetylporphyrin, and statistical reduction to obtain the α-hydroxyethyl and acetyl substituent pattern.8 These unavoidably difficult syntheses point up the limitations of existing methodology for the placement of small oxygenic functional groups close to the porphyrin perimeter, particularly in the presence of otherwise identical groups in distinct oxidation states.
Chart 1.
All of the aforementioned studies and applications would benefit from efficient synthetic access to porphyrinic analogues bearing a broad range of one-carbon substituents. One-carbon oxygenic substituents (hydroxymethyl, aldehyde, ketone and ester) offer low molecular weight, small size, limited hydrophobicity, and the capacity for further synthetic elaboration. More recent approaches to such porphyrins have employed 5-substituted dipyrromethanes bearing one-carbon substituents (or protected variants) such as ethoxycarbonyl,2 1,3-dithian-2-yl,19 1,3-dithiolan-2-yl,32 5,5-dimethyl-1,3-dioxan-2-yl,32 hydroxymethyl,4 S-acetylthiomethyl,4 and Se-acetylselenomethyl.4 We have extended these approaches to the more general problem of introducing multiple one-carbon substituents at the porphyrin meso positions.
In this paper, we describe our studies concerning the synthesis of A4-, trans-A2-, trans-A2B2-, trans-AB- and trans-AB2C-porphyrins bearing primarily hydroxymethyl groups, but also alkoxycarbonyl and formyl groups at meso positions. The general synthetic approach relies on dipyrromethane building blocks that bear various one-carbon units. Key building blocks in this regard include the tert-butyldimethylsilyl (TBDMS)-protected 5-hydroxymethyldipyrromethane and S-2-pyridyl hydroxythioacetate, which allow introduction of protected hydroxymethyl groups at designated meso positions. TBDMS protection was employed to avoid possible side reactions by the hydroxymethyl group during the synthesis. A long-term goal is to extend these approaches established with porphyrins to hydroporphyrins such as chlorins and bacteriochlorins.
2. Results and discussion
2.1. Synthesis of precursors for porphyrins
2.1.1. Dipyrromethanes bearing one-carbon substituents
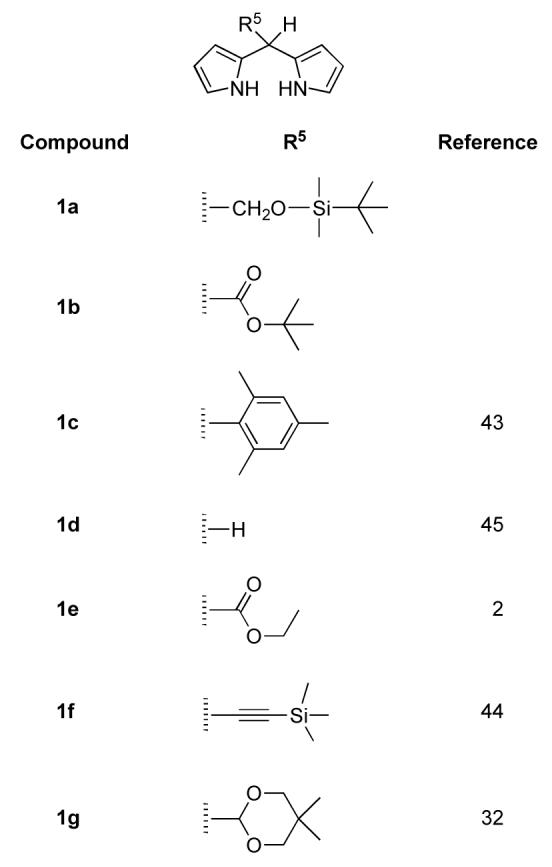
Seven dipyrromethanes (1a-g) were employed in the porphyrin syntheses described herein (Chart 2). Four 5-substituted dipyrromethanes (1a, 1b, 1e, and 1g) bear various one-carbon groups in protected form. Dipyrromethanes 1a and 1b are new compounds. The new dipyrromethanes bear a tert-butyldimethylsilyl (TBDMS) protected hydroxymethyl group (1a) or a tert-butoxycarbonyl group (1b). The 5-ethoxycarbonyldipyrromethane (1e)2 and acetal-dipyrromethane (1g)32 have been reported previously. The remaining dipyrromethanes (1c,43,44 1d,44,45 1f44) also are known compounds.
Chart 2.
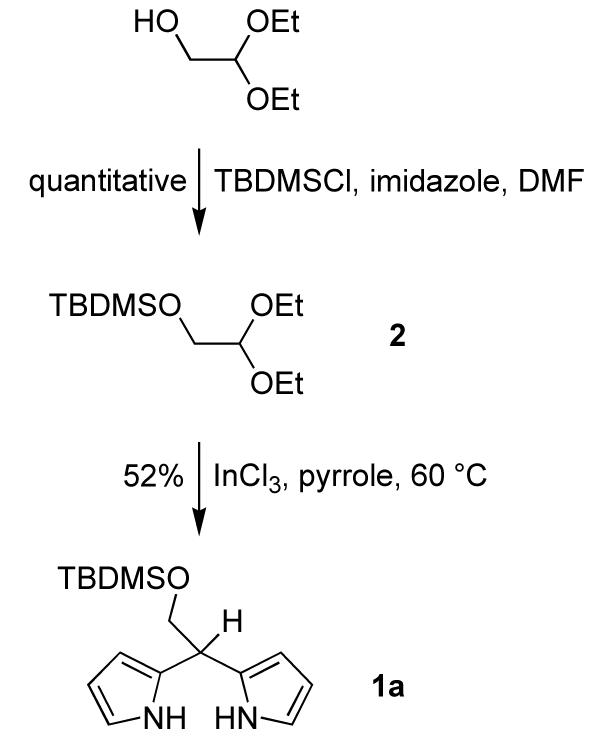
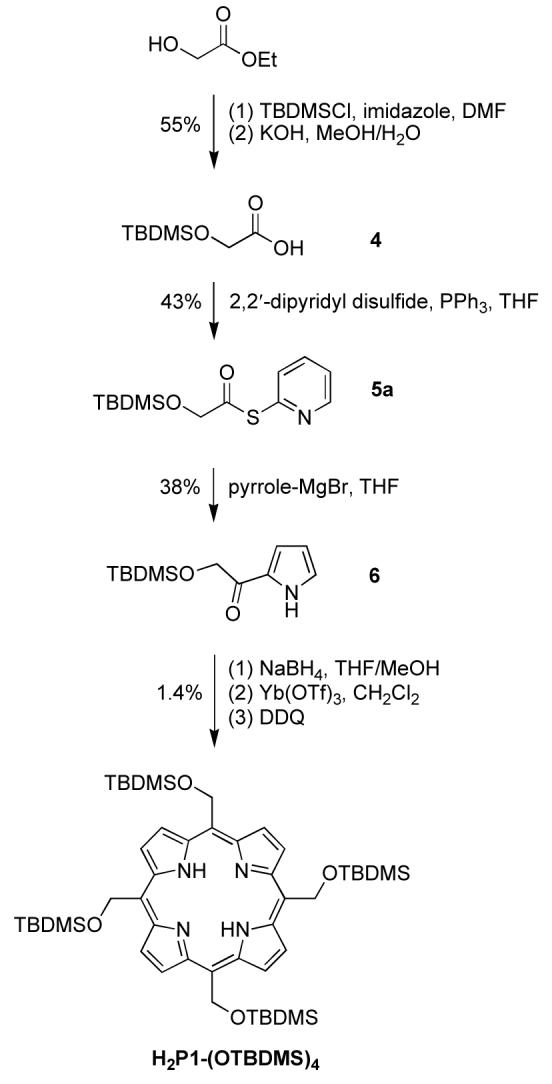
The synthesis of 1a started with the protection of glycolaldehyde diethyl acetal with tert-butyldimethylsilyl chloride (TBDMSCl). The TBDPS-protected analogue is known.46 The protection reaction yielded diethyl acetal 2 quantitatively. The reaction of acetal 2 and pyrrole was carried out under the same conditions employed for aldehyde and pyrrole condensations44 except that higher temperature was required. Thus, a sample of 2 was treated with pyrrole and InCl3 at 60 °C to afford 5-(tert-butyldimethylsiloxymethyl)dipyrromethane (1a) as a colorless oil in 52% yield (Scheme 1).
Scheme 1.
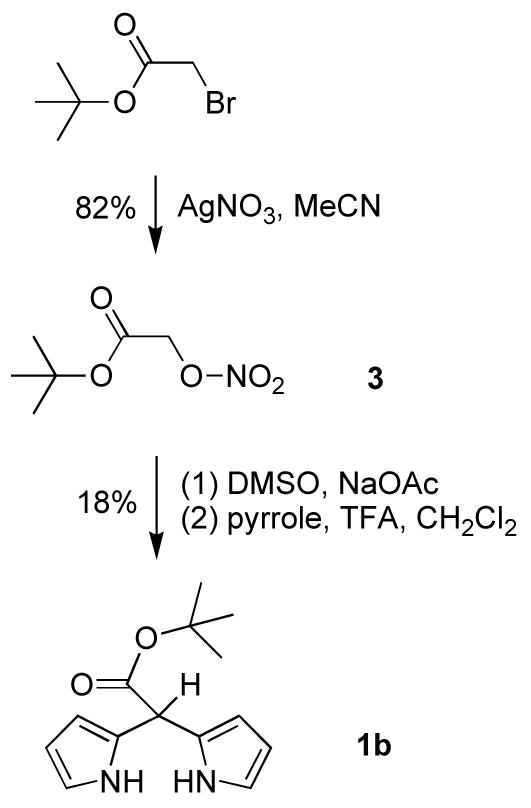
Compared to 5-ethoxycarbonyldipyrromethane (1e), 5-tert-butoxycarbonyldipyrromethane (1b) possesses a more base-resistant ester moiety due to the bulkiness of the tert-butyl group. tert-Butyl bromoacetate reacted47 with excess silver nitrate in acetonitrile to afford nitrate ester 3 in 82% yield (Scheme 2). Treatment of 3 with sodium acetate in DMSO gave tert-butyl glyoxalate, which in crude form was condensed2 with pyrrole in the presence of TFA in CH2Cl2 to yield tert-butyl ester dipyrromethane 1b in 18% yield (from 3).
Scheme 2.
2.1.2. A Mukaiyama reagent bearing a hydroxymethyl moiety
Mukaiyama reagents (S-2-pyridyl thioacyl compounds) are valuable species in porphyrin synthesis. The acylation of a dipyrromethane with a Mukaiyama reagent selectively introduces the desired acyl group at the 1-position of the dipyrromethane.48 An attempt to synthesize S-2-pyridyl hydroxythioacetate without TBDMS protection was not successful. The synthesis of TBDMS-protected glycolic acid 4 from glycolic acid and TBDMSCl has been reported but with limited data.49 However, in our hands the yield of the reaction varied and sometimes was lower than 10%. Alternatively, ethyl glycolate was treated with TBDMSCl to afford the TBDMS-protected glycolate (Scheme 3). Without purification, the ester was hydrolyzed to afford TBDMS-protected glycolic acid 4 in 55% yield, a known compound with limited characterization data.49,50 Reaction51 of 4 with 2,2′-dipyridyl disulfide gave the corresponding pyridyl thioester 5a in 43% yield.
Scheme 3.
2.2. Syntheses of hydroxymethylporphyrins
2.2.1. A4-Porphyrins with four hydroxymethyl groups
Our initial approach to meso-tetrakis(tert-butyldimethylsiloxymethyl)porphyrin [H2P1-(OTBDMS)4] relied on condensation of acetal 2 and pyrrole. This approach failed to give a workable yield of porphyrin. An alternative approach relied on a more advanced intermediate, namely the self-condensation of a pyrrole-carbinol bearing a protected hydroxymethyl group. Treatment51 of pyrrole at -78 °C with EtMgBr followed by Mukaiyama reagent 5a gave TBDMS-protected α-hydroxyacetylpyrrole 6 in 38% yield. Compound 6 was then reduced by NaBH4 and treated with the catalyst52 Yb(OTf)3 followed by DDQ oxidation to afford TBDMS-protected tetrakis(hydroxymethyl)porphyrin H2P1-(OH)4 in 1.4% yield.
The low yield prompted examination of an even more advanced intermediate, the dipyrromethane-1-carbinol bearing two protected hydroxymethyl groups. Thus, dipyrromethane 1a was treated with EtMgBr followed by Mukaiyama reagent 5a with a known procedure48 to give 1-acyldipyrromethane 7 in 62% yield (Scheme 4). Acyldipyrromethane 7 was reduced by NaBH4 to give the dipyrromethane-1-carbinol, which upon self-condensation and oxidation gave the desired A4-porphyrin H2P1-(OTBDMS)4 in 8% yield. Treatment of the latter with Zn(OAc)2 gave the zinc chelate ZnP1-(OTBDMS)4. The TBDMS protecting groups were removed by treatment with TBAF to give 5,10,15,20-tetrakis(hydroxymethyl)porphyrin [ZnP1-(OH)4] in 63% yield.
Scheme 4.
