

Abstract
A growing body of evidence suggests that a preclinical phase of Alzheimer’s disease (AD) exists several years or more prior to the overt manifestation of clinical symptoms and is characterized by subtle neuropsychological and brain changes. Identification of individuals prior to the development of significant clinical symptoms is imperative in order to have the greatest treatment impact by maintaining cognitive abilities and preserving quality of life. Functional magnetic resonance imaging (fMRI) offers considerable promise as a non-invasive tool for detecting early functional brain changes in asymptomatic adults. In fact, evidence to date indicates that functional brain decline precedes structural decline in preclinical samples. Therefore, fMRI may offer the unique ability to capture the dynamic state of change in the degenerating brain. This review examines the clinical utility of blood oxygen level dependent (BOLD) fMRI in those at risk for AD as well as in early AD. We provide an overview of fMRI findings in at-risk groups by virtue of genetic susceptibility or mild cognitive decline followed by an appraisal of the methodological issues concerning the diagnostic usefulness of fMRI in early AD. We conclude with a discussion of future directions and propose that BOLD-fMRI in combination with cerebral blood flow or diffusion techniques will provide a more complete accounting of the neurovascular changes that occur in preclinical AD and thus improve our ability to reliably detect early brain changes prior to disease onset.
Keywords: BOLD-fMRI, Preclinical Alzheimer’s disease, APOE ε4, Mild cognitive impairment, Arterial spin labeling, Cerebral blood perfusion
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by the accrual of neuritic plaques and neurofibrillary tangles that result in a disruption of neuronal function and which causes cognitive and behavioral dysfunction (Braak & Braak, 1991). Although the progression of the neuropathological changes of AD are not fully known, studies support the notion that medial temporal lobe (MTL) structures such as the hippocampus and entorhinal cortex are involved in the earliest stage of the disease, and that frontal, temporal, and parietal cortices become increasingly affected as the disease progresses (Braak & Braak, 1991; Brewer & Moghekar, 2002). Consistent with this proposed progression, numerous studies have shown that MTL-dependent episodic memory measures are usually affected earliest in the disease and are quite effective in differentiating between very mildly demented patients with clinically diagnosed AD and normal older adults (Salmon & Bondi, 1999, in press). As AD progresses, the dementia syndrome is characterized by a prominent amnesia with rapid forgetting of information, marked executive dysfunction, and additional deficits in certain aspects of language, visuospatial abilities, and attention.
Unfortunately, early diagnosis of AD is difficult since neuronal hallmarks are not yet detectable in vivo, although promising advances are being made in imaging the underlying pathologic burden in mild cognitive impairment (MCI) and AD (e.g., see Edison et al., 2007; Small et al., 2006). In addition, in recent years there has been a strong emphasis on identifying preclinical markers of AD, given that pathologic changes begin years to decades before clinical features are apparent, suggesting that a latent phase of the disease exists (Braak & Braak, 1991). Thus, it is imperative to reliably identify individuals preclinically (i.e., prior to the development of significant clinical symptoms) in order to treat them during a period that will have the greatest impact in maintaining cognitive abilities and ultimately preserving quality of life.
From a neuropsychological perspective, evidence suggests that subtle declines in episodic memory may presage the development of the dementia syndrome associated with AD (Fig. 1; Albert et al., 2001; Bondi et al., 1999; Collie & Maruff, 2000; Grober & Kawas, 1997; Jacobs et al., 1995). Indeed, subtle deficits in a broad range of neuropsychological domains have been implicated during the preclinical period (see Twamley et al., 2006, for review), and the combination of visuospatial learning and semantic memory deficits have been found to accurately detect cognitive dysfunction characteristic of preclinical AD (Blackwell et al., 2004).
Fig. 1.
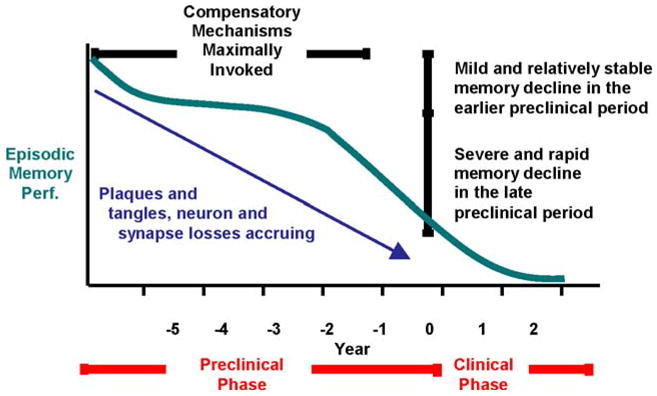
Proposed model of nonlinear pattern of episodic memory decline during the preclinical period of Alzheimer’s disease (adapted from Twamley, Ropacki, & Bondi, 2006 and based on data of Backman et al., 2001; Chen et al., 2001; Lange et al., 2002)
Currently the most widely used neuroimaging techniques in clinical practice include structural magnetic resonance imaging (MRI) and positron emission tomography (PET). Structural morphometric analyses reveal relatively consistent reductions in medial temporal lobe volumes in adults with AD and MCI (see Mosconi et al., 2006 for review). PET metabolic alterations in the temporoparietal cortices and in the posterior cingulate have been reported in MCI and AD (Desgranges et al., 1998; Matsuda, 2001; Reiman et al., 1996; Small et al., 2000a) as well as in nondemented young and middle-aged adults at genetic risk for AD (Reiman et al., 1996, 2004, 2005; for review, see Petrella et al., 2003; Wolf et al., 2003). Structural neuroimaging techniques are traditionally favored for their ability to relatively easily rule out potentially treatable causes of cognitive dysfunction such as neoplasms or normal pressure hydrocephalus (Knopman et al., 2001). However, evidence reviewed herein indicates that functional brain decline precedes structural decline and thereby suggests that functional magnetic resonance imaging (fMRI) offers considerable promise as a non-invasive technique for detecting such early and oftentimes subtle functional brain changes that occur in asymptomatic adults. Specifically, fMRI may offer the unique ability to capture the dynamic state of change in the degenerating brain. Early detection of such preclinical markers may also facilitate studies of early intervention to slow or prevent disease progression (Petrella et al., 2003) by providing a proxy indicator for the effectiveness of new therapies (Masdeu et al., 2005). Although the few available pharmacological treatments for AD have limited efficacy (Mayeux & Sano, 1999; Tariot & Federoff, 2003), preliminary evidence suggests that the greatest benefit is achieved with early introduction of treatments (Tariot & Federoff, 2003; Petersen et al., 2006). However, at our VA medical center—and likely elsewhere across the U.S.—patients must be diagnosed with dementia and obtain a range of impaired scores on the Mini-Mental State Examination (Folstein et al., 1976) to obtain eligibility for initiation of cholinesterase inhibitor (e.g., donepezil) treatment. Therefore, identifying preclinical markers of AD and initiating preventative treatment during this asymptomatic period ultimately may prove more beneficial than current practices.
In an effort to identify functional preclinical markers of AD, much attention has focused on at-risk groups that ostensibly provide a model of possible early pathological changes that predate clinical symptoms. The most commonly studied risk factors for the development of sporadic late-onset AD include genetic susceptibility markers (e.g., possession of the apolipoprotein E epsilon-4 [APOE ε4] allele, positive family history of AD in a first-degree relative) or diagnosis of MCI, traditionally thought to reflect a transitional state between healthy aging and frank dementia (Petersen & Morris, 2005). Since individuals at genetic risk for AD are typically studied before developing cognitive impairments, the presence of the susceptibility genes are generally thought to reflect presymptomatic AD in but a subgroup of these risk samples, whereas the presence of MCI is thought to have stronger predictive power for conversion to AD and has therefore been described as prodromal AD (Dickerson & Sperling, 2005). That functional brain changes are identified despite obvious a priori commingling of preclinical and disease-free individuals within “risk” groups is compelling and argues for more exacting work in successfully predicting and separating such subgroup risk categories.
Thus, the aims of this review are to provide an overview of findings regarding the functional changes in at-risk adults and in patients diagnosed with AD. We follow with an appraisal of the diagnostic utility of fMRI methods as a clinical tool to detect early AD. Future directions for the clinical use of fMRI will be suggested in the context of integrating the BOLD signal with measures of blood perfusion acquired using arterial spin labeling (ASL) or with diffusion tensor imaging (DTI) techniques to better capture functional neurovascular and neuroanatomic changes associated with preclinical AD.
Functional MRI patterns of activity associated with risk of Alzheimer’s disease
In general, fMRI findings of at-risk adults report both increases and decreases in brain activity that vary according to level of risk or cognitive decline. We will attempt to report the general consensus and highlight discrepancies between nondemented low-risk adults (e.g., no copies of the APOE ε4 allele, negative family history for AD), nondemented high-risk adults (e.g., APOE ε4 carriers, positive family history, MCI), and adults with AD. Although our review will focus primarily on APOE-related and family-history associated risk, there is growing recognition of fMRI-based distinctions with a host of risk factors (e.g., brain-derived neurotrophic factor gene [Egan et al., 2003; Hariri et al., 2003], Presenilin gene mutations [Mondadori et al., 2006a]), and this area will continue to show tremendous growth with the further identification of as yet undiscovered risk factors for AD.
Before proceeding, a brief introduction and discussion of the compensatory hypothesis as an explanation of differential regional activity seen in at-risk groups is warranted. The compensatory hypothesis holds that as AD-related degenerative processes progress the brain employs various compensatory measures to overcome these pathologic encroachments until the decline in cognitive abilities becomes apparent and, ultimately, daily functioning is disrupted. Accordingly, increased functional activity has been interpreted as a compensatory process whereby greater cognitive effort—usually reflected by increases in BOLD activity—is required to perform at an equivalent level compared to healthy control groups, and it assumes that enough healthy tissue remains to support performance. Similarly, decreased activity in healthy functioning has been interpreted as evidence of efficient processing (Mondadori et al., 2006b), whereas decreased activity in neurodegenerative diseases appears to reflect cortical compromise or disconnection.
However, a potential distinction that is often overlooked but may not only clarify discrepant findings but possibly stage disease progression is the ability to differentiate between compensatory “reserve”—that is, increased activity in regions typically involved in cognitive processing in healthy adults—and compensatory “recruitment”—namely, involvement of additional regions not normally activated in healthy individuals (Bookheimer et al., 2000; Petrella et al., 2003). In the absence of correlations between functional activity and behavioral performance, the assumption that increased activity is inherently beneficial is premature since increased activity may in fact reflect a number of possibilities, including (a) decreased processing efficiency due to disinhibition of nonspecialized networks, (b) inefficient recruitment of specialized neural mechanisms to perform a task, (c) dedifferentiation of function due to reduced cortical specialization of cognitive functions (Li & Lindenberger, 1999; Rajah & Esposito, 2005), (d) BOLD activity differences in baseline conditions that in turn give rise to differences in functional versus baseline condition contrasts, or (e) disruption of resting state activity irrespective of the particular tasks performed during scanning (Greicius et al., 2004). We recently reported that increased recruitment of right hemisphere frontal regions in healthy older adults was not universally beneficial to behavioral performance (Wierenga et al., 2006). Taken together, findings across studies auger not only for the correlation between BOLD response and behavioral performance, but also for sensitivity to the nature of the atypical patterns of activity in the interpretation of regional differences in functional activity between groups.
fMRI in cognitively intact adults at risk for AD by virtue of the APOE ε4 allele
The apolipoprotein E ε4 allele on chromosome 19 is a well-known susceptibility gene for late-onset AD, presumably due to its association with small vessel arteriolosclerosis, microinfarcts of the deep nuclei, neuritic plaque density, and amyloid angiopathy in autopsy-confirmed patients with AD (Yip et al., 2005; Tiraboschi et al., 2004). However, the ε4 allele has been shown to have relatively low positive predictive value for the diagnosis of AD (Devanand et al., 2005; Slooter et al., 1996) and accounts for less than 40% of AD cases (Saunders et al., 1993). Although individuals who possess the ε4 allele have been termed “presymptomatic” and are typically studied while still cognitively intact (Dickerson & Sperling, 2005), our neuropsychological studies have demonstrated that nondemented older adults who are at risk for developing AD by virtue of the APOE ε4 allele perform below those without this risk factor on tests of episodic memory. Episodic memory measures are more effective for differentiating between these groups than are other cognitive measures of executive functions, attention, constructional ability, or psychomotor speed (Bondi et al., 1999; Lange et al., 2002). These results suggest that a preclinical phase of detectable cognitive decline (Bondi et al., 1999; Lange et al., 2002) may precede clinical diagnosis of AD by several years, suggesting corresponding changes in underlying brain functions in nondemented older adults at risk for AD by virtue of the APOE ε4 allele (Bondi et al., 2005; Han et al., 2007). Notably, these cognitive declines and functional changes in APOE ε4 carriers tend to become most evident after age 65 (Jorm et al., 2007; Mondadori et al., 2006b), suggesting that the negative effects of the APOE ε4 allele accelerate with age. In fact, young adult ε4 carriers may demonstrate more efficient neural processing as evidenced by better episodic memory performance and reduced learning- and retrieval-related activity (Mondadori et al., 2006b).
Given that the earliest neuropathologic changes resulting from AD occur in the MTL that support memory encoding (Squire, 1992), and episodic memory complaints are among the first cognitive symptoms of AD, the majority of fMRI studies have investigated neural substrates of episodic memory encoding, recall, or recognition in genetically at-risk adults. In general, these fMRI studies have found increased BOLD brain response in the MTL during episodic learning tasks in non-demented adults with versus without the APOE ε4 allele, consistent with a compensatory response during learning (e.g., over-recruitment of typical brain resources to achieve comparable levels of memory-related performance in the face of incipient cortical compromise) (Bondi et al., 2005; Bookheimer et al., 2000; Dickerson et al., 2004, 2005; Han et al., 2006). In addition, regardless of the nature of the stimuli (e.g., verbal, pictoral, etc.), laterality effects have been reported within the MTL whereby ε4 carriers not only activate the right hippocampus to a greater degree than the left hippocampus (Bondi et al., 2005; Han et al., 2007), but the right hippocampus has been shown to be less sensitive to the distinction between familiar and novel items (e.g., does not show the expected reduction in activity for familiar items; Lind et al., 2006b). Given evidence that the hippocampus functions as a novelty detector (Roberts et al., 1962; Vinogradova, 2001) and that AD pathology targets regions of the medial temporal lobe including the entorhinal cortex and hippocampus through the accumulation of β-amyloid plaques and neurofibrillary tangles (Braak & Braak, 1991), neural dysfunction within the medial temporal cortex likely contributes to abnormal novelty detection seen in APOE ε4 carriers (Lind et al., 2006b).
Notably, increased activity is not restricted to the MTL but appears to extend to other cortical regions. For instance, Bookheimer et al. (2000) reported greater extent and intensity of activity in the temporal and frontal regions in the APOE ε4 group compared to the non-ε4 group during encoding and recall compared to rest. In another study, Wishart et al. (2006) demonstrated equivalent performance on an auditory verbal working memory task, ε4 carriers nonetheless demonstrated greater activity in the medial frontal and parietal regions bilaterally and in the right dorsolateral prefrontal cortex compared to non-ε4 peers. Similarly, during a visual working memory task, another study showed increased frontal and cingulate gyri activity in ε4 positive adults (Filbey et al., 2006).