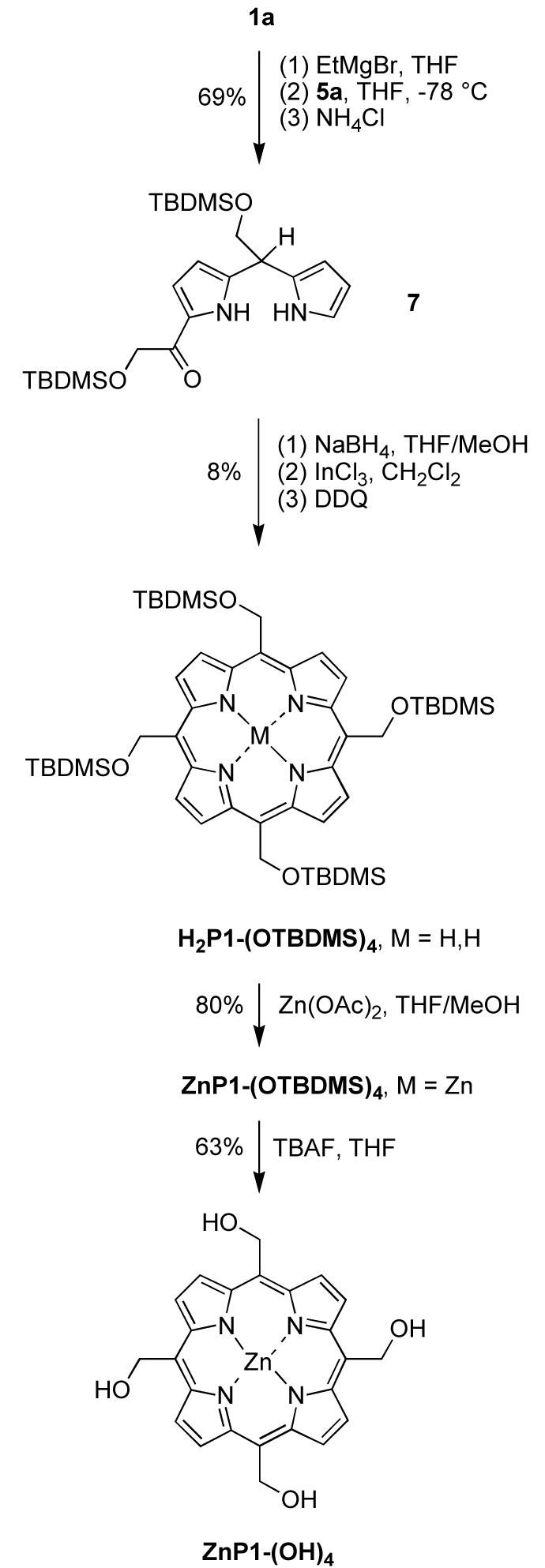
Porphyrin ZnP1-(OH)4 was difficult to purify by chromatography due to its poor solubility in organic solvents such as CH2Cl2, ethyl ether, MeOH and THF. The reaction mixture from the deprotection reaction was washed with water, acetone and THF. The structure of the resulting purple solid was confirmed by mass spectrometry and NMR spectroscopy (in DMSO-d6). The molecule ion of ZnP1-(OH)4 was observed by laser-desorption mass spectrometry (LD-MS) with 1,4-bis(5-phenyloxazol-2-yl)benzene (POPOP) as the matrix. Measurement without a matrix, which typically affords excellent results with porphyrins,53 or with other matrixes (dithranol, α-cyano-4-hydroxycinnamic acid, 3,5-dihydroxybenzoic acid) failed to display the expected mass signal. High-resolution experiments by electrospray ionization mass spectrometry (ESI-MS) and fast atom bombardment mass spectrometry (FAB-MS) were not successful.
2.2.2. trans-A2- and trans-A2B2-Porphyrins with two hydroxymethyl groups
Trans-A2-porphyrins or A2B2-porphyrins can be synthesized by the condensation of a dipyrromethane and an aldehyde.54 However, alkyl-substituted dipyrromethanes are somewhat susceptible to scrambling.54-56 An alternative synthetic route to a trans-A2- or A2B2-porphyrin is via a 1-acyldipyrromethane.48 Two 1-acyldipyrromethanes bearing one hydroxymethyl moiety were obtained from the corresponding dipyrromethanes in a manner similar to that of the synthesis for H2P1-(OTBDMS)4. 5-Mesityldipyrromethane (1c) or unsubstituted dipyrromethane (1d) reacted48 with EtMgBr and pyridyl thioester 5a to afford TBDMS-protected 1-acyldipyrromethane 8 or 9 in 74% or 85% yield, respectively (Scheme 5). 1-Acyl-5-mesityldipyrromethane 8 was reduced by NaBH4 and the resulting dipyrromethane-1-carbinol was self-condensed48,52,57 in the presence of InCl3 followed by oxidation with DDQ to afford TBDMS-protected bis(hydroxymethyl)porphyrin H2P2-(OTBDMS)2 in 13% yield (Scheme 5). Cleavage of the TBDMS group by TBAF58 afforded porphyrin H2P2-(OH)2 quantitatively.
Scheme 5.
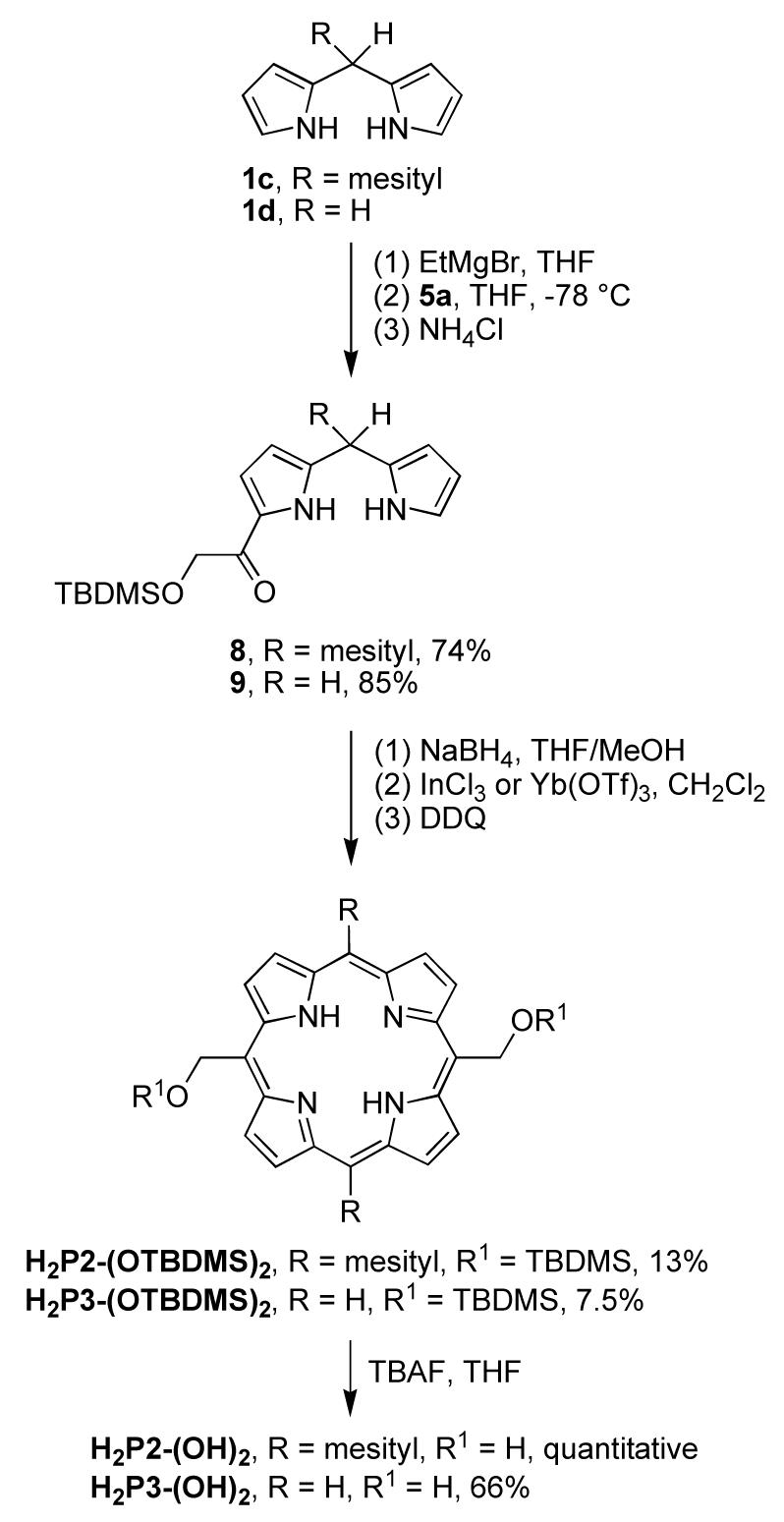
A trans-A2-porphyrin was synthesized in a similar manner. The self-condensation of the carbinol derived from 1-acyldipyrromethane 9 was catalyzed by Yb(OTf)3 instead of InCl3. The protected bis(hydroxymethyl)porphyrin H2P3-(OTBDMS)2 was obtained in 7.5% yield. Deprotection with TBAF afforded the trans-A2-porphyrin H2P3-(OH)2 in 66% yield. Similar to ZnP1-(OH)4, H2P3-(OH)2 also showed poor solubility and was purified by washing with water, acetone and THF.
We also explored a route in which 1-acyl-5-mesityldipyrromethane 8 was treated with TBAF to remove the TBDMS group prior to porphyrin formation. The resulting 1-(α-hydroxyacetyl)dipyrromethane 8′ was subjected to reduction, and the resulting unprotected hydroxymethyl-substituted dipyrromethane-1-carbinol was treated to self-condensation and oxidation. The desired trans-A2B2-porphyrin H2P2-(OH)2 was obtained along with scrambled porphyrin species (18% total spectroscopic yield). This result establishes the importance of protecting the hydroxymethyl group during the porphyrin-forming process.
2.2.3. trans-AB-Porphyrins bearing one or two one-carbon substituents
Trans-AB-porphyrins present a compact architecture for use in various applications or further synthetic elaboration. Two meso positions without substituents can be derivatized for a wide variety of applications. We recently described the synthesis of trans-AB-porphyrins via condensation of a 1,9-bis(N,N-dimethylaminomethyl)dipyrromethane and a dipyrromethane.59 By following such a method, three trans-AB-porphyrins with distinct substitution patterns were synthesized (Scheme 6). The substituents include hydroxymethyl, ethoxycarbonyl, tert-butoxycarbonyl, and ethynyl groups.
Scheme 6.

Thus, 1,9-bis(N,N-dimethylaminomethyl)dipyrromethanes were synthesized from the corresponding dipyrromethanes (1b, 1e and 1f) by treatment with Eschenmoser’s reagent.60 The resulting crude products were used in the porphyrin syntheses without further purification. The condensation reaction was performed in refluxing ethanol containing Zn(OAc)2 followed by oxidation with DDQ. Zinc porphyrins ZnP4-OTBDMS, ZnP5-OTBDMS and ZnP6-OTBDMS were purified by chromatography and obtained in pure form but in low yield. The zinc porphyrins were then treated with TBAF to remove the TBDMS group, affording hydroxymethylporphyrins ZnP4-OH, ZnP5-OH and ZnP6-OH. Porphyrin ZnP6-OH contains a free hydroxymethyl group and an ethynyl group owing to removal of both the TBDMS and TMS groups, respectively. The molecule ion of ZnP6-OH was observed by LD-MS with POPOP as the matrix. Similar to ZnP1-(OH)4, high-resolution mass measurements for ZnP6-OH were not successful.