Despite the majority of evidence showing increased BOLD response in the MTL, frontal, and parietal regions in APOE ε4 adults, an upsurge in activity does not occur uniformly throughout the brain, and several regions have been shown a decrease in BOLD response in genetically at-risk adults. For example, Lind et al. (2006a) reported decreased functional brain activity in cortical regions including the inferior parietal cortex, and bilateral anterior cingulate region during a semantic categorization task. Notably, gene-dose effects were also found, whereby ε4 homozygotes showed greater reduction in BOLD response compared to their heterozygote peers. Additionally, no difference was found between ε3 and ε4 carriers on a modified digit-span (forwards) working memory test, suggesting that perhaps the substrates of attention remain intact in at-risk adults (Burggren et al., 2002).
Taken together, these findings suggest a functional deterioration of the hippocampal system for encoding new information as well as recognizing previously encountered stimuli as familiar can be identified in presymtomatic adults at genetic risk for developing AD as an upsurge in BOLD response. These functional changes may reflect early hippocampal pathology consistent with findings of greater right hippocampal volume loss in APOE ε4 carriers (Tohgi et al., 1997).
Functional MRI of cognitively intact at-risk adults with a positive family history of Alzheimer’s disease
A positive family history of AD in a first-degree relative is known to increase the prevalence of neurofibrillary tangles and amyloid plaques (Fratiglioni et al., 1993) in older adults. The presence of one first-degree relative with AD approximately doubles the individual’s lifetime risk of AD whereas the presence of two or more family members with AD may increase risk by eight-fold (Devi et al., 2000; Fratiglioni, 1993; van Duijn et al., 1991). Although the biologic mechanisms of late-onset sporadic AD risk associated with a positive family history are not well understood, a recent study reported that 45% of adult children of AD patients possessed the ε4 allele (Sager et al., 2005), indicating that familial history and APOE ε4 genotype co-occur. One recent study found that first-degree family history of AD (+FH) was associated with an increased risk of dementia only in adult ε4 carriers over the age of 75 (Huang et al., 2004). For instance, ε3/3 homozygotes with a +FH have a 30% chance of developing AD, whereas ε3/4 heterozygotes with a +FH have a 46% lifetime risk and ε4/4 homozygotes with +FH have a 61% likelihood of developing AD (Trivedi et al., 2006). Cognitively, several studies have shown that nondemented older adults with a positive family history for AD perform significantly worse than those with a negative family history on tests of episodic memory (Bondi et al., 1994; Caselli et al., 2004; La Rue et al., 1992; Levy et al., 2004).
In a large sample of asymptomatic offspring of autopsy-confirmed late-onset familial AD cases, Bassett et al. (2006) found greater extent and magnitude of BOLD response in the frontal and temporal lobes including the hippocampus during auditory paired-associates memory encoding but decreased activity in the thalamus and anterior and posterior cingulate during recall compared to an age-matched control group despite comparable behavioral performance. Notably, 39% of the at-risk adults and 22% of the control subjects possessed at least one ε4 allele. However, no differences were found for comparisons of ε4 carriers to non-ε4 carriers with respect to FH, though the possibility of an interaction of FH and APOE status was not discussed. However, when the role of APOE genotype was examined within a sample of middle-aged adults, all of whom had a +FH, ε4 carriers displayed reduced activity in the right hippocampus and MTL compared to ε3 homozygote peers during an encoding novelty detection task (Trivedi et al., 2006). In a similar novelty detection study comparing high-risk (+FH, ε4 carrier) and low-risk (−FH, non-ε4 carrier) middle aged adults during verbal paired associate learning, we reported that high-risk adults demonstrated increased activity in the right posterior middle temporal area, left lingual area, and bilateral anterior cingulate for the comparison of novel versus repeated words (Fleisher et al., 2005). No group differences were found for this comparison in bilateral hippocampal and parahippocampal regions of interest, although when novel words were compared to a resting fixation, high-risk adults demonstrated greater activity in the left parahippocampal gyrus. When high-risk adults (+FM, ε4 carriers) were compared to a low-risk middle aged group (−FH, non-ε4 carriers) on language tasks (verbal fluency, object naming), the high-risk group showed increased activity in the left parietal lobe (verbal fluency) but reduced activity in the inferotemporal cortex during object-naming (Smith et al., 1999, 2002, 2005) with greater longitudinal decline. Unfortunately, performance during scanning was not monitored.
Interpretation of these studies is complicated by the assumption that possession of multiple AD risk factors act in concert resulting in additive disruptive effects. In response to such concerns, recent attempts have been made to clarify the relative contribution of the APOE ε4 allele and positive family history. For example, Johnson et al. (2006) found greater response to novel items in the MTL and fusiform gyrus in adults with a −FH (collapsed across APOE genotype). The −FH ε4 carriers showed greatest signal change in the hippocampus and the ε4 carriers with a +FH demonstrated the least signal change during novel versus learned picture detection, suggesting that FH may modulate the effect of APOE ε4 in middle age adults. Unfortunately, the ability to integrate these genetic findings with the broader APOE literature that does not take into account familial history effects is limited by the fact that studies of family history tend to include younger samples, thereby neglecting the aging component. Given that age is the strongest risk factor for the development of AD (Kawas & Katzman, 1999), genetic risk factors for AD may interact with age, and conceivably their negative effects on MTL function may accelerate with age.
Functional MRI in at-risk adults with mild cognitive impairment
Mild cognitive impairment is thought to represent a transitional stage between healthy and pathological aging and has been posited to represent a prodromal phase of AD. The original MCI classification of Petersen et al. (1995, 1999) has been modified since its initial characterization and currently conceptualizes MCI as a heterogeneous construct (Petersen & Morris, 2005). Specifically, current views hold that the original construct of the amnestic MCI subtype may not characterize all of the prodromal states of dementia, and therefore recent diagnostic criteria divide MCI into two main subdivisions of amnestic and nonamnestic MCI with further subclassifications into single- or multi-domain categories (Petersen & Morris, 2005). These subtypes are thought to aid the determination of the proposed etiology of cognitive decline. For example, conversion from single domain amnestic MCI to AD ranges from 12% to 15% per year (Petersen et al., 1999), whereas multiple domain MCI may signal concomitant vascular compromise. Recent structural neuroimaging and PET imaging of amyloid deposition using the [(11)C] PIB-ligand provide support for differential underlying neuropathology of these MCI subtypes (Delano-Wood et al., 2006; Frisoni, 2007; Small et al., 2006).
Despite the fact that amnestic MCI may represent prodromal AD, relatively few studies have investigated functional changes in MCI using fMRI, let alone attempted to distinguish between the proposed subtypes. Thus, it is not surprising that the majority of fMRI studies of MCI involve memory processes. Investigations of the MTL generally demonstrate that less impaired MCI subjects show increased BOLD response in the hippocampus compared to control groups, whereas more impaired MCI subjects demonstrate decreased BOLD response similar to the levels observed in mild AD patients (Celone et al., 2006; Dickerson et al., 2004, 2005; Hamalainen et al., in press; Johnson et al., 2006; Machulda et al., 2003). For instance, Dickerson et al. (2004) reported a positive correlation between extent of parahippocampal and hippocampal activation with memory performance in MCI but, in a paradoxical fashion, found that greater clinical impairment, as determined by the Dementia Rating Scale, was associated with recruitment of a larger region of the right parahippocampal gyrus during encoding. Johnson et al. (2004) provided further evidence for hippocampal dysfunction in MCI, whereby a reduction in hippocampal adaptation during repetition of a picture encoding task was found in an amnestic MCI group with compromised learning (Johnson et al., 2004), suggesting that adults with MCI do not habituate to increasingly familiar items in the same manner as healthy older adults who show expected reductions in BOLD response to repeated items over time.