2.2.4. trans-AB2C-Porphyrins bearing one or two one-carbon substituents
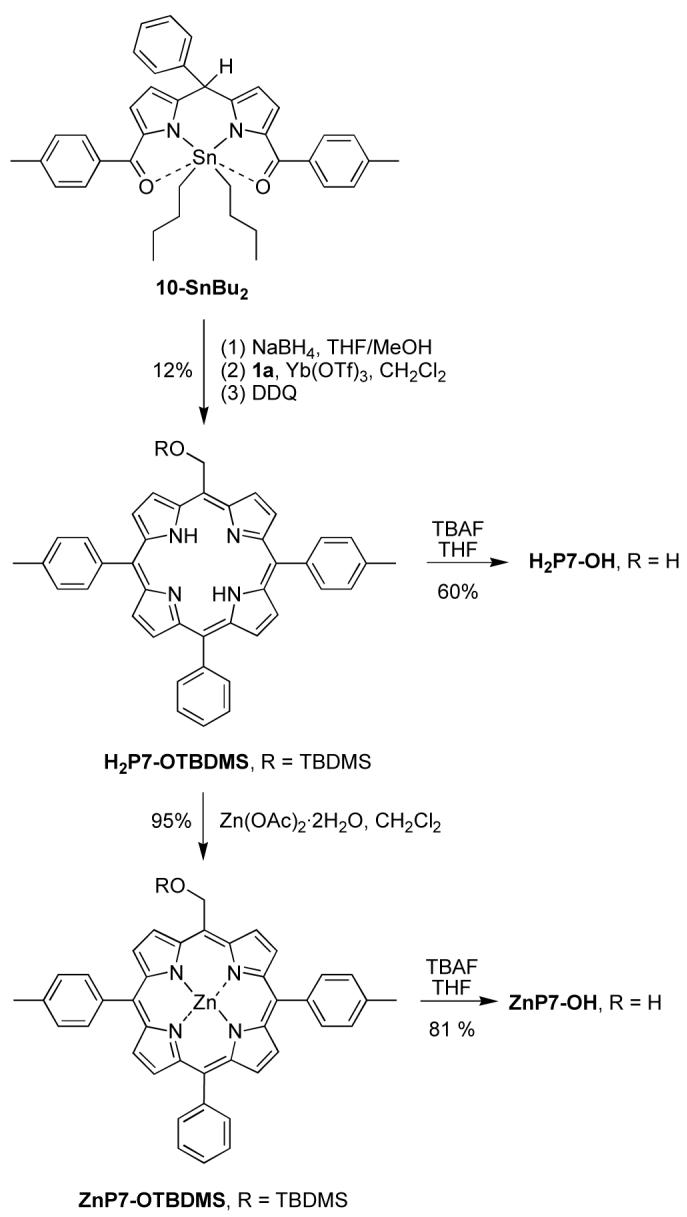
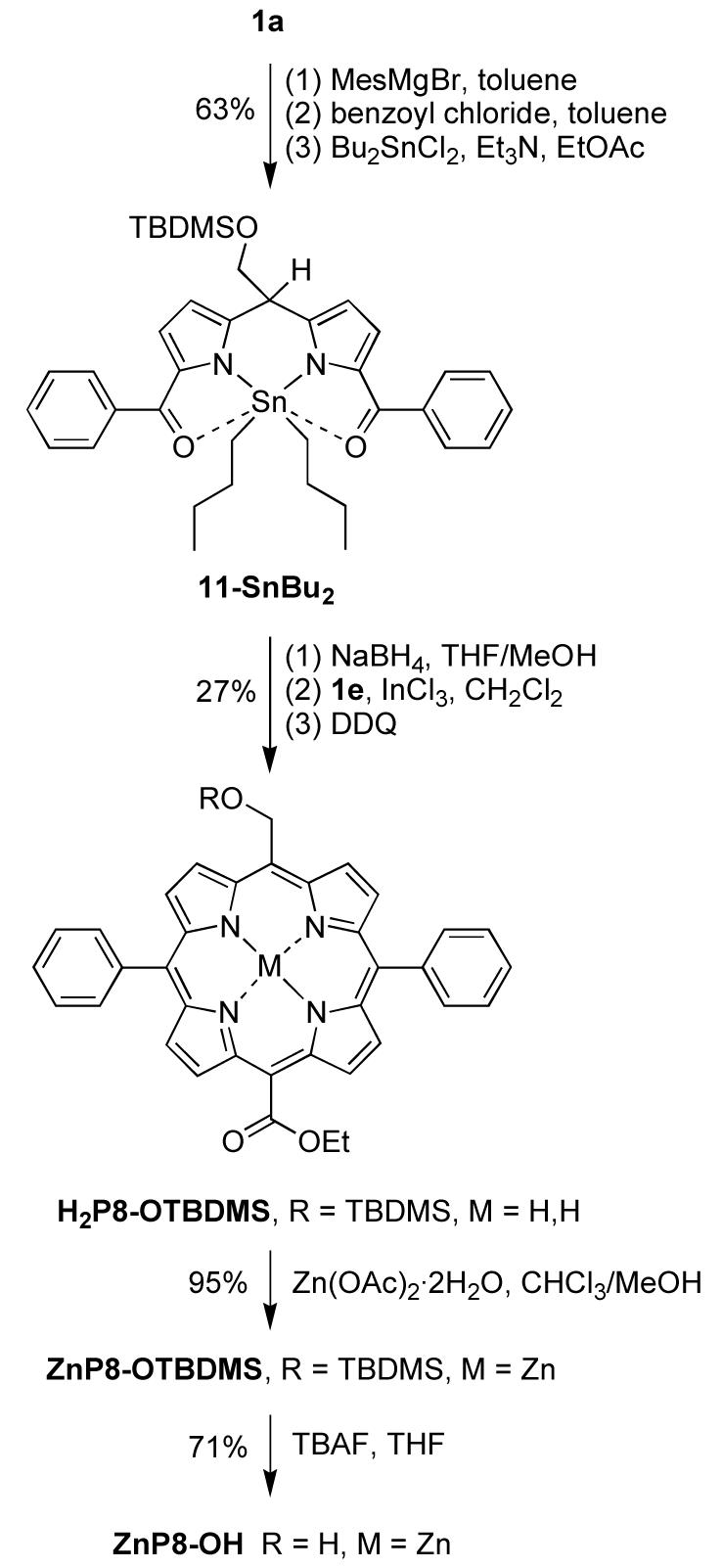
Three trans-AB2C-porphyrins with different patterns of one-carbon substitution were synthesized following literature procedures for the condensation of a dipyrromethane-1,9-dicarbinol and a dipyrromethane.52,61 A porphyrin with one hydroxymethyl and three aryl groups was first synthesized. Tin complex 10-SnBu262 was reduced by NaBH4. The resulting dipyrromethane-1,9-dicarbinol was condensed with TBDMS-protected hydroxymethyldipyrromethane 1a to afford free base porphyrin H2P7-OTBDMS in 12% yield (Scheme 7). Treatment of H2P7-OTBDMS with Zn(OAc)2·2H2O afforded ZnP7-OTBDMS in 95% yield. The TBDMS group of H2P7-OTBDMS and ZnP7-OTBDMS was removed by treatment with TBAF to yield H2P7-OH and ZnP7-OH in 60% and 81% yield, respectively. The synthesis of H2P7-OH and ZnP7-OH was previously carried out with the unprotected 5-hydroxymethyldipyrromethane.4 Although the yields are comparable in the two routes, the presence of a protecting group opens access to synthetic routes that are not possible with the free hydroxy group. An example is provided for porphyrins bearing two different one-carbon groups, such as ZnP8-OH, which bears one hydroxymethyl group and one ester group at opposite meso positions.
Scheme 7.
The synthesis of ZnP8-OH entailed the acylation of dipyrromethane 1a (Scheme 8). Treatment of 1a with EtMgBr and benzoyl chloride followed by tin complexation62 with Bu2SnCl2 yielded tin complex 11-SnBu2 in 63% yield. The latter was then reduced by NaBH4. The resulting dipyrromethane-1,9-dicarbinol was condensed with ester-dipyrromethane 1e in the presence of InCl3 followed by oxidation with DDQ to afford the free base porphyrin H2P8-OTBDMS in 27% yield. Metalation gave zinc porphyrin ZnP7-OTBDMS in 95% yield, and deprotection with TBAF yielded ZnP7-OH in 71% yield.
Scheme 8.
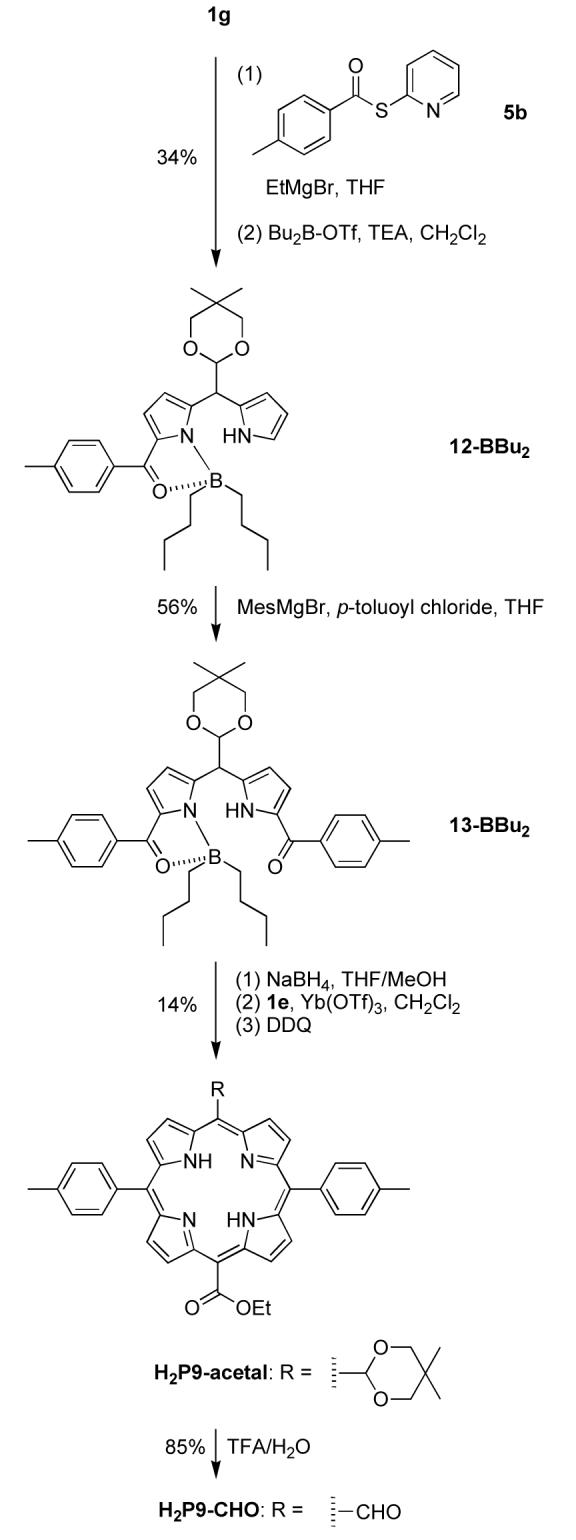
A porphyrin bearing a formyl and an ester group at trans-meso positions was synthesized from a 1,9-diacyl-5-acetaldipyrromethane and an ester-dipyrromethane (Scheme 9). The direct diacylation of the acetal-dipyrromethane 1g32 with EtMgBr and p-toluoyl chloride was not successful. We then tried to introduce the two 4-methylbenzoyl substituents in separate steps. Following known procedures,32,63,64 reaction of dipyrromethane 1g with EtMgBr followed by Mukaiyama reagent 5b48 gave the crude 1-p-toluoyl-5-(5,5-dimethyl-1,3-dioxan-2-yl)dipyrromethane, which upon dialkylboron complexation with Bu2B-OTf gave the boron complex 12-BBu2 in 34% yield. The boron complex 12-BBu2 was treated with MesMgBr and p-toluoyl chloride to form the 1,9-diacyldipyrromethane 13-BBu2 in 56% yield. The latter was reduced by NaBH4 and the resulting dipyrromethane-1,9-dicarbinol was condensed with ester-dipyrromethane 1e to afford acetal/ester-porphyrin H2P9-acetal. A small amount (∼1%) of aldehyde-porphyrin (from deprotection of H2P9-acetal) was also observed in the reaction mixture by LD-MS. The purified acetal/ester-porphyrin H2P9-acetal was obtained in 14% yield. Treatment of H2P9-acetal in the biphasic mixture of CH2Cl2 and TFA/H2O32,65 gave the aldehyde/ester-porphyrin H2P9-CHO in 85% yield.
Scheme 9.
2.3. Characterization
The porphyrins were generally characterized by 1H NMR and 13C NMR spectroscopy, laser-desorption mass spectrometry (LD-MS), high-resolution fast atom bombardment mass spectrometry (FAB-MS), and absorption spectroscopy. In some cases solubility limitations precluded 13C NMR spectroscopy or FAB-MS. The hydroxymethylporphyrins exhibited the expected resonances upon 1H NMR spectroscopy, including the doublet (6.82 to 7.29 ppm) for the methylene protons and the triplet (5.14 to 6.31 ppm) for the hydroxyl proton. One outlier was the resonance of the methylene at 6.18 ppm for trans-hydroxymethyl/ethynyl-porphyrin ZnP6-OTBDMS. Such multiplets were clearly observed when deuterated THF or DMSO was used as solvent, but were less distinguishable with CDCl3. The carbon of the hydroxymethyl group (TBDMS-protected or free) gave a characteristic resonance in the region 61 - 66 ppm upon 13C NMR spectroscopy. The porphyrins that contained an ethyl ester moiety also gave a resonance in the same region.
2.4. Solubility
Most of the TBDMS-protected hydroxymethylporphyrins are soluble in CH2Cl2. Two exceptions were ZnP4-OTBDMS and ZnP5-OTBDMS, which are zinc trans-AB-porphyrins bearing a protected hydroxymethyl group and an ester group. Such porphyrins were soluble in THF. In general, the presence of the ester group tends to decrease the solubility of the porphyrins in solvents such as CH2Cl2. The unprotected hydroxymethylporphyrins generally required a more polar solvent such as DMF or THF for dissolution. Indeed, ZnP8-OH, which bears a hydroxymethyl group, an ester group and an apical zinc site in a linear alignment, is soluble in THF but not in CH2Cl2. It has been reported that in bacteriochlorophyll c, the hydroxymethyl group, the carbonyl group, the central zinc ion and the linear alignment of the three elements are essential for self-assembly.6,7 The low solubility of porphyrins such as ZnP8-OH in less polar solvents likely stems from analogous self-assembly, as reported by Balaban and coworkers for compounds I and II.