However, upsurges in BOLD response appear to be regionally specific to the MTL in MCI patients. Cortical findings indicate greater memory-related deactivation (e.g., reduction in brain activity) in medial and lateral parietal regions in less impaired MCI adults and a loss of deactivation in more impaired MCI and mild AD adults (Celone et al., 2006). In fact, a “default mode network” has been identified comprising regions with a high resting state metabolism including the medial frontal, medial parietal, and posterior cingulate cortex to explain decreases in brain activity that occur during task performance (Greicius et al., 2003; Raichle et al, 2001; Rombouts et al., 2005a). Essentially, it has been argued that regions in the default mode network in healthy adults are more involved during resting states due to engagement in attending to environmental stimuli, reviewing past knowledge and planning of future behavior, spontaneous semantic processing and allocation of resources to perform a task. Alterations in the default mode network have been reported in both MCI and AD patients (Rombouts et al., 2005a) in the anterior frontal regions, whereby MCI patients show deactivation in the same network but to a lesser degree as the AD patients (Greicius et al., 2004; Lustig et al., 2003; Wang et al., 2006).
Functional MRI patterns of activity associated with Alzheimer’s disease
In order to determine whether such functional changes in at-risk adults represent an endophenotype specific to AD, an understanding of the functional changes in clinically evident AD is necessary. Indeed, there is mounting evidence that Alzheimer’s disease represents a cortico-cortical disconnection syndrome. Anatomically, neurofibrillary tangle pathology has shown a strong predilection for cortical layers and cell types of the entorhinal cortex that disconnect the hippocampus from neocortex, affecting cortical connections within and between limbic and neocortical regions (Festa et al., 2005). Similarly, neurofibrillary tangles in pyramidal neurons in layers III and V selectively disrupt functionally related cortical association areas. Given these findings, it is plausible that changes in functional activity seen during memory performance in at-risk adults may reflect early evidence of a breakdown in the communication between the medial temporal lobe and neocortex.
Therefore, it is not surprising that, in general, patients with AD show reduced MTL activity (Dickerson et al., 2005; Gron & Reipe, 2004; Machulda et al., 2003; Pariente et al., 2005; Rombouts et al., 2000; Small et al., 2000b; Sperling et al., 2003). Notably, Golby et al. (2005) reported a graded deficit in activity along the ventral visual stream approaching the hippocampal complex, consistent with increasing cortical disconnection between the medial temporal lobe and its posterior afferents. Similarly, Dickerson (2005) examined extent versus magnitude of activity in AD patients and healthy elderly subjects with and without memory complaints and found adults with memory complaints had increased activity in the hippocampus and entorhinal cortex compared to adults without memory complaints and AD patients showed reduced activity in these MTL regions compared to both healthy groups.
Alternatively, more diffuse cortical activity has been shown in AD patients in additional regions such as the lateral temporal lobe for semantic memory (Grossman et al., 2003), occipital lobe for visuospatial processing (Kato et al., 2001; Small et al., 1999; Corkin et al., 1997), left inferior frontal gyrus and right prefrontal cortex during semantic decision-making (Johnson et al., 2000; Saykin et al., 1999), and bilateral frontoparietal regions during associative memory (Pariente et al., 2005). Several studies have interpreted increased cortical activity in AD to reflect engagement of top-down attentional systems to improve performance (Backman et al., 1999; Becker et al., 1996; Pariente et al., 2005; Saykin et al., 1999; Woodard et al., 1998) since there tends to be a selective recruitment of frontal and parietal regions. However, altered prefrontal activity has not been linked to performance in these studies, and there are some reports of decreased prefrontal activity during retrieval in AD patients despite equal performance to their healthy peers (Corkin et al., 1997). Johnson et al. (2000) also found that greater brain atrophy corresponded to increased brain activity in the left inferior frontal gyrus in AD patients during an auditory semantic decision task. Discrepant findings in the AD literature likely reflect differences in cognitive task demands, level of performance, cognitive reserve capacity, or stage of AD. Alternatively, as seen in the MCI literature, functional activation may be bimodal, increasing with a slight or moderate neuronal dysfunction and decreasing with greater disease progression, as the cortical neuronal networks become more severely impaired, as in the entorhinal cortex (Masdeu et al., 2005). Figure 2 presents a proposed model of the nonlinear trajectory of alterations in brain activity along the spectrum of disease risk in the medial temporal lobe, frontal lobe, and inferior posterior regions based on a review of the literature.
Fig. 2.

Proposed model of nonlinear trajectory of brain activity alterations during the evolution of Alzheimer’s disease in several brain regions according to literature reviewed in the text. CI e4 = cognitively intact e4-carrier; mAD = mild Alzheimer’s disease; mMCI = mildly impaired mild cognitive impairment; MTL = medial temporal lobe; sMCI = more severely impaired mild cognitive impairment; NC = normal control
Although the majority of fMRI studies have compared regional activation between AD and healthy control groups, perhaps an even more relevant neuroimaging finding to the clinical utility of fMRI relates to differences in the dynamics of the BOLD response in AD. Specifically, there appears to be an increasing delay of the BOLD response along the continuum from normal aging to MCI to AD (Rombouts et al., 2005b), especially in the occipital lobe during a face encoding task, providing additional support that MCI reflects a transitional period between normal aging and dementia (Rombouts et al., 2005b). In fact, AD patients demonstrated altered BOLD response in widespread areas. Taken together, results auger for the incorporation of analyses of BOLD dynamics (e.g., time to peak, post-stimulus undershoot) in patient populations in addition to traditional comparisons of regional differences in BOLD response.
Assessment of the clinical utility of fMRI to identify preclinical Alzheimer’s disease
Given that functional differences often occur in the absence of volumetric loss in at-risk asymptomatic adults (Bassett et al., 2006; Bondi et al., 2005; Reiman et al., 1996, 1998), functional assays may be more sensitive to early biomarkers that precede cognitive decline. That being said, there is a strong need for sensitive, standardized, validated, and reliable fMRI techniques to identify patterns of brain response at the individual subject level that heralds preclinical dysfunction. To achieve clinical applicability, fMRI techniques must also demonstrate prognostic value. Unfortunately, several methodological issues clearly present a challenge to the clinical utility of fMRI in preclinical AD, given that differences in methodology across studies can account for discrepant findings in the literature. Such methodological issues include task selection and baseline comparisons, performance confounds, and the ability to translate group findings to single-subject designs due to the paucity of longitudinal and case-study designs. These methodological issues will be briefly highlighted before we conclude with a discussion of future directions for fMRI in clinical settings.
Task selection and baseline comparison
The clinical utility of fMRI to detect early preclinical changes depends on the ability to sensitively and reliably identify “signatures” of brain response at the individual subject level. Since such signatures are expected to be task-dependent (e.g., upsurge in BOLD response in MTL during encoding), selection of the appropriate task depends on identifying the cognitive process of interest and choosing a sensitive yet specific task to elicit the cognitive function. It remains unclear whether the best approach is to challenge the neural substrates that may be compromised by the early disease process or to elicit cognitive functions expected to remain relatively preserved early in the course of the disease. In general, the tendency has been to examine aspects of memory or encoding thought to reside in the medial temporal lobe and therefore likely more susceptible to early disruption, although evidence of cortical changes may be equally informative.
Currently there is no consensus regarding the optimal task-baseline comparison to use in preclinical AD. Trivedi et al. (2006) argue that an active baseline is more beneficial in light of several problems associated with a resting baseline (e.g., inability to control mental processes, demonstrations of increased activity during rest). To complicate matters, resting state activity may be disrupted in MCI and AD (Greicius et al., 2003; Greicius et al., 2004; Rombouts et al., 2005a; Wang et al., 2006). However, when attempting to elicit a response in a compromised brain, we have previously found that a less constrained baseline task is preferred in order to increase sensitivity to the BOLD response in traditional cognitive subtraction paradigms, especially when statistical power is limited due to single-case studies (Peck et al., 2004). However, in some cases the comparison between levels of a cognitive task can be quite informative. For example, several fMRI studies comparing novel and familiar items report reductions in the expected differential brain response, suggesting decreased ability to distinguish novel from familiar, which may be an important distinction regarding hippocampal functioning. Consistent with Peck et al. (2004), these same studies report increased brain response when comparing novel items to fixation (Fleisher et al., 2005).
Assessment of performance
Although it has been common practice to challenge the MTL, given its primacy in the pathologic decline in AD, this raises the question of whether decreased performance drives group differences in brain response. Strategies to avoid the potential confound of performance differences include incorporation of tasks that AD patients can still perform accurately or selectively including only accurate trials in data analysis. For example, in a study by Gould et al. (2005), after matching performance across groups by examining only successful encoding and retrieval attempts with adjustment for task difficulty on an individual basis, thereby equating group performance and relative levels of difficulty, no group differences were found. However, across groups the BOLD response increased linearly with task difficulty.
Need for case studies and longitudinal study designs
The applicability of fMRI to clinical practice relies on the ability to translate findings from group studies to individual patients. Surprisingly, although fMRI case studies are relatively prevalent across other neurological disorders (e.g., stroke), there is a paucity of fMRI case studies in the Alzheimer literature. However, in an elegant design that investigated five nondemented family members at genetic risk for the development of familial AD, two of whom were carriers of the PSEN1 mutation (C410Y), and 21 control participants, Mondadori et al. (2006a) reported alterations in functional activity on the single-subject level ostensibly decades before the clinical manifestations of AD are apparent. Specifically, of the two carriers of the PSEN1 mutation, the younger adult (age 20) demonstrated subtle episodic memory problems and the middle-aged adult (age 45) met criteria for MCI. Consistent with previous group studies, the younger mutation carrier demonstrated increased, whereas the middle-aged mutation carrier showed decreased, brain activity within memory-related neural networks specific to episodic learning and retrieval but not working memory (Mondadori et al., 2006a).
Similarly, since the functional correlates in asymptomatic adults at risk for AD are not yet agreed upon, the study of such patterns of activation as prognostic indicators for AD necessitates a longitudinal process to follow the progression or conversion of these signatures to classic phenotypes of AD in order to achieve prognostic value (Sunderland et al., 2006). At this point, the longitudinal component of most fMRI studies involves relating future cognitive changes back to a previously acquired functional scan. Notably, these studies have demonstrated some predictive power in the ability to detect patterns of activity that correspond to eventual cognitive decline (Bookheimer et al., 2000; Dickerson et al., 2004; Lind et al., 2006a; Smith et al., 2005). As techniques for increasing the reliability of the fMRI signal at an individual subject level and equating sensitivity of signal detection across scans continue to be developed, it is anticipated that longitudinal studies at the individual-subject level will become more feasible.
Future directions
In a recent consensus paper reviewing neuroimaging tools to assist dementia diagnosis (Frisoni et al., 2006), it is remarkable that fMRI was not presented or discussed. Rather, the review focused on structural volumetric techniques, perfusion, and CSF biomarkers (e.g., tau and amyloid β proteins). This omission perhaps highlights the nascent state with which fMRI is regarded and uncertainty over fMRI’s value—either alone or in combination with other techniques—in the early identification of AD. Determining fMRI’s added value vis-à-vis existing neuroimaging techniques will continue to be an important area of research. In addition, as fMRI begins to gain acceptance, further clarification of the evolution of brain and behavioral signatures of preclinical AD through the use of fMRI combined with other techniques as well as with expanded use in additional risk factors will be needed.
Combining BOLD-fMRI with arterial spin labeling
One promising area for future efforts entails the use of arterial spin labeling/blood oxygen level dependent [ASL/BOLD] imaging in order to advance our ability to understand the unique features associated with the development of AD in those at risk. With the increasing application of fMRI techniques to the study of AD, there is a growing need for quantitative measures that can more accurately reflect neural activity and its antecedent changes during the preclinical period. fMRI using ASL/BOLD requires the acquisition and joint analysis of quantitative measure of functional cerebral blood flow and BOLD response to a functional stimulus like episodic memory encoding and a separate hypercapnic challenge (such as a CO2 inhalation task) and may be most useful to address the confounds of using BOLD measures alone to characterize brain function in older adults at risk for AD.
Most fMRI studies—including our own—treat the BOLD response as an indirect qualitative measure of neural activity and interpret BOLD signal differences as differences in neural activity. However, the BOLD signal reflects local changes in deoxyhemoglobin content, which in turn exhibits a complex dependence on changes in cerebral blood flow (CBF), cerebral blood volume (CBV) and the cerebral metabolic rate of oxygen consumption (CMRO2) (Buxton et al., 2004). Of these quantities, CMRO2 is thought to be most tightly linked to neural activity, reflecting the notion that neurons necessarily expend energy to accomplish their work (Hyder, 2004). The positive BOLD response observed in most fMRI experiments reflects the fact that CBF increases relatively more than CMRO2, so that local capillary and venous blood are more oxygenated during increased brain activity. In general, the actual amplitude of the BOLD response reflects a delicate balance between the relative increases in CBF and CMRO2 (Brown et al., this issue).
Factors that affect the coupling between CBF and CMRO2—as is possible with the degenerating AD brain or cerebrovascular changes or both—may therefore alter the BOLD response even when neural activity is unchanged. For example, there is growing evidence that changes in the cerebrovascular system due to age and disease can significantly alter the BOLD signal and complicate its interpretation (D’Esposito et al., 2003). Age-related factors include altered cerebrovascular ultrastructure, reduced elasticity of vessels, increased atherosclerosis, reduced resting state CBF, decreased resting CMRO2, and reduced vascular reactivity to chemical modulators (Bentourkia et al., 2000; Claus et al., 1998; D’Esposito et al., 2003; Kawamura et al., 1993; Markus et al., 2001; Takada et al., 1992; Yamaguchi et al., 1986; Yamamoto et al., 1980). In fMRI studies of the effects of aging, researchers have found a significant age-related decrease in the BOLD signal amplitude (Buckner et al., 2000; Gopinath et al., 2006; Tekes et al., 2005), possibly reflecting age-related decreases in the elasticity of the cerebrovascular system (D’Esposito et al., 2003; Uspenskaia et al., 2004).
As reviewed above, studies of nondemented older (Bondi et al., 2005; Han et al., 2007) or middle-aged adults (Fleisher et al., 2005) have demonstrated BOLD response differences by APOE genotype, positive family history of AD (Johnson et al., 2006), or MCI (Dickerson et al., 2004, 2005; Johnson et al., 2004). For example, we have found that nondemented older adults with the APOE ε4 genotype exhibit an increased BOLD response in comparison to matched non-ε4 counterparts (Bondi et al., 2005; Han et al., 2007), whereas decreased MTL activity has been reported in poorer performing adults with MCI or AD. Increases and/or decreases in BOLD response can be explained by changes both in neural activity or in cerebrovascular functioning. A straightforward interpretation of these findings is shown in Fig. 3. The top row of Fig. 3 demonstrates that a compensatory increase in neural activity in combination with normal coupled increases in both CBF and CMRO2, results in an increase in the BOLD response. However, in the face of altered neurovascular coupling or impaired CBF response, a decrease in the BOLD response may be seen. Alternatively, a decrease in neural activity along with normal coupling of CBF and CMRO2 may lead to a decrease in BOLD response whereas a decrease in neural activity and with normal coupling of CBF but lower baseline CBF would lead to an increase in the measured BOLD response (Fig. 3, row 2). Other interpretations of the increased BOLD response that do not necessarily relate to neural activity are also plausible. As an example, the third row shows another scenario in which an increase in vasoreactivity leads to a relatively greater percent change in CBF with relatively little change in neural activity and change in the CMRO2; this combination would also lead to an increase in the BOLD response. Finally, the fourth row shows a scenario in which baseline CBF is reduced, leading to an increase in the baseline deoxyhemoglobin content. In this case, if percent change in CBF and CMRO2 were relatively unaffected, an increase in BOLD signal would also be seen.
Fig. 3.