3. Conclusions
A set of rational synthetic methods has been explored to gain access to meso-substituted porphyrins bearing distinct patterns of hydroxymethyl and related one-carbon oxygenic substituents. The patterns include A4-, trans-A2-, trans-A2B2-, trans-AB- and trans-AB2C-porphyrins where A is hydroxymethyl. For porphyrins bearing exclusively one-carbon units such as A4-, trans-AB- and trans-A2-porphyrins, the current rational methods afforded low yields (< 10%). On the other hand, trans-AB2C-porphyrins bearing two different one-carbon groups (hydroxymethyl/ester, formyl/ester) were obtained in somewhat better yields (14%-27%). While much remains to be done in porphyrin chemistry to increase yields in a global manner, the existing routes provide access in a rational manner to porphyrins bearing substituent patterns of interest across a range of fields encompassing light-harvesting, life sciences, and materials chemistry. The work presented herein also may provide the foundation for extension to the synthesis of chlorins and bacteriochlorins that bear similar substituents.
4. Experimental section
4.1. General methods
All 1H NMR spectra (400 MHz) and 13C NMR spectra (100 MHz) were recorded in CDCl3 unless noted otherwise. Mass spectra of porphyrins were obtained by high-resolution FAB-MS or by LD-MS, the latter without or with a matrix.53 Absorption spectra were collected in CH2Cl2 at room temperature unless noted otherwise. Melting points are uncorrected. Silica gel (40 μm average particle size) and alumina (80-200 mesh) were used for column chromatography. All reagents were used as received. Dry THF was obtained by distillation over Na/benzophenone. Room temperature was determined to be 21-22 °C using a calibrated thermometer.
4.2. Noncommercial compounds
Dipyrromethanes 1c,43 1d,45 1f,44 and 1g32 were prepared using a literature method that entails reaction of an aldehyde in 100 equiv of pyrrole containing a Lewis acid (e.g., InCl3).44 Dipyrromethane 1e,2 Mukaiyama reagent 5b,48 and the diacyldipyrromethane-tin complex 10-SnBu262 were prepared as described in the literature.
4.3. Synthesis of precursors
4.3.1. 5-(tert-Butyldimethylsiloxymethyl)dipyrromethane (1a)
Following a literature procedure44 with modification, a mixture of 2 (12.4 g, 50.0 mmol) and pyrrole (347 mL, 5.00 mol) was treated with InCl3 (1.11 g, 5.00 mmol) at 60 °C under argon for 2 h. The reaction was quenched with TEA (5 mL) and stirred for 15 min at room temperature. The reaction mixture was then concentrated. Chromatography [silica, hexanes/ethyl acetate (4:1)] yielded a light yellow oil (7.5 g, 52%): 1H NMR δ 0.04 (s, 6H), 0.91 (s, 9H), 4.06 (d, J = 5.2 Hz, 2H), 4.24 (t, J = 5.2 Hz, 1H), 5.99 (m, 2H), 6.12-6.17 (m, 2H), 6.69 (m, 2H), 8.40-8.60 (br, 2H); 13C NMR δ -5.3, 18.4, 26.2, 40.1, 67.7, 106.1, 108.3, 117.0, 131.8; ESI-TOF obsd 313.17076, calcd 313.17066 [(M + Na)+, M = C16H26N2OSi]; Anal. Calcd for C16H25N2OSi: C 66.16, H 9.02, N 9.64. Found: C 66.12, H 9.16, N 9.61.
4.3.2. 5-tert-Butoxycarbonyldipyrromethane (1b)
Following a literature procedure47 with modification, a solution of 3 (2.50 g, 14.1 mmol) in anhydrous DMSO (30 mL) was treated with anhydrous sodium acetate (1.00 g, 12.2 mmol) at room temperature for 20 min. The reaction mixture was poured into a mixture of brine and ice (150 mL). The mixture was extracted with ether (10 × 30 mL). The organic layers were combined, washed with saturated aqueous Na2CO3 and concentrated. The resulting oil was dissolved in CH2Cl2 and washed with brine. The organic layer was separated, dried (Na2SO4) and concentrated to yield crude tert-butyl glyoxalate as a thick colorless oil (1.36 g). The latter was dissolved in CH2Cl2 (50 mL). Pyrrole (17.4 mL, 250 mmol) was added to the solution. The mixture was degassed with argon for 10 min. TFA (77 μL, 1.0 mmol) was added. The mixture was stirred at room temperature under argon for 15 h. TEA (1 mL) was added to the mixture. Stirring was continued for 30 min. The reaction mixture was poured into CH2Cl2 (100 mL). The mixture was washed with water and brine, dried (Na2SO4) and concentrated. Chromatography [silica, hexanes/CH2Cl2 (1:1)] yielded a light yellow oil, which was contaminated with residual pyrrole. The crude sample was concentrated and entrained with hexanes (3 × 20 mL). Volatile materials were removed to yield an off-white powder (0.64 g, 18%): mp 75-77 °C; 1H NMR δ 1.49 (s, 9H), 4.99 (s, 1H), 6.06-6.09 (m, 2H), 6.13-6.16 (m, 2H), 6.70-6.72 (m, 2H), 8.44 (br, 2H); 13C NMR δ 28.2, 44.8, 82.5, 107.0, 108.6, 118.0, 127.5, 170.9; Anal. Calcd for C14H18N2O2: C, 68.27; H, 7.37; N 11.37. Found: C, 68.30; H, 7.40, N 11.31.
4.3.3. 1-(tert-Butyldimethylsiloxy)-2,2-diethoxyethane (2)
Following a literature procedure46 with modification, a solution of glycolaldehyde diethyl acetal (6.70 g, 50.0 mmol) in DMF (150 mL) was treated with tert-butyldimethylsilyl chloride (18.8 g, 125 mmol) and imidazole (17.0 g, 250 mmol) at room temperature under argon for 14 h. The reaction mixture was poured into water (5 mL). The mixture was extracted with ethyl acetate. The organic layer was washed with water and brine, dried (Na2SO4) and concentrated to yield a light yellow liquid (12.4 g, quantitative): 1H NMR δ 0.07 (s, 6H), 0.90 (s, 9H), 1.22 (t, J = 7.2 Hz, 6H), 3.56-3.76 (m, 4H), 3.63 (d, J = 5.2 Hz, 2H), 4.49 (t, J = 5.2 Hz, 1H); 13C NMR δ -5.1, 15.6, 18.6, 26.1, 63.0, 64.8, 103.2; ESI-MS obsd 271.16894, calcd 271.16999 [(M + Na)+, M = C12H28NO3Si].
4.3.4. tert-Butoxycarbonylmethyl nitrate (3)
Following a literature procedure,47 a solution of tert-butyl bromoacetate (4.60 g, 23.6 mmol) in anhydrous acetonitrile (20 mL) was treated with AgNO3 (7.31 g, 47.2 mmol) at room temperature in the dark for 48 h. The solvent was removed. The resulting residue was extracted with Et2O (3 × 50 mL). The Et2O extracts were combined, washed with water and brine, dried (Na2SO4) and concentrated to yield a colorless oil (3.4 g, 82%): 1H NMR δ 1.49 (s, 9H), 4.77 (s, 2H); 13C NMR δ 28.1, 67.8, 84.1, 165.0; Anal. Calcd for C6H11NO5: C 40.69, H 6.26, N 7.91. Found: C 40.95, H 6.41, N 7.80.
4.3.5. (tert-Butyldimethylsiloxy)acetic acid (4)
Following literature procedures,49,50 a solution of ethyl glycolate (5.21 g, 50.0 mmol) and imidazole (4.08 g, 60.0 mmol) in DMF (50 mL) was treated with tert-butyldimethylsilyl chloride (9.04 g, 60.0 mmol) at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred for 1 h. Water (50 mL) was added to the reaction mixture. The mixture was extracted with ether (2 × 100 mL). The combined organic layer was washed with water (2 × 100 mL), dried (Na2SO4) and concentrated in vacuo to yield a viscous liquid. The resulting ester was dissolved in 30 mL THF. A solution of KOH (2.95 g, 52.5 mmol) in MeOH/water (1:2, 18 mL) was slowly added to the solution at -10 °C. The reaction mixture was allowed to warm up to 5 °C over 30 min and diluted with 175 mL of water and ether (125 mL). The mixture was acidified with aqueous HCl (5.5 mL, 55 mmol) at 0 °C and extracted with ether (3 × 125 mL). The combined organic layer was washed with water, dried (Na2SO4) and concentrated to yield a white solid (5.2 g, 55%): mp 48-49 °C; 1H NMR (300 MHz) δ 0.15 (s, 6H), 0.94 (s, 9H), 4.22 (s, 2H), the carboxylic acid proton was not observed; 13C NMR δ -5.3, 18.5, 25.9, 61.6, 175.3; Anal. calcd. for C8H18O3Si: C, 50.49; H, 9.53. Found: C, 50.40; H, 9.56.
4.3.6. S-2-Pyridyl (tert-butyldimethylsiloxy)thioacetate (5a)
Following a literature procedure,51 a solution of 4 (3.81 g, 20.0 mmol) in anhydrous THF (40 mL) was treated with 2,2′-dipyridyl disulfide (8.80 g, 39.9 mmol) and triphenylphosphine (10.5 g, 40.0 mmol) at room temperature under argon for 24 h. The reaction mixture was concentrated. Chromatography [silica, hexanes/ethyl acetate (9:1) then hexanes/ethyl acetate (3:2)] yielded a yellow solid (2.4 g, 43%): mp 50-52 °C; 1H NMR δ 0.18 (s, 6H), 0.99 (s, 9H), 4.38 (s, 2H), 7.28-7.34 (m, 1H), 7.58-7.62 (m, 1H), 7.72-7.78 (m, 1H), 8.64-8.68 (m, 1H); 13C NMR δ -5.6, 18.2, 25.7, 68.9, 123.4, 130.6, 137.1, 150.5, 151.6, 200.3; FAB-MS obsd 284.1126, calcd 284.1141 [(M + H)+, M = C13H21NO2SSi].
4.3.7. 2-(tert-Butyldimethylsiloxyacetyl)pyrrole (6)
Following a general procedure,51 a solution of EtMgBr (1.0 M solution in THF, 17 mL, 17 mmol) was slowly added to a stirred solution of pyrrole (1.17 mL) in dry THF (17.2 mL) under Ar. The mixture was stirred at room temperature for 10 min and then cooled to -78 °C. A solution of 5a (2.41 g, 8.49 mmol) in THF (8.5 mL) was quickly added to the mixture. The reaction mixture was stirred at -78 °C for 20 min and quenched with saturated aqueous NH4Cl. The mixture was extracted with CH2Cl2. The organic layer was separated, washed with water, dried (Na2SO4) and concentrated. Chromatography [silica, hexanes/CH2Cl2(1:4)] yielded a dark purple oil (0.77 g, 38%): 1H NMR δ 0.14 (s, 6H), 0.96 (s, 9H), 4.72 (s, 2H), 6.27-6.29 (m, 1H), 7.06-7.08 (m, 2H), 10.20-10.60 (br s, 1H); 13C NMR δ -5.2, 18.6, 26.0, 67.0, 110.7, 117.0, 125.5, 129.6 188.6; FAB-MS obsd. 240.1425, calcd 240.1420 [(M + H)+, M = C12H21NO2Si]; Anal. calcd. for C12H21NO2Si: C, 60.21; H, 8.84; N, 5.85. Found: C, 60.26; H, 8.81; N, 5.84.
4.3.8. 1-[(tert-Butyldimethylsiloxy)acetyl]-5-(tert-butyldimethylsiloxymethyl)dipyrromethane (7)
Following a literature procedure,48 a solution of 1a (265 mg, 0.914 mmol) in dry THF (1 mL) was treated with EtMgBr (1.0 M in THF, 2.28 mL, 2.28 mmol) at room temperature under argon for 15 min. The reaction mixture was cooled to -78 °C. A solution of 5a (285 mg, 1.01 mmol) in dry THF (1 mL) was slowly added. The reaction mixture was stirred at -78 °C for 10 min, and then allowed to warm to room temperature. Stirring was continued for 45 min. Saturated aqueous NH4Cl solution was added. The mixture was extracted with ethyl acetate. The organic layer was separated, dried (Na2SO4) and concentrated. Chromatography [silica, CH2Cl2/ethyl acetate (49:1)] yielded a viscous yellow oil. The oil turned to a yellow solid upon standing (290 mg, 69%): mp 121-122 °C (blackened at 118 °C); 1H NMR (300 MHz) δ 0.03 (s, 6H), 0.11 (s, 6H), 0.90 (s, 9H), 0.93 (s, 9H), 4.05-4.10 (m, 2H), 4.41-4.47 (m, 1H), 4.61 (s, 2H), 5.96-6.02 (m, 1H), 6.02-6.08 (m, 1H), 6.11-6.18 (m, 1H), 6.67-6.74 (m, 1H), 6.95-7.02 (m, 1H), 8.65 (br, 1H), 9.63 (br, 1H); 13C NMR (75 MHz) δ -5.4, -5.1, 18.4, 18.7, 26.1, 26.2, 40.5, 67.1, 67.6, 106.7, 108.6, 109.3, 117.3, 117.5, 129.4, 130.2, 139.6, 187.6; Anal. calcd for C24H42N4O3Si2: C, 62.29; H, 9.15; N, 6.05. Found: C, 62.33; H, 9.28, N, 6.01.