Possible neural and perfusion mechanisms resulting in either an increase or decrease in the BOLD response. BOLD = blood oxygen level dependent; CBF = cerebral blood flow; CMRO2 = cerebral metabolic rate of oxygen consumption
Therefore, ASL/BOLD imaging would have the potential to identify each of these components to the hemodynamic response function and thereby provide a more complete accounting of the functional changes occurring in specified brain regions during the performance of cognitive or other tasks. With these combined measures, one could begin to characterize the impact of advancing age, MCI, genetic risk, cerebrovascular compromise, etc., on the CMRO2 response to a cognitive function such as episodic memory. In short, quantitative fMRI with combined ASL/BOLD can address questions that cannot be answered with BOLD measures alone (Brown et al., 2007).
Combining BOLD-fMRI with diffusion tensor imaging
On its own, fMRI offers considerable promise as a non-invasive technique for detecting early functional brain changes in AD. However, the functional neuroanatomy of preclinical AD could also be explored more comprehensively if the integrity of neural connectivity between active cortical regions could also be incorporated. Diffusion tensor imaging (DTI) provides a useful tool for assessing microstructural changes in white matter, and the combination of fMRI with DTI would provide valuable complementary information in establishing the cortical networks—and their anatomical connections—mediating associative encoding. Valdés-Sosa, Kötter, and Friston (2005) indicate that the “comparison of in vivo diffusion MRI-based tractography information with physiological connectivity measures in the same subjects has not been carried out systematically” and suggest this as a promising new direction.
For example, within the area of memory, much of the research on the anatomical substrates of memory dysfunction in AD risk has rightly focused on the integrity of gray matter structures (e.g., hippocampus, association cortices), although there is increasing recognition of the importance of white matter changes as well. Baxter et al. (2006) demonstrate that global cognitive decline in AD relates to both gray and white matter reductions, and Stoub et al. (2006) have shown that specific white matter volume decreases in the parahippocampal region contribute to memory decline. Diffusion tensor imaging is another MRI technique that shows high sensitivity for detecting microscopic structural changes in white matter and holds promise for the early detection of AD. For example, researchers have shown that assessing microstructural changes in white matter with DTI may be more sensitive to the detection of early AD than macrostructural measures of brain volume alone (Kantarci et al., 2005; Muller et al., 2007; Persson et al., 2006; Rose et al., 2006). Relatively few studies have used DTI to characterize the microstructural white matter changes that occur in those at risk for AD (cf. Medina et al., 2006; Wang et al., 2006; Delano-Wood et al., 2007), and none of these prior efforts have correlated diffusion parameters with fMRI. Nevertheless, a growing number of studies demonstrate that the combined use of fMRI and DTI to characterize structure-function relationships in visual (Toosy et al., 2004) and motor systems (Guye et al., 2003), as well as higher-order cognitive systems such as attention (Madden et al., 2006) and encoding (Takahashi et al., 2007) is possible.
No study has yet combined both imaging approaches to compare fMRI activity with alterations in white matter integrity in those at risk for AD, and future research designed to combine these measures in an effort to correlate conditions in the underlying brain substrata with risk factors for early cognitive decline in AD could provide important new information. For example, is the BOLD-fMRI response to episodic memory encoding constrained by white matter disconnectivity between the subserving cortical regions involved? It may be the case that white matter integrity between active cortical regions will mediate risk-related increases in activation in an encoding network or identify ways in which such a network is faulty.
Combining BOLD-fMRI with novel methods to identify AD risk
Aside from fMRI techniques, future efforts may also benefit from other genetic and behavioral susceptibilities by which to identify older adults who may later convert to AD. Expansion of fMRI studies with newly identified risk factors for AD could provide for important new as well as confirmatory findings to the studies reviewed. For example, our research group has also characterized risk groups and those who convert to AD through the use of cognitive discrepancies between pairs of tests, rather than through traditional methods of analyzing overall group means on individual cognitive tasks (Houston et al., 2005; Jacobson et al., 2002, 2005a, b; Wetter et al., 2005, 2006). Although the episodic memory decline described above appears to be one of the most salient markers of preclinical AD, our group has also shown that mild asymmetric cognitive decline may also detect preclinical AD. In an initial case-control study, we (Jacobson et al., 2002) demonstrated that the initial presentation of cognitive deficits in AD may have asymmetrical involvement as a common feature (i.e., language decrements significantly greater than visuospatial decrements, or vice versa). Measures of asymmetric cognitive profiles were derived using difference scores on tests of verbal and visuospatial ability. Although both groups performed similarly on the individual cognitive tests (i.e., mean score analyses between groups), the use of difference scores measuring asymmetric cognitive performance yielded consistent evidence of subtle differences in cognition in a subgroup of preclinical AD patients. The preclinical AD group showed significantly larger discrepancies between naming and visuoconstructive skills relative to matched control participants, and a higher frequency of asymmetric cognitive profiles compared to a larger normative group.
We have further demonstrated such cognitive discrepancies on tests of auditory and spatial attention (Jacobson et al., 2005b), verbal and design fluency (Houston et al., 2005), global versus local item processing (Jacobson et al., 2005a), response inhibition and cognitive flexibility (Wetter et al., 2005), as well as heterogeneity in verbal memory (Wetter et al., 2006). Taken together, this line of research suggests that cognitive discrepancy measures not only appear to be a useful method for identifying individuals at risk for cognitive deficits, but they also show promise in predicting those who decline. Therefore, utilization of cognitive discrepancies—either alone or in combination with other risk factors—may yield important improvements in the positive predictive value of conversion to AD and thus would be of interest for fMRI efforts in this area. For example, we would expect that cognitive asymmetries might correspond to hemispheric asymmetries in structure or function and, when both are present, improvements in prediction of conversion to AD might result than either alone.
Brief case example
To demonstrate the feasibility of performing quantitative fMRI in the MTL and to examine whether age-related differences exist in the decoupling of the CBF and CMRO2 ratio, we performed a preliminary study in which we extended an fMRI episodic memory encoding experiment to include a hypercapnic task (three minutes of 5% CO2; two repeats) on one young (age 40) and one healthy older adult (age 67) with a positive family history of AD. Participants performed a picture encoding task in which they viewed a series of novel and familiar landscape scenes, presented in a block design and repeated over three 4-minute runs. Data were also acquired at rest for quantification of baseline CBF. To calibrate the BOLD and CBF measurements, a 5% CO2 challenge was administered consisting of two minutes of room air followed by three minutes of CO2 and two minutes of room air for two runs. We used a breathing circuit for the hypercapnia study, in which the subjects breathed through a mouthpiece connected to a low-deadspace non-rebreathing valve (Hans Rudolph or similar). A three-way valve on the inspired limb of the circuit was used to switch between room air (0% CO2) and CO2 enriched gas (5% CO2, 21% O2, balance N2). Inspired gas was dispensed into a small Douglas bag connected to the inspired limb of the breathing circuit.
For each subject, a hippocampal region of interest was defined to include both the hippocampus and parahippocampal gyrus using a high-resolution anatomic T1-weighted image. From the combination of the CBF and BOLD responses to the memory encoding and hypercapnic tasks, we calculated estimates of the percent change in the cerebral metabolic rate of oxygen utilization in response to memory encoding and the neurovascular coupling ratio (n) using the method proposed by Davis et al. (1998) (see Brown et al., 2007, this issue for more details). As shown in Fig. 4, the older adult demonstrated greater BOLD response, suggesting compensatory reserve. The increased BOLD response was observed, even though the two individuals showed similar percent change in the cerebral metabolic rate of oxygen utilization (%ΔCMRO2) and cerebral blood flow (%ΔCBF) during picture encoding. The %ΔCBF and %ΔCMRO2 results imply normal brain activation rather than a compensatory reserve. The apparently disparate findings between the BOLD results, on the one hand, and %ΔCBF and %ΔCMRO2, on the other, were due to differences in the proportionality constant, M, from the Davis deoxyhemoglobin dilution model (See Brown et al., 2007, Eq. (1)). The proportionally constant was 0.07 for the younger adult and 0.14 for the older adult. Differences in M imply differences in baseline fractional blood volume or differences in baseline O2 extraction fraction. According to the deoxyhemoglobin wash out model, BOLD contrast can give misleading information about the underlying metabolic activity of brain cells, when differences in baseline CBV or CMRO2 are present. When baseline CBV or metabolism vary among individuals, %ΔCMRO2 might more accurately reflect brain activation. No age-related differences were found in neurovascular coupling. The coupling factor between CBF and CMRO2 changes (n = %ΔCBF/%Δ CMRO2) was found to be 1.6 for both the younger and older adults. These values are in good agreement with typical values found in the hippocampus (Restom et al., 2007) and slightly lower than values found in quantitative fMRI studies of visual and motor cortices (Hoge et al., 1999; Stefanovic et al., 2005). Taken together, these findings highlight the ambiguity of the BOLD response and bring to light the problems of interpreting fMRI data at the individual subject level without calibrated quantification, as several neurovascular variables can contribute to an increased BOLD response.
Fig. 4.
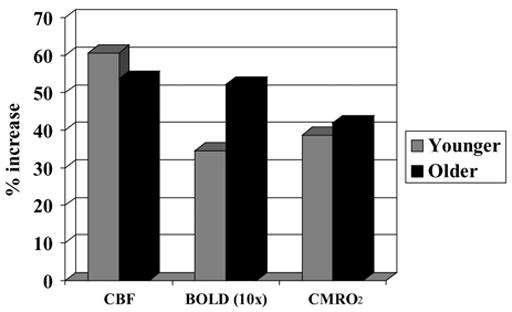
Quantified CBF, BOLD (magnified 10 ×), and CMRO2 responses to picture encoding in a young adult (gray) and an older adult (black). BOLD = blood oxygen level dependent; CBF = cerebral blood flow; CMRO2 = cerebral metabolic rate of oxygen consumption
Summary
Practical considerations such as availability, accessibility, cost, and technical feasibility will continue to influence practitioners’ decisions regarding the use of brain imaging techniques. The National Institute on Aging consensus conference meeting in 1998 defined the ideal characteristics of a diagnostic marker as ease of use, simplicity, and cost effectiveness. Clearly, the non-invasive nature and relatively high temporal and spatial resolution of BOLD-fMRI have made it an essential research tool for studies of the working human brain. Moreover, potential benefits of fMRI include the ability to detect disease-related changes in the brain and monitor their progression in vivo. Whereas structural neuroimaging modalities are well-equipped to detect pathologic anatomy, functional neuroimaging modalities such as fMRI may be better at identifying and assessing surrogate features of AD, such as possible compensatory upsurges in BOLD response in the MTL in asymptomatic adults with subsequent decrease in BOLD as clinical symptoms become apparent and worsen (Celone et al., 2006). It has been argued that functional and structural changes identified by neuroimaging methods are more closely associated with cognitive symptoms than are histopathologic disease markers and therefore may serve as more sensitive correlates of ensuing cognitive decline. This may also serve to explain why level of AD plaque pathology, for example, is not well correlated with the clinical symptoms of AD (see Terry et al., 1999, for discussion) and highlights the importance of examining and accounting for various contributions to cognitive reserve such as genetic factors, age, education, and co-morbid cerebrovascular pathology that may alter an individual’s reserve capacity (Wolf et al., 2003). This poor association between plaque pathology and cognition will also have important bearing on emerging PET amyloid imaging techniques (Edison et al., 2007; Small et al., 2006) and their ability to accurately predict those individuals who will later convert to AD. What is clear is the multitude of risk factors and neurodegenerative and neuroprotective factors co-occurring in the degenerating brain of an individual with AD and fMRI appears well positioned to provide one of the most complete accountings of the brain and behavioral changes during this dynamic period.
Acknowledgments
This work was supported by NIH R01 AG12674 and P50 AG05131. The authors thank Nikki Horne, Thomas Liu, and Khalid Restom for their invaluable assistance in conducting the case example.
Contributor Information
Christina E. Wierenga, Department of Psychiatry, University of California San Diego, La Jolla, California, USA
Mark W. Bondi, Department of Psychiatry, University of California San Diego and Psychology Service, VA San Diego Healthcare System, La Jolla, California, USA
References
- Albert MS, Moss MB, Tanzi R, Jones K. Preclinical prediction of AD using neuropsychological tests. Journal of the International Neuropsychological Society. 2001;7:631–639. doi: 10.1017/s1355617701755105. [DOI] [PubMed] [Google Scholar]
- Backman L, Andersson JL, Nyberg L, Winblad B, Nordberg A, Almkvist O. Brain regions associated with episodic retrieval in normal aging and Alzheimer’s disease. Neurology. 1999;52:1861–1870. doi: 10.1212/wnl.52.9.1861. [DOI] [PubMed] [Google Scholar]
- Bangen KJ, Restom K, Liu TT, Jak AJ, Han SD, Fleisher AS, Salmon DP, Thal LJ, Bondi MW. Hippocampal perfusion during picture encoding: A comparison between younger and older adults. Human Brain Mapping. 2006 (abstract) [Google Scholar]
- Bassett SS, Yousem DM, Cristinziio C, Kusevic I, Yassa MA, Caffo BS, Zeger SL. Familial risk for Alzheimer’s disease alters fMRI activation patterns. Brain. 2006;129:1229–1239. doi: 10.1093/brain/awl089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter LC, Sparks DL, Johnson SC, Lenoski B, Lopez JE, Connor DJ, Sabbagh MN. Relationship of cognitive measures and gray and white matter in Alzheimer’s disease. Journal of Alzheimers Disorder. 2006;9:253–260. doi: 10.3233/jad-2006-9304. [DOI] [PubMed] [Google Scholar]
- Becker JT, Mintun MA, Aleva K, Wiseman MB, Nichols T, DeKosky ST. Compensatory reallocation of brain resources supporting verbal episodic memory in Alzheimer’s disease. Neurology. 1996;46:692–700. doi: 10.1212/wnl.46.3.692. [DOI] [PubMed] [Google Scholar]
- Bentourkia M, Bol A, Ivanoiu A, Labar D, Sibomana M, Coppens A, Michel C, Cosnard G, De Volder AG. Comparison of regional cerebral blood flow and glucose metabolism in the normal brain: Effect of aging. Journal of Neurological Sciences. 2000;181:19–28. doi: 10.1016/s0022-510x(00)00396-8. [DOI] [PubMed] [Google Scholar]
- Blackwell AD, Sahakian BJ, Vessey R, Semple JM, Robbins TW, Hodges JR. Detecting dementia: Novel neuropsychological markers of preclinical Alzheimer’s disease. Dementia and Geriatric Cognitive Disorders. 2004;17:42–48. doi: 10.1159/000074081. [DOI] [PubMed] [Google Scholar]
- Bondi MW, Houston WW, Eyler LT, Brown GG. fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer’s disease. Neurology. 2005;64:501–508. doi: 10.1212/01.WNL.0000150885.00929.7E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondi MW, Monsch AU, Galasko D, Butters N, Salmon DP, Delis DC. Preclinical cognitive markers of dementia of the Alzheimer type. Neuropsychology. 1994;8:374–384. [Google Scholar]
- Bondi MW, Salmon DP, Galasko D, Thomas RG, Thal LJ. Neuropsychological function and apolipoprotein E genotype in the preclinical detection of Alzheimer’s disease. Psychology and Aging. 1999;14:295–303. doi: 10.1037//0882-7974.14.2.295. [DOI] [PubMed] [Google Scholar]
- Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, Small GW. Patterns of brain activation in people at risk for Alzheimer’s disease. New England Journal of Medicine. 2000;343:450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Brewer JB, Moghekar A. Imaging the medial temporal lobe: Exploring new dimensions. Trends in Cognitive Science. 2002;6:217–223. doi: 10.1016/s1364-6613(02)01881-8. [DOI] [PubMed] [Google Scholar]
- Brown GG, Perthen J, Liu TT, Buxton R. A primer on functional magnetic resonance imaging. Neuropsychology Review. 2007 doi: 10.1007/s11065-007-9028-8. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Sanders AL, Raichle ME, Morris JC. Functional brain imaging of young, nondemented, and demented older adults. Journal of Cognitive Neuroscience. 2000;12(Suppl 2):24–34. doi: 10.1162/089892900564046. [DOI] [PubMed] [Google Scholar]