4.3.9. 1-[(tert-Butyldimethylsiloxy)acetyl]-5-mesityldipyrromethane (8)
Following the procedure for 7, the reaction of 1c (630 mg, 2.38 mmol) and Mukaiyama reagent 5a (754 mg, 2.66 mmol) followed by chromatography [silica, CH2Cl2 then CH2Cl2/ethyl acetate (9:1)] yielded a brown solid (0.77 g, 74%): mp 40-42 °C; 1H NMR δ 0.10 (s, 6H), 0.91 (s, 9H), 2.05 (s, 6H), 2.28 (s, 3H), 4.60 (s, 2H), 5.90 (s, 1H), 6.06-6.10 (m, 2H), 6.18-6.21 (m, 1H), 6.65-6.68 (m, 1H) 6.88 (s, 2H), 7.02-7.05 (m, 1H), 7.74-7.84 (br, 1H), 9.06-9.22 (br, 1H); 13C NMR δ -5.4, 18.4, 20.6, 20.8, 25.6, 25.8, 38.5, 67.3, 107.1, 108.9, 109.6, 116.8, 117.7, 128.9, 130.5, 133.0, 137.2, 137.4, 140.0, 187.8; Anal. calcd for C26H36N2O2Si: C, 71.51; H, 8.31; N, 6.42. Found: C, 71.59; H, 8.09; N, 6.25.
4.3.10. 1-(tert-Butyldimethylsiloxy)acetyldipyrromethane (9)
Following the procedure for 7, the reaction of 1d (0.29 g, 2.0 mmol) and Mukaiyama reagent 5a (0.62 g, 2.2 mmol) followed by chromatography [silica, CH2Cl2 then CH2Cl2/ethyl acetate (10:1)] yielded a brown solid (0.54 g, 85%): mp 64-66 °C; 1H NMR δ 0.1 (s, 6H), 0.94 (s, 9H), 4.10 (s, 2H), 4.75 (s, 2H), 5.99-6.02 (m, 1H), 6.10-6.13 (m, 2H), 6.61-6.64 (m, 1H), 6.96-6.99 (m, 1H), 9.15-9.25 (br s, 1H), 10.50-10.60 (br s, 1H); 13C NMR δ -5.0, 19.0, 26.3, 66.2, 106.2, 106.3, 108.4, 109.8, 109.9, 117.4, 119.2, 128.4, 141.6, 188.0; Anal. calcd for C17H26N2O2Si; C, 64.11; H, 8.23; N, 8.80. Found: C, 64.10; H, 8.18; N, 8.69.
4.3.11. Dibutyl[5,10-dihydro-1,9-dibenzoyl-5-(tert-butyldimethylsiloxymethyl)dipyrrinato]tin(IV) (11-SnBu2)
Following a literature procedure,62 EtMgBr (1.0 M in THF, 10 mL, 10 mmol) was added dropwise to a solution of 1a (700 mg, 2.41 mmol) in toluene (5 mL) in an ice bath. The reaction was stirred at 0 °C for 30 min. Benzoyl chloride (590 μL, 5.06 mmol) was added dropwise, and the mixture was stirred at 0 °C for 1 h. The reaction mixture was poured into a mixture of saturated aqueous NH4Cl solution and ethyl acetate. The organic layer was separated, washed with brine, dried (Na2SO4) and concentrated. The resulting yellow oil was dissolved in ethyl acetate (20 mL) and treated with TEA (1 mL) and Bu2SnCl2 (733 mg, 2.41 mmol) at room temperature for 30 min. The reaction mixture was washed with water and brine, dried (Na2SO4) and concentrated. Chromatography [silica, hexanes/CH2Cl2 (1:2 with 0.5% TEA)] yielded a yellow viscous oil (1.1 g, 63%): 1H NMR δ -0.12 (s, 6H), 0.62 (t, J = 7.2 Hz, 3H), 0.85 (s, 9H), 0.94-1.40 (m, 3H), 1.12-1.20 (m, 2H), 1.30-1.38 (m, 4H), 1.56-1.64 (m, 4H), 1.76-1.82 (m, 2H), 3.79 (d, J = 7.2 Hz, 2H), 4.46 (d, J = 7.2 Hz, 1H), 6.45 (d, J = 4.0 Hz, 2H), 7.12 (d, J = 4.0 Hz, 2H), 7.48-7.60 (m, 6H), 7.88-7.94 (m, 4H); 13C NMR δ -5.6, 13.71, 13.86, 18.6, 23.2, 25.6, 25.9, 26.1, 26.8, 27.2, 27.8, 43.7, 71.3, 116.1, 123.9, 128.6, 129.2, 131.7, 136.3, 137.9, 150.1, 184.6; FAB-MS obsd 731.2721, calcd 730.2613 (C38H50N2O3SiSn); Anal. Calcd for C38H50N2O3SiSn: C, 62.69; H, 7.06; N, 3.83; Found: C, 62.55; H, 6.91; N, 3.84.
4.3.12. 10-(Dibutylboryl)-1-p-toluoyl-5-(5,5-dimethyl-1,3-dioxan-2-yl)dipyrromethane (12-BBu2)
Following a general procedure,63 a solution of 1g (260 mg, 1.00 mmol) in THF (1 mL) was treated with EtMgBr (1.0 M in THF, 2.5 mL, 2.5 mmol) at -78°C for 10 min. A solution of 5b (230 mg, 1.00 mmol) in THF (1 mL) was added dropwise to the reaction mixture. The mixture was stirred at room temperature for 10 min. Saturated aqueous NH4Cl solution was added, and the reaction mixture was extracted with ethyl acetate. The organic layer was washed with brine, dried (Na2SO4) and concentrated. The resulting solid was dissolved in CH2Cl2 (2 mL) and treated with TEA (335 μL, 2.40 mmol) and Bu2B-OTf (2.00 mL, 2.00 mmol) at room temperature for 30 min. The solvent was removed under reduced pressure. Chromatography [silica, hexanes/CH2Cl2 (1:4)] yielded a yellow-greenish oil (0.17 g, 34%): 1H NMR δ 0.50-1.30 (m, 24H), 2.43 (s, 3H), 3.40-3.48 (m, 2H), 3.63-3.69 (m, 2H), 4.54 (d, J = 3.6 Hz, 1H), 4.79 (d, J = 3.6 Hz, 1H), 5.88-5.93 (m, 1H), 6.07-6.12 (m, 1H), 6.64 (d, J = 4.4 Hz, 1H), 6.70-6.74 (m, 1H), 7.22 (d, J = 4.4 Hz, 1H), 7.32 (d, J = 8.0 Hz, 2H), 8.09 (d, J = 8.0 Hz, 2H), 8.95 (br, 1H); 13C NMR δ 14.3, 14.5, 21.9, 22.0, 22.8 (br), 23.2 (br), 23.28, 26.2, 26.4, 27.0, 27.2, 30.4, 42.9, 77.51, 77.56, 103.1, 108.03, 108.07, 117.3, 117.6, 119.8, 128.1, 128.4, 129.86, 129.90, 133.8, 145.1, 147.1, 175.4; LD-MS obsd 500.3; FAB-MS 503.3426, calcd 503.3445 [(M + H)+, M = C31H43BN2O3].
4.3.13. 10-(Dibutylboryl)-1,9-di-p-toluoyl-5-(5,5-dimethyl-1,3-dioxan-2-yl)dipyrromethane (13-BBu2)
Following a general precedure,64 a solution of 12-BBu2 (172 mg, 343 μmol) in THF (343 μL) was treated with MesMgBr (1.0 M in THF, 0.69 mL, 0.69 mmol) at room temperature for 5 min. A solution of p-toluoyl chloride (99.7 μL, 754 μmol) in THF was added dropwise to the mixture. The reaction was stirred at room temperature for 10 min. Saturated aqueous NH4Cl solution was added and the reaction mixture was extracted with ethyl acetate. The organic layer was washed with brine, dried (Na2SO4) and concentrated. Chromatography (silica, CH2Cl2) yielded an orange solid (0.12 g, 56%): 1H NMR δ 0.50-1.30 (m, 24H), 2.41 (s, 3H), 2.47 (s, 3H), 3.40-3.52 (m, 2H), 3.68-3.78 (m, 2H), 4.59 (d, J = 4.0 Hz, 1H), 4.83 (d, J = 4.0 Hz, 1H), 6.00-6.06 (m, 1H), 6.70 (d, J = 4.0 Hz, 1H), 6.73-6.78 (m, 1H), 7.22-7.28 (m, 3H), 7.35 (d, J = 8.0 Hz, 2H), 7.80 (d, J = 8.0 Hz, 2H), 8.12 (d, J = 8.0 Hz, 2H), 10.1 (br, 1H); 13C NMR δ 14.3, 14.5, 21.8, 21.9, 22.1, 22.7 (br), 23.1 (br), 23.3, 26.2, 26.4, 27.14, 27.21, 30.4, 43.0, 77.67, 77.76, 102.4, 111.4, 117.7, 119.3, 119.8, 128.0, 129.11, 129.24, 130.02, 130.04, 131.1, 134.1, 126.1, 136.4, 142.3, 145.1, 145.5, 176.4, 184.3; FAB-MS obsd 621.3864, calcd 621.3864 [(M + H)+, M = C39H49BN2O4].
4.4. Synthesis of porphyrins
4.4.1. Synthesis of A4-, trans-A2 or trans-A2B2-porphyrins from 1-acyldipyrromethanes, exemplified for 5,10,15,20-tetrakis(tert-butyldimethylsiloxymethyl)porphyrin [H2P1-(OTBDMS)4]
Following a literature procedure,48,57 a solution of 7 (206 mg, 0.446 mmol) in dry THF (9 mL) was treated with NaBH4 (421 mg, 11.2 mmol) at room temperature. A sample of MeOH (3 mL) was added dropwise. The reaction mixture was stirred for 1 h at room temperature. The reaction mixture was poured into saturated aqueous NH4Cl solution. The mixture was extracted with ethyl acetate. The organic layer was separated, washed with water, and dried (Na2SO4). The solvents were removed to yield the dipyrromethane-1-carbinol as a brownish oil. The dipyrromethane-1-carbinol was dissolved in CH2Cl2 (90 mL). The solution was treated with InCl3 (49.3 mg, 0.223 mmol) at room temperature under argon for 1 h. A sample of DDQ (75.9 mg, 0.335 mmol) was added. The reaction mixture was stirred for 15 min. A sample of TEA (5 mL) was added. The mixture was stirred for 5 min. The reaction mixture was concentrated. The resulting black solid was passed through a silica pad with CH2Cl2 as eluant. The filtrate was concentrated. Chromatography [silica, hexanes/CH2Cl2 (1:4)] yielded a red solid (15.0 mg, 8%): 1H NMR δ -3.07 to -3.03 (br, 2H), 0.19 (s, 24H), 0.96 (s, 36H), 6.97 (s, 8H), 9.65 (s, 8H); 13C NMR δ -4.4, 18.6, 26.2, 64.8, 115.8 (the α and β carbons were not observed presumably due to tautomerization); LD-MS obsd 886.3; FAB-MS obsd 886.5059, calcd 886.5100 (C49H78N4O4Si4); λabs 414, 512, 589, 644 nm.
4.4.2. Zinc metalation of a free-base porphyrin, exemplified for Zn(II)-5,10,15,20-tetrakis(tert-butyldimethylsiloxymethyl)porphyrin [ZnP1-(OTBDMS)4]
Following a literature procedure4 with modification, a solution of H2P1-(OTBDMS)4 (14.0 mg, 15.8 μmol) in CH2Cl2/MeOH (4:1, 5 mL) was treated with Zn(OAc)2 (14.4 mg, 78.9 μmol) at room temperature for 30 min. The reaction mixture was concentrated. Chromatography (silica, CH2Cl2) yielded a red solid (12.0 mg, 80%): 1H NMR (300 MHz) δ 0.16 (s, 24H), 0.96 (s, 36H), 6.89 (s, 8H), 9.61 (s, 8H); 13C NMR (75 MHz) δ -4.5, 18.7, 26.3, 64.9, 116.1, 129.6, 150.1; LD-MS obsd 948.3; FAB-MS obsd 948.4158, calcd 948.4235 (C49H76N4O4Si4Zn); λabs 416, 548 nm.
4.4.3. TBDMS removal with TBAF (method A), exemplified for Zn(II)-5,10,15,20-tetrakis(hydroxymethyl)porphyrin [ZnP1-(OH)4]
Following a literature procedure58 with modification, a solution of ZnP1-(OTBDMS)4 (14.9 mg, 15.7 μmol) in THF (3 mL) was treated with TBAF (1.0 M in THF, 157 μL, 157 μmol) at room temperature for 2 h. The reaction mixture was concentrated to dryness. The resulting dark solid was suspended in acetone and filtered through a Buchner filter. The filtered material was washed with water, THF and acetone. The resulting purple solid was washed from the filter by DMF. Removal of DMF under reduced pressure yielded a purple solid (4.9 mg, 63%): 1H NMR (300 MHz, DMSO-d6) δ 6.10 (t, J = 5.2 Hz, 4H), 6.84 (d, J = 5.2 Hz, 8H), 9.80 (s, 8H); 13C NMR (75 MHz, DMSO-d6) δ 62.6, 116.8, 129.5, 149.6; LD-MS (POPOP) obsd 492.5, calcd 492.1 (C24H20N4O4Zn). A solution of the title compound in DMSO was diluted in THF. The diluted solution was subjected to absorption measurement: λabs = 420, 556 nm.
4.4.4. 5,15-Bis(tert-butyldimethylsiloxymethyl)-10,20-dimesitylporphyrin [H2P2-(OTBDMS)2]
Following the procedure for H2P1-(OTBDMS)4, the self-condensation of the carbinol derived from 1-acyldipyrromethane 8 (88 mg, 0.20 mmol) followed by chromatography (silica, CH2Cl2) yielded a purple solid (11 mg, 13%): 1H NMR δ -2.90 to -2.70 (br, 2H), 0.21 (s, 12H), 0.94 (s, 18H), 1.82 (s, 12H), 2.65 (s, 6H), 6.93 (s, 4H), 7.29 (s, 4H), 8.76 (d, J = 4.7 Hz, 4H), 9.5 (d, J = 4.7 Hz, 4H); 13C NMR δ -4.5, 18.7, 21.8, 26.2, 29.9, 64.4, 115.6, 118.0, 127.9, 137.9, 138.9, 139.7 (the α and β carbons were not observed presumably due to tautomerization); LD-MS obsd 834.6; FAB-MS obsd 834.4772, calcd 834.4724 (C52H66N4O2Si2); λabs (toluene) 418, 514 nm; λem (toluene) 650, 725 nm.
4.4.5. TBDMS removal with TBAF (method B), exemplified for 5,15-bis(hydroxymethyl)-10,20-dimesitylporphyrin [H2P2-(OH)2]
Following a literature procedure58 with modification, a solution of H2P2-(OTBDMS)2 (7.0 mg, 8.3 μmol) in THF (2.0 mL) was treated with TBAF (1.0 M in THF, 80 μL, 80 μmol) at room temperature under argon for 9 h. The reaction was quenched with water. The mixture was extracted with CH2Cl2 and with ethyl acetate. The combined organic extract was dried (Na2SO4) and concentrated to yield the crude product as a solid. Chromatography [silica, CH2Cl2 then CH2Cl2/ethyl acetate (9:1)] yielded a purple solid (5.0 mg, quantitative): 1H NMR (THF-d8) δ -2.75 to -2.58 (br, 2H), 1.83 (s, 12H), 2.64 (s, 6H), 5.26 (t, J = 5.6 Hz, 2H), 6.82 (d, J = 5.6 Hz, 4H), 7.34 (s, 4H), 8.71 (d, J = 4.4 Hz, 4H), 9.67 (d, J = 4.4 Hz, 4H); LD-MS obsd 606.1; FAB-MS obsd 606.3017, calcd 606.2995 (C40H38N4O2); λabs 415, 513, 544, 589 nm; λem 645, 720 nm.
4.4.6. 5,15-Bis(tert-butyldimethylsiloxymethyl)porphyrin [H2P3-(OTBDMS)2]
Following the procedure for H2P1-(OTBDMS)4, the self-condensation of the carbinol derived from 9 (0.51 g, 1.6 mmol) followed by chromatography (silica, CH2Cl2) yielded a purple solid (36 mg, 7.5%): 1H NMR (300 MHz) δ -3.35 to -2.85 (br, 2H), 0.19 (s, 12H), 0.97 (s, 18H), 7.00 (s, 4H), 9.41 (d, J = 4.4 Hz, 4H), 9.69 (d, J = 4.4 Hz, 4H), 10.21 (s, 2H); 13C NMR (75 MHz) δ -4.4, 18.7, 26.2, 64.2, 105.1, 115.6, 128.7, 132.2, 145.1, 147.9; LD-MS obsd 598.3; FAB-MS obsd 598.3158, calcd 598.3159 (C34H46N4O2Si2); λabs 402, 500, 532, 573 nm.
4.4.7. 5,15-Bis(hydroxymethyl)porphyrin [H2P3-(OH)2]
Following the procedure for H2P1-(OH)4, the reaction of H2P2-(OTBDMS)2 (12.3 mg, 20.5 μmol) yielded a purple solid (5.0 mg, 66%): 1H NMR (DMSO-d6) δ -3.39 (br, 2H), 6.31 (t, J = 5.8 Hz, 2H), 6.87 (d, J = 5.8 Hz, 4H), 9.69 (d, J = 4.7 Hz, 4H), 9.94 (d, J = 4.7 Hz, 4H), 10.5 (s, 2H); LD-MS obsd 370.2; 13C NMR (DMSO-d6) δ 61.4, 104.6, 116.6, 129.1, 132.1, 144.2, 147.1; LD-MS 370.2; ESI-TOF obsd 371.15025, calcd 371.15020.1430 [(M + H)+, M = C22H18N4O2]. A solution of the title compound in DMSO was diluted in THF. The diluted solution was subjected to absorption measurement: λabs 401, 499, 531, 574 nm.
4.4.8. Synthesis of trans-AB-porphyrins from 1,9-(N,N-dimethylaminomethyl)dipyrromethanes and dipyrromethanes, exemplified for Zn(II)-5-(tert-butoxycarbonyl)-15-(tert-butyldimethylsiloxymethyl)porphyrin [ZnP4-OTBDMS]
Following a literature procedure,59 a solution of 1b (460 mg, 1.87 mmol) in anhydrous CH2Cl2 (25 mL) was treated with Eschenmoser’s salt (692 mg, 3.74 mmol) at room temperature for 3 h. Saturated aqueous NaHCO3 (20 mL) was added to the reaction mixture. The organic layer was separated, washed with brine, dried (Na2SO4) and concentrated to give a thick orange oil. The crude product was dissolved in EtOH (178 mL). Dipyrromethane 1a (543 mg, 1.87 mmol) was added to the solution. The solution was treated with anhydrous Zn(OAc)2 (3.43 g, 18.7 mmol) and refluxed exposed to air for 5 h. The mixture was cooled to room temperature. DDQ (1.28 g, 5.61 mmol) was added to the reaction mixture. The reaction mixture was stirred for 20 min. TEA (1 mL) was then added. The reaction mixture was concentrated. The crude product was filtered through a silica pad with CH2Cl2 as the eluant. After a fast-moving orange fraction was discarded, the eluate was collected and concentrated. Chromatography [silica, hexanes/CH2Cl2 (4:1)] yielded a purple solid (46 mg, 4.0%): 1H NMR (THF-d8) δ 0.22 (s, 6H), 0.91 (s, 9H), 2.10 (s, 9H), 7.21 (s, 2H), 9.47-9.49 (m, 4H), 9.64 (d, J = 4.4 Hz, 2H), 9.88 (d, J = 4.4 Hz, 2H), 10.28 (s, 2H); LD-MS obsd 617.0; FAB-MS obsd 616.1873, calcd 616.1848 (C32H36N4SiO3Zn); λabs (toluene) 409, 538, 575.
4.4.9. Zn(II)-5-(tert-Butoxycarbonyl)-15-hydroxymethylporphyrin (ZnP4-OH)
Following the procedure for H2P2-(OH)2, the reaction of ZnP4-OTBDMS (38 mg, 62 μmol) followed by chromatography [silica, CH2Cl2/ethyl acetate (9:1)] yielded a purple solid (16 mg, 51%): 1H NMR (THF-d8) δ 2.11 (s, 9H), 5.23 (t, J = 5.6 Hz, 1H), 7.00 (d, J = 5.6 Hz, 2H), 9.48 (d, J = 4.4 Hz, 4H), 9.65 (d, J = 4.4 Hz, 2H), 9.93 (d, J = 4.4 Hz, 2H), 10.27 (s, 2H); 13C NMR (THF-d8) δ 29.1, 64.5, 83.4, 106.8, 111.8, 119.8, 131.2, 131.7, 132.6, 133.1, 149.2, 150.5, 150.9, 151.6, 172.1; LD-MS obsd 503.4; FAB-MS obsd 502.0960, calcd 502.0983 (C26H22N4O3Zn); λabs (toluene) 409, 538, 573.
4.4.10. Zn(II)-5-(tert-Butyldimethylsiloxymethyl)-15-ethoxycarbonylporphyrin (ZnP5-OTBDMS)
Following the procedure for ZnP4-OTBDMS, the reaction of 1e (109 mg, 500 μmol) and 1a (145 mg, 500 μmol) followed by chromatography [silica, CH2Cl2/THF (99:1)] yielded a purple solid (22 mg, 7.5%): 1H NMR (300 MHz, THF-d8) δ 0.22 (s, 6H), 0.91 (s, 9H), 1.82 (t, J = 7.2 Hz, 3H), 5.07 (q, J = 7.2 Hz, 2H), 7.21 (s, 2H), 9.44-9.51 (m, 4H), 9.65 (t, J = 4.7 Hz, 2H), 9.88 (d, J = 4.4 Hz, 2H), 10.3 (s, 2H); 13C NMR (75 MHz, THF-d8) δ -4.3, 15.4, 19.1, 26.5, 63.1, 65.7, 106.9, 109.8, 118.5, 131.0, 132.1, 133.2, 149.8, 150.7, 150.9, 151.3; LD-MS obsd 588.2; FAB-MS obsd 588.1539, calcd 588.1535 (C30H32N4O3Zn); λabs 404, 536, 574 nm.
4.4.11. Zn(II)-5-Ethoxycarbonyl-15-hydroxymethylporphyrin (ZnP5-OH)
Following the procedure for H2P2-(OH)2, the reaction of ZnP5-OTBDMS (21.0 mg, 35.0 μmol) followed by chromatography [silica, CH2Cl2/MeOH (10:1)] yielded a purple solid (11 mg, 66%): 1H NMR (THF-d8) δ 1.82 (t, J = 6.5 Hz, 3H), 5.08 (q, J = 6.5 Hz, 2H), 5.28 (t, J = 4.0 Hz, 1H), 7.00 (d, J = 4.0 Hz, 2H), 9.45-9.49 (m, 4H), 9.65 (d, J = 5.0 Hz, 2H), 9.93 (d, J = 5.0 Hz, 2H), 10.28 (s, 2H); LD-MS obsd 474.3; FAB-MS obsd 474.0695, calcd 474.0670 (C24H18N4O3Zn); λabs 404, 536 nm.
4.4.12. Zn(II)-5-(tert-Butyldimethylsiloxymethyl)-15-[2-(trimethylsilyl)ethynyl]porphyrin (ZnP6-OTBDMS)
Following the procedure for ZnP4-OTBDMS, the reaction of 1f (1.38 g, 5.70 mmol) and 1a (1.65 g, 5.70 mmol) followed by chromatography [silica, hexanes/CH2Cl2 (1:3)] yielded a purple solid (85 mg, 2.4%): 1H NMR (300 MHz) δ 0.21 (s, 6H), 0.78 (s, 9H), 0.97 (s, 9H), 6.18 (s, 2H), 8.69 (d, J = 4.4 Hz, 2H), 8.88-8.96 (m, 4H), 9.31 (s, 2H), 9.44 (d, J = 4.4 Hz, 2H); 13C NMR (75 MHz, THF-d8) δ -4.3, 0.7, 19.1, 26.5, 65.6, 99.0, 100.4, 107.3, 109.8, 118.2, 130.9, 131.9, 132.7, 132.9, 150.5, 150.8, 151.6, 153.2; LD-MS obsd 612.5; FAB-MS obsd 612.1760, calcd 612.1719 (C32H36N4OSi2Zn); λabs 417, 551, 588 nm.
4.4.13. Zn(II)-5-Ethynyl-15-hydoxymethylporphyrin (H2P6-OH)
Following the procedure for H2P2-(OH)2, the reaction of ZnP6-OTBDMS (12.1 mg, 19.7 μmol) followed by chromatography [silica, CH2Cl2/ethyl acetate (4:1)] yielded a purple solid (4.1 mg, 49%): 1H NMR (THF-d8) δ 4.62 (s, 1H), 5.24 (t, J = 6.0 Hz, 1H), 6.96 (d, J = 6.0 Hz, 2H), 9.42-9.45 (m, 4H), 9.81 (d, J = 4.4 Hz, 2H), 9.88 (d, J = 4.4 Hz, 2H), 10.2 (s, 2H); LD-MS (POPOP) obsd 426.0, calcd 426.0 (C23H14N4OZn); λabs 413, 549, 586 nm.
4.4.14. Synthesis of trans-AB2C-porphyrins from 1,9-diacyldipyrromethane and dipyrromethane, exemplified for 5-(tert-butyldimethylsiloxymethyl)-15-phenyl-10,20-di-p-tolylporphyrin (H2P7-OTBDMS)
Following a literature procedure52,62 with modifications, a solution of 10-SnBu2 (588 mg, 852 μmol) in dry THF/MeOH (10:1, 33 mL) was treated with NaBH4 (642 mg, 17.0 mmol) for 2 h. A second batch of NaBH4 (642 mg, 17.0 mmol) was added, and the reaction mixture was stirred for another 2 h. Saturated aqueous NH4Cl was added, and the mixture was extracted with CH2Cl2. The organic layer was separated, dried (K2CO3) and concentrated. The resulting dicarbinol was dissolved in CH2Cl2 (340 mL) and treated with 1a (247 mg, 852 μmol) and Yb(OTf)3 (675 mg, 1.09 mmol) under argon for 30 min. DDQ (579 mg, 2.55 mmol) was added. The mixture was stirred for 1 h. TEA (2 mL) was added. The reaction mixture was concentrated. Chromatography [silica, hexanes/CH2Cl2 (1:3)] yielded a purple solid (77 mg, 12%): 1H NMR δ -2.87 to -2.80 (br, 2H), 0.20 (s, 6H), 0.95 (s, 9H), 2.72 (s, 6H), 7.01 (s, 2H), 7.56 (d, J = 8.0 Hz, 4H), 7.71-7.78 (m, 3H), 8.10 (d, J = 8.0 Hz, 4H), 8.19 (dd, 1J = 7.6 Hz, 2J = 1.6 Hz, 2H), 8.80 (d, J = 4.8 Hz, 2H), 8.83 (d, J = 4.8 Hz, 2H), 8.98 (d, J = 4.8 Hz, 2H), 9.59 (d, J = 4.8 Hz, 2H); 13C NMR (75 MHz) δ -4.4, 18.7, 21.7, 26.2, 64.8, 115.8, 120.2, 120.7, 126.9, 127.6, 127.9, 134.8, 137.6, 139.7, 142.5 (one carbon resonance apparently was overlapped; the α and β carbons were not observed presumably due to tautomerization); LD-MS obsd 710.7; FAB-MS obsd 710.3429, calcd 710.3441 (C47H46N4OSi); λabs 417, 515, 549, 590, 646 nm.
4.4.15. Zn(II)-5-(tert-Butyldimethylsiloxymethyl)-15-phenyl-10,20-di-p-tolylporphyrin (ZnP7-OTBDMS)
Following the procedure for ZnP1-(OTBDMS)4, the reaction of H2P7-OTBDMS (70.0 mg, 98.5 μmol) followed by chromatography (silica, CH2Cl2) yielded a purple solid (72 mg, 95%): 1H NMR δ 0.18 (s, 6H), 0.96 (s, 9H), 2.71 (s, 6H), 6.87 (s, 2H), 7.54 (d, J = 8.0 Hz, 4H), 7.69-7.78 (m, 3H), 8.07 (d, J = 8.0 Hz, 4H), 8.16-8.22 (m, 2H), 8.90 (d, J = 4.0 Hz, 2H), 8.93 (d, J = 4.0 Hz, 2H), 8.99 (d, J = 4.8 Hz, 2H), 9.55 (d, J = 4.8 Hz, 2H); 13C NMR (75 MHz) δ -4.4, 18.7, 21.7, 26.3, 65.1, 116.6, 121.2, 121.8, 126.7, 127.5, 127.7, 129.5, 132.0, 132.1, 132.9, 134.6, 134.7, 137.4, 140.2, 143.2, 150.2, 150.5, 150.7, 151.3; LD-MS obsd 772.8; FAB-MS obsd 772.2573, calcd 772.2576 (C47H44N4OSiZn); λabs 419, 548 nm.
4.4.16. 5-Hydroxymethyl-15-phenyl-10,20-di-p-tolylporphyrin (H2P7-OH)
Following the procedure for H2P2-(OH)2, the reaction of H2P7-OTBDMS (50.2 mg, 70.7 μmol) followed by chromatography [silica, CH2Cl2/ethyl acetate (4:1)] yielded a purple solid (25 mg, 60%): 1H NMR (THF-d8) δ -2.80 to -2.72 (br, 2H), 2.68 (s, 6H), 5.34 (t, J = 5.6 Hz, 1H), 6.86 (d, J = 5.6 Hz, 2H), 7.57 (d, J = 7.6 Hz, 4H), 7.68-7.78 (m, 3H), 8.07 (d, J = 7.6 Hz, 4H), 8.14-8.22 (m, 2H), 8.74-8.84 (m, 4H), 8.92 (d, J = 4.0 Hz, 2H), 9.73 (d, J = 4.0 Hz, 2H); 13C NMR (75 MHz, THF-d8) δ 21.7, 64.3, 118.4, 120.8, 121.3, 127.7, 128.4, 128.7, 129.1, 135.4, 138.4, 140.8, 143.5 (the α and β carbons were not observed due to tautomerization); LD-MS obsd 596.8; FAB-MS obsd 596.2570, calcd 596.2576 (C41H32N4O); λabs 417, 515, 549, 590, 646 nm.
4.4.17. Zn(II)-5-Hydroxymethyl-15-phenyl-10,20-di-p-tolylporphyrin (ZnP7-OH)
Following the procedure for H2P2-(OH)2, the reaction of ZnP7-OTBDMS (10.8 mg, 14.0 μmol) followed by chromatography [silica, CH2Cl2/ethyl acetate (4:1)] yielded a red purple solid (7.5 mg, 81%): 1H NMR (THF-d8) δ 2.70 (s, 6H), 5.14 (t, J = 6.0 Hz, 1H), 6.94 (d, J = 6.0 Hz, 2H), 7.57 (d, J = 8.0 Hz, 4H), 7.69-7.75 (m, 3H), 8.08 (d, J = 8.0 Hz, 4H), 8.14-8.19 (m, 2H), 8.79 (d, J = 4.8 Hz, 2H), 8.83 (d, J = 4.8 Hz, 2H), 8.95 (d, J = 4.8 Hz, 2H), 9.76 (d, J = 4.8 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ 21.1, 62.7, 117.7, 119.8, 120.3, 126.5, 127.2, 127.4, 129.9, 131.2, 131.7, 134.0, 134.1, 136.5, 140.0, 142.9, 148.8, 149.29, 149.33, 150.5; LD-MS obsd 658.4; FAB-MS obsd 658.1736, calcd 658.1711 (C41H30N4OZn); λabs 419, 548 nm.
4.4.18. 5-(tert-Butyldimethylsiloxymethyl)-15-ethoxycarbonyl-10,20-diphenylporphyrin (H2P8-OTBDMS)
Following the procedure for H2P7-OTBDMS, a sample of NaBH4 (151 mg, 4.00 mmol) was added in portions to a stirred solution of 11-SnBu2 (100 mg, 200 μmol) in THF/methanol (10:1, 8 mL). The progress of the reaction was followed by TLC. The reaction was complete in 40 min, at which point the reaction mixture was quenched with water (8 mL) and then poured into CH2Cl2 (30 mL). The organic phase was separated, washed with water, dried (Na2SO4), and concentrated to give the dipyrromethane-1,9-dicarbinol as a yellow oil. The latter was immediately subjected to condensation with dipyrromethane 1e (44.0 mg, 200 μmol) in the presence of InCl3 (5.70 mg, 25.0 μmol) in CH2Cl2 (80 mL) for 45 min. DDQ (136 mg, 0.60 mmol) was added to the reaction mixture. The reaction mixture was stirred for 20 min. TEA (1 mL) was added. The crude mixture was purified by chromatography [silica, hexanes/CH2Cl2 (1:3)] to obtain a purple solid (37 mg, 27%): 1H NMR δ -2.94 to -2.85 (br, 2H), 0.23 (s, 6H), 0.96 (s, 9H), 1.79 (t, J = 7.0 Hz, 3H), 5.08 (q, J = 7.0 Hz, 2H), 6.94 (s, 2H), 7.26-7.82 (m, 6H), 8.18-8.22 (m, 4H), 8.93 (t, J = 4,8 Hz, 4H), 9.43 (d, J = 4.8 Hz, 2H), 9.58 (d, J = 4.8 Hz, 2H); 13C NMR δ -4.5, 15.0, 18.6, 26.2, 63.2, 64.6, 109.3, 118.4, 120.9, 127.0, 128.1, 134.7, 142.2, 171.5 (the α and β carbons were not observed presumably due to tautomerization); LD-MS obsd 679.1; FAB-MS obsd 678.3051, calcd 678.3026 (C42H42N4O3Si); λabs 415, 512, 546, 586 nm.
4.4.19. Zn(II)-5,15-Diphenyl-10-ethoxycarbonyl-20-(tert-butyldimethylsiloxymethyl)porphyrin (ZnP8-OTBDMS)
Following the procedure for ZnP1-(OTBDMS)4, the reaction of H2P8-OTBDMS (37.0 mg, 54.0 μmol) followed by chromatography [silica, hexanes/CH2Cl2 (3:4)] yielded a purple solid (38 mg, 95%): 1H NMR (THF-d8) δ 0.22 (s, 6H), 0.89 (s, 9H), 1.79 (t, J = 7.0 Hz, 3H), 5.01 (q, J = 7.0 Hz, 2H), 7.14 (s, 2H), 7.76-7.80 (m, 6H), 8.19-8.22 (m, 4H), 8.90 (d, J = 4.8 Hz, 2H), 8.93 (d, J = 4.8 Hz, 2H), 9.43 (d, J = 4.8 Hz, 2H), 9.73 (d, J = 4.8 Hz, 2H); 13C NMR (75 MHz, THF-d8) δ -4.9, 14.4, 17.8, 25.7, 62.3, 64.1, 109.7, 118.1, 120.4, 126.4, 127.4, 129.9, 130.0, 131.6, 132.2, 134.0, 142.2, 147.0, 149.1, 149.5, 149.7, 171.3; LD-MS obsd 740.5; FAB-MS obsd 740.2169, calcd 740.2161 (C42H40N4O3SiZn); λabs 416, 547, 580 nm.
4.4.20. Zn(II)-5-Ethoxycarbonyl-15-hydroxymethyl-10,20-diphenylporphyrin (ZnP8-OH)
Following the procedure for H2P2-(OH)2, the reaction of ZnP8-OTBDMS (50.0 mg, 67.0 μmol) followed by chromatography [silica, CH2Cl2/ethyl acetate (10:1)] yielded a purple solid (30 mg, 71%): 1H NMR (300 MHz, DMSO-d6) δ 1.69 (t, J = 7.0 Hz, 3H), 5.01 (q, J = 7.3 Hz, 2H), 6.20 (t, J = 5.8 Hz, 1H), 6.95 (d, J = 5.9 Hz, 2H), 7.60-7.80 (m, 6H), 8.15-8.25 (m, 4H), 8.84 (d, J = 2.2 Hz, 2H), 8.86 (d, J = 2.3 Hz, 2H), 9.38 (d, J = 4.8 Hz, 2H), 9.82 (d, J = 4.8 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ 14.4, 62.3, 62.4, 109.4, 120.2, 120.3, 125.5, 127.4, 130.0, 130.3, 131.4, 132.2, 134.0, 142.3, 147.0, 148.9, 149.6, 149.9, 171.3; LD-MS obsd 626.4; FAB-MS obsd 626.1306, calcd 626.1296 (C36H26N4O3Zn); λabs 416, 547, 582 nm.
4.4.21. 5-Ethoxycarbonyl-15-(5,5-dimethyl-1,3-dioxan-2-yl)-10,20-di-p-tolylporphyrin (H2P9-acetal)
The general procedure for H2P7-(OTBDMS) was followed for the reaction of 13-BBu2 (119 mg, 192 μmol) and 1e (41.9 mg, 192 μmol). LD-MS analysis showed the presence of a small amount (∼1%) of an aldehyde-porphyrin. Chromatography (silica, CH2Cl2) yielded the title compound as a purple solid (18 mg, 14%): 1H NMR δ -3.00 to -2.94 (br, 2H), 1.11 (s, 3H), 1.77 (t, J = 7.2 Hz, 3H), 1.91 (s, 3H), 2.72 (s, 6H), 4.27-4.36 (m, 4H), 5.05 (q, J = 7.2 Hz, 2H), 7.57 (d, J = 8.0 Hz, 4H), 7.94 (s, 1H), 8.06 (d, J = 8.0 Hz, 4H), 8.91-8.99 (m, 4H), 9.42 (d, J = 4.8 Hz, 2H), 9.93 (d, J = 4.8 Hz, 2H); 13C NMR δ 15.0, 21.8, 22.8, 25.2, 31.1, 63.2, 80.4, 106.3, 109.6, 114.3, 121.2, 127.6, 129.5, 129.9, 132.3, 132.7, 134.7, 137.8, 139.4, 171.4 (the α carbons were not observed presumably due to tautomerization); LD-MS obsd 676.4; FAB-MS obsd 676.3049, calcd 676.3050 (C43H40N4O4); λabs 415, 512, 546, 588, 642 nm.
4.4.22. 5-Ethoxycarbonyl-15-formyl-10,20-di-p-tolylporphyrin (H2P9-CHO)
Following a general procedure,32,65 solution of H2P9-acetal (7.2 mg, 11 μmol) in CH2Cl2 (1 mL) was treated with a mixture of TFA/H2O (8:1, 900 μL) at room temperature for 28 h. The reaction mixture was diluted in CH2Cl2, washed with saturated aqueous NaHCO3 and water, dried (Na2SO4) and concentrated. Chromatography (silica, CH2Cl2) yielded a purple solid (5.5 mg, 85%): 1H NMR δ -2.56 to -2.51 (br, 2H), 1.77 (t, J = 7.2 Hz, 3H), 2.73 (s, 6H), 5.07 (q, J = 7.2 Hz, 2H), 7.58 (d, J = 7.6 Hz, 4H), 8.04 (d, J = 7.6 Hz, 4H), 8.88 (d, J = 4.8 Hz, 2H), 9.01(d, J = 4.8 Hz, 2H), 9.35 (d, J = 4.8 Hz, 2H), 10.00 (d, J = 4.8 Hz, 2H), 12.51 (s, 1H); 13C NMR δ 15.0, 21.8, 63.5, 109.3, 113.7, 123.2, 127.9, 129.1 (br) 130.5 (br), 132.5 (br), 134.6 (br), 170.8, 195.2 (the α carbons were not observed presumably due to tautomerization); LD-MS obsd 590.4; FAB-MS obsd 590.2321, calcd 590.2318 (C38H30N4O3); λabs 422, 525, 567, 604, 661 nm.
Acknowledgements
This work was supported by the NIH (GM36238). Mass spectra were obtained at the Mass Spectrometry Laboratory for Biotechnology at North Carolina State University. Partial funding for the facility was obtained from the North Carolina Biotechnology Center and the National Science Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Pardridge WM. In: In Introduction to the Blood-Brain Barrier. Pardridge WM, editor. Cambridge University Press; Cambride, UK: 1998. pp. 1–8. [Google Scholar]
- 2.Trova MP, Gauuan PJF, Pechulis AD, Bubb SM, Bocckino SB, Crapo JD, Day BJ. Bioorg. Med. Chem. 2003;11:2695–2707. doi: 10.1016/s0968-0896(03)00272-4. [DOI] [PubMed] [Google Scholar]
- 3.Lahaye D, Muthukumaran K, Gryko D, Hung C-H, Spasojevic I, Batinic-Haberle I, Fridovich I, Lindsey JS.Bioorg. Med. Chem 2007. in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carcel CM, Laha JK, Loewe RS, Thamyongkit P, Schweikart K-H, Misra V, Bocian DF, Lindsey JS. J. Org. Chem. 2004;69:6739–6750. doi: 10.1021/jo0498260. [DOI] [PubMed] [Google Scholar]
- 5(a).Yasseri AA, Syomin D, Loewe RS, Lindsey JS, Zaera F, Bocian DF. J. Am. Chem. Soc. 2004;126:15603–15612. doi: 10.1021/ja045243w. [DOI] [PubMed] [Google Scholar]; (b) ErratumJ. Am. Chem. Soc 20051279308 [Google Scholar]
- 6.Balaban TS, Tamiaki H, Holzwarth AR. Top. Curr. Chem. 2005;258:1–38. [Google Scholar]
- 7.Balaban TS. Acc. Chem. Res. 2005;38:612–623. doi: 10.1021/ar040211z. [DOI] [PubMed] [Google Scholar]
- 8.Balaban TS, Bhise AD, Fischer M, Linke-Schaetzel M, Roussel C, Vanthuyne N. Angew. Chem. Int. Ed. 2003;42:2140–2144. doi: 10.1002/anie.200250465. [DOI] [PubMed] [Google Scholar]
- 9.Balaban TS, Linke-Schaetzel M, Bhise AD, Vanthuyne N, Roussel C. Eur. J. Org. Chem. 2004:3919–3930. [Google Scholar]
- 10.Balaban TS, Linke-Schaetzel M, Bhise AD, Vanthuyne N, Roussel C, Anson CE, Buth G, Eichhöfer A, Foster K, Garab G, Gliemann H, Goddard R, Javorfi T, Powell AK, Rösner H, Schimmel T. Chem. Eur. J. 2005;11:2267–2275. doi: 10.1002/chem.200400664. [DOI] [PubMed] [Google Scholar]
- 11.Balaban TS, Eichhöfer A, Lehn J-M. Eur. J. Org. Chem. 2000:4047–4057. [Google Scholar]
- 12.Borbas KE, Mroz P, Hamblin MR, Lindsey JS. Bioconjugate Chem. 2006;17:638–653. doi: 10.1021/bc050337w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dogutan DK, Ptaszek M, Lindsey JS.J. Org. Chem 200772 in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14(a).Ptaszek M, McDowell BE, Taniguchi M, Kim H-J, Lindsey JS. Tetrahedron. 2007;63:3826–3839. doi: 10.1016/j.tet.2007.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Taniguchi M, Ptaszek M, McDowell BE, Lindsey JS. Tetrahedron. 2007;63:3840–3849. doi: 10.1016/j.tet.2007.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Taniguchi M, Ptaszek M, McDowell BE, Boyle PD, Lindsey JS. Tetrahedron. 2007;63:3850–3863. doi: 10.1016/j.tet.2007.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laha JK, Muthiah C, Taniguchi M, McDowell BE, Ptaszek M, Lindsey JS. J. Org. Chem. 2006;71:4092–4102. doi: 10.1021/jo060208o. [DOI] [PubMed] [Google Scholar]
- 16.Muthiah C, Bhaumik J, Lindsey JS. J. Org. Chem. 2007;72:5839–5842. doi: 10.1021/jo0707885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Senge MO, Hatscher SS, Wiehe A, Dahms K, Kelling A. J. Am. Chem. Soc. 2004;126:13634–13635. doi: 10.1021/ja045223u. [DOI] [PubMed] [Google Scholar]
- 18.Dahms K, Senge MO, Bakar MB. Eur. J. Org. Chem. 2007:3833–3848. [Google Scholar]
- 19.Hong S-K, Jeoung E, Lee C-H. J. Porphyrins Phthalocyanines. 2005;9:285–289. [Google Scholar]
- 20.Johnson AW, Oldfield D. J. Chem. Soc. C. 1966:794–798. [Google Scholar]
- 21.Witte L, Fuhrhop J-H. Angew. Chem. Int. Ed. 1975;14:361–363. doi: 10.1002/anie.197503612. [DOI] [PubMed] [Google Scholar]
- 22.Schlözer R, Fuhrhop J-H. Angew. Chem. Int. Ed. 1975;14:363. [Google Scholar]
- 23.Arnold D, Johnson AW, Winter M. J. Chem. Soc., Perkin Trans. I. 1977:1643–1647. doi: 10.1039/p19770001643. [DOI] [PubMed] [Google Scholar]
- 24.Smith KM, Bisset GMF. J. Org. Chem. 1979;44:2077–2081. [Google Scholar]
- 25.Smith KM, Bisset GMF. J. Chem. Soc., Perkin Trans. I. 1981:2625–2630. [Google Scholar]
- 26.Ponomarev GV. Chem. Heterocycl. Comp. 1994;30:1444–1465. [Google Scholar]
- 27.Torpey JW, de Montellano PRO. J. Org. Chem. 1995;60:2195–2199. [Google Scholar]
- 28.Yashunsky DV, Ponomarev GV, Arnold DP. Tetrahedron Lett. 1995;36:8485–8488. [Google Scholar]
- 29.Ishkov, Yu V, Zhilina ZI, Krivushko VA. Russ. J. Org. Chem. 1997;33:1346–1350. [Google Scholar]
- 30.Higuchi H, Shimizu K, Takeuchi M, Ojima J, Sugiura K-I, Sakata Y. Bull. Chem. Soc. Jpn. 1997;70:1923–1933. [Google Scholar]
- 31.Yeh C-Y, Miller SE, Carpenter SD, Nocera DG. Inorg. Chem. 2001;40:3643–3646. doi: 10.1021/ic001387+. [DOI] [PubMed] [Google Scholar]
- 32.Balakumar A, Muthukumaran K, Lindsey JS. J. Org. Chem. 2004;69:5112–5115. doi: 10.1021/jo049819b. [DOI] [PubMed] [Google Scholar]
- 33.Odobel F, Blart E, Lagrée M, Villieras M, Boujtita H, Murr NE, Caramori S, Bignozzi CA. J. Mater. Chem. 2003;13:502–510. [Google Scholar]
- 34.Harris D, Johnson AW. J. Chem. Soc., Chem. Commun. 1977:771. [Google Scholar]
- 35.Lin JJ, Gerzevske KR, Liddell PA, Senge MO, Olmstead MM, Khoury RG, Weeth BE, Tsao SA, Smith KM. J. Org. Chem. 1997;62:4266–4276. doi: 10.1021/jo9619138. [DOI] [PubMed] [Google Scholar]
- 36.Yashunsky DV, Ponomarev GV, Arnold DP. Tetrahedron Lett. 1996;37:7147–7150. [Google Scholar]
- 37.Ponomarev GV. Chem. Heterocycl. Comp. 1996;32:1263–1280. [Google Scholar]
- 38.Nishino N, Wagner RW, Lindsey JS. J. Org. Chem. 1996;61:7534–7544. doi: 10.1021/jo9611576. [DOI] [PubMed] [Google Scholar]
- 39.Arnold DP, Johnson AW, Mahendran M. J. Chem. Soc., Perkin Trans. I. 1978:366–370. [Google Scholar]
- 40.Yashunsky DV, Morozova, Yu V, Ponomarev GV. Chem. Heterocycl. Comp. 2000;36:485–486. [Google Scholar]
- 41.Takanami T, Hayashi M, Chijimatsu H, Inoue W, Suda K. Org. Lett. 2005;7:3937–3940. doi: 10.1021/ol0514294. [DOI] [PubMed] [Google Scholar]
- 42.Morozova, Yu V, Starikova ZA, Maksimov BI, Yashunskii DV, Ponomarev GV. Russ. Chem. Bull., Int. Ed. 2004;53:2192–2195. [Google Scholar]
- 43.Lee C-H, Lindsey JS. Tetrahedron. 1994;50:11427–11440. doi: 10.1016/j.tet.2008.08.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laha JK, Dhanalekshmi S, Taniguchi M, Ambroise A, Lindsey JS. Org. Process Res. Dev. 2003;7:799–812. [Google Scholar]
- 45.Wang QM, Bruce DW. Synlett. 1995:1267–1268. [Google Scholar]
- 46.Taylor EC, Goswami S. Tetrahedron Lett. 1991;32:7357–7360. [Google Scholar]
- 47.Kornblum N, Frazier HW. J. Am. Chem. Soc. 1966;88:865–866. [Google Scholar]
- 48.Rao PD, Littler BJ, Geier GR, III, Lindsey JS. J. Org. Chem. 2000;65:1084–1092. doi: 10.1021/jo9915473. [DOI] [PubMed] [Google Scholar]
- 49.Kobayashi S, Horibe M, Saito Y. Tetrahedron. 1994;50:9629–9642. [Google Scholar]
- 50.Bischofberger N, Waldmann H, Saito T, Simon ES, Lees W, Bednarski MD, Whitesides GM. J. Org. Chem. 1988;53:3457–3465. [Google Scholar]
- 51.Nicolaou KC, Claremon DA, Papahatjis DP. Tetrahedron Lett. 1981;22:4647–4650. [Google Scholar]
- 52.Geier GR, III, Callinan JB, Rao PD, Lindsey JS. J. Porphyrins Phthalocyanines. 2001;5:810–823. [Google Scholar]
- 53.Srinivasan N, Haney CA, Lindsey JS, Zhang W, Chait BT. J. Porphyrins Phthalocyanines. 1999;3:283–291. [Google Scholar]
- 54.Littler BJ, Ciringh Y, Lindsey JS. J. Org. Chem. 1999;64:2864–2872. doi: 10.1021/jo982452o. [DOI] [PubMed] [Google Scholar]
- 55.Geier GR, III, Littler BJ, Lindsey JS. J. Chem. Soc., Perkin Trans. 2. 2001:701–711. [Google Scholar]
- 56.Geier GR, III, Littler BJ, Lindsey JS. J. Chem. Soc., Perkin Trans. 2. 2001:712–718. [Google Scholar]
- 57.Zaidi SHH, Loewe RS, Clark BA, Jacob MJ, Lindsey JS. Org. Process Res. Dev. 2006;10:304–314. [Google Scholar]
- 58.Corey EJ, Venkateswarlu A. J. Am. Chem. Soc. 1972;94:6190–6191. [Google Scholar]
- 59.Fan D, Taniguchi M, Yao Z, Dhanalekshmi S, Lindsey JS. Tetrahedron. 2005;61:10291–10302. [Google Scholar]
- 60.Schreiber J, Maag H, Hashimoto N, Eschenmoser A. Angew. Chem. Int. Ed. Eng. 1971;10:330–331. [Google Scholar]
- 61.Rao PD, Dhanalekshmi S, Littler BJ, Lindsey JS. J. Org. Chem. 2000;65:7323–7344. doi: 10.1021/jo000882k. [DOI] [PubMed] [Google Scholar]
- 62.Tamaru S-I, Yu L, Youngblood WJ, Muthukumaran K, Taniguchi M, Lindsey JS. J. Org. Chem. 2004;69:765–777. doi: 10.1021/jo035622s. [DOI] [PubMed] [Google Scholar]
- 63.Muthukumaran K, Ptaszek M, Noll B, Scheidt WR, Lindsey JS. J. Org. Chem. 2004;69:5354–5364. doi: 10.1021/jo0492620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zaidi SHH, Muthukumaran K, Tamaru S-I, Lindsey JS. J. Org. Chem. 2004;69:8356–8365. doi: 10.1021/jo048587d. [DOI] [PubMed] [Google Scholar]
- 65.Lindsey JS, Brown PA, Siesel DA. Tetrahedron. 1989;45:4845–4866. [Google Scholar]