- Burggren AC, Small GW, Sabb FW, Bookheimer SY. Specificity of brain activation patterns in people at genetic risk for Alzheimer disease. American Journal of Geriatric Psychiatry. 2002;10:44–51. [PubMed] [Google Scholar]
- Buxton RB, Uludag K, Dubowitz DJ, Liu TT. Modeling the hemodynamic response to brain activation. Neuroimage. 2004;23(Suppl 1):S220–33. doi: 10.1016/j.neuroimage.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Caselli RJ, Reiman EM, Osborne D, Hentz JG, Baxter LC, Hernandez JL, Alexanter GG. Longitudinal changes in cognition and behavior in asymptomatic carriers of the APOE e4 allele. Neurology. 2004;62:1990–1995. doi: 10.1212/01.wnl.0000129533.26544.bf. [DOI] [PubMed] [Google Scholar]
- Celone KA, Calhoun VD, Dickerson BC, Atri A, Chua EF, Miller SL, DePeau K, Rentz DM, Selkoe DJ, Blacker D, Albert MS, Sperling RA. Journal of Neuroscience. 2006;26:10222–10231. doi: 10.1523/JNEUROSCI.2250-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus JJ, Breteler MM, Hasan D, Krenning EP, Bots ML, Grobbee DE, Van Swieten JC, Van Harskamp F, Hofman A. Regional cerebral blood flow and cerebrovascular risk factors in the elderly population. Neurobiology of Aging. 1998;19:57–64. doi: 10.1016/s0197-4580(98)00004-9. [DOI] [PubMed] [Google Scholar]
- Collie A, Maruff P. The neuropsychology of preclinical Alzheimer’s disease and mild cognitive impairment. Neuroscience and Biobehavioral Reviews. 2000;24:365–374. doi: 10.1016/s0149-7634(00)00012-9. [DOI] [PubMed] [Google Scholar]
- Corkin S, Kennedy AM, Bucci J, et al. Relation between recognition performance and fMRI data in Alzheimer’s disease and older normal subjects. Society for Neuroscience. 1997;23:193–195. [Google Scholar]
- Davis TL, Kwong KK, Weisskoff RM, Rosen BR. Calibrated functional MRI: Mapping the dynamics of oxidative metabolism. Proceedings of the National Academy of Sciences, USA. 1998;95:1834–1839. doi: 10.1073/pnas.95.4.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano-Wood L, Bozoki A, Abeles N. Evidence for heterogeneity in mild cognitive impairment: Differences in neuropsychological profile and associated white matter lesion pathology. International Neuropsychological Society. 2006 doi: 10.1017/S1355617709990257. (published abstract) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano-Wood L, Jak AJ, Schweinsburg BC, Wierenga CE, Horne NR, Salmon DP, Thal LJ, Frank LR, Bondi MW. Posterior white matter changes in MCI: Associations with cognition and stroke risk. Journal of the International Neuropsychological Society. 2007 (abstract) [Google Scholar]
- Desgranges B, Baron JD, de la Sayette V, Petit-Taboue MC, Benali K, Landeau B, Lechevalier B, Eustache F. The neural substrates of memory systems impairment in Alzheimer’s disease. A PET study of resting brain glucose utilization. Brain. 1998;121:611–631. doi: 10.1093/brain/121.4.611. [DOI] [PubMed] [Google Scholar]
- D’Esposito M, Deouell LY, Gazzaley A. Alterations in the BOLD-fMRI signal with ageing and disease: A challenge for neuroimaging. Nature Reviews Neuroscience. 2003;4:863–72. doi: 10.1038/nrn1246. [DOI] [PubMed] [Google Scholar]
- Devanand DP, Pelton GH, Zamora D, Liu X, Tabert MH, Goodkind M, Scarmeas N, Braun I, Stern Y, Mayeux R. Predictive utility of apolipoprotein E genotype for Alzheimer disease in outpatients with mild cognitive impairment. Archives of Neurology. 2005;62:975–980. doi: 10.1001/archneur.62.6.975. [DOI] [PubMed] [Google Scholar]
- Devi G, Ottman R, Tang MX, Marder K, Stern Y, Mayeux R. Familial aggregation of Alzheimer disease among whites, African Americans, and Caribbean Hispanics in northern Manhattan. Archives of Neurology. 2000;57:72–77. doi: 10.1001/archneur.57.1.72. [DOI] [PubMed] [Google Scholar]
- Dickerson BC, Salat DH, Bates JF, Atiya M, Killiany RJ, Greve DN, Dale AM, Stern CE, Blacker D, Albert MS, Sperling RA. Medial temporal lobe function and structure in mild cognitive impairment. Annals of Neurology. 2004;56:27–35. doi: 10.1002/ana.20163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, Bertram L, Mullin K, Tanzi RE, Blacker D, Albert MS, Sperling RA. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005;65:404–411. doi: 10.1212/01.wnl.0000171450.97464.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, Sperling RA. Neuroimaging biomarkers for clinical trials of disease-modifying therapies in Alzheimer’s disease. NeuroRx. 2005;2:348–360. doi: 10.1602/neurorx.2.2.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, Hotton G, Cutler D, Fox N, Kennedy A, Rossor M, Brooks DJ. Amyloid, hypometabolism, and cognition in Alzheimer disease: An [11C]PIB and [18F]FDG PET study. Neurology. 2007;68:501–508. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Festa EK, Insler RZ, Salmon DP, Paxton J, Hamilton JM, Heindel WC. Neocortical disconnectivity disrupts sensory integration in Alzheimer’s disease. Neuropsychology. 2005;19:728–738. doi: 10.1037/0894-4105.19.6.728. [DOI] [PubMed] [Google Scholar]
- Filbey FM, Slack KJ, Sunderland TP, Cohen RM. Functional magnetic resonance imaging and magnetoencephalography differences associated with APOE epsilon 4 in young healthy adults. Neuroreport. 2006;17:1585–1590. doi: 10.1097/01.wnr.0000234745.27571.d1. [DOI] [PubMed] [Google Scholar]
- Fleisher AS, Houston WS, Eyler LT, Frye S, Jenkins C, Thal LJ, Bondi MW. Identification of Alzheimer disease risk by functional magnetic resonance imaging. Archives of Neurology. 2005;62:1881–1888. doi: 10.1001/archneur.62.12.1881. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. ‘Mini-mental State.’ A practical method for grading the cognitive status of patients for the clinician. Journal of Psychiatry Research. 1976;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Fratiglioni L. Epidemiology of Alzheimer’s disease. Issues of etiology and validity. Acta Neurologica Scandinavia Supplement. 1993;145:1–70. [PubMed] [Google Scholar]
- Frisoni GB. Dementia: Important advances in research in 2006. Lancet Neurology. 2007;6:4–5. doi: 10.1016/S1474-4422(06)70659-7. [DOI] [PubMed] [Google Scholar]
- Frisoni GB, Scheltens P, Galluzzi S, Nobili FM, Fox NC, Robert PH, Soininen H, Wahlund L-O, Waldemar G, Salmon E. Neuroimaging tools to rate regional atrophy, subcortical cerebrovascular disease, and regional cerebral blood flow and metabolism: Consensus paper of the EADC. Journal of Neurology, Neurosurgery, and Psychiatry. 2006;74:1371–1381. doi: 10.1136/jnnp.74.10.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golay X, Hendrikse J, Lim TC. Perfusion Imaging Using Arterial Spin Labeling. Topics in Magnetic Resonance Imaging. 2004;15:10–27. doi: 10.1097/00002142-200402000-00003. [DOI] [PubMed] [Google Scholar]
- Golby A, Silverberg F, Race E, Gabrieli S, O’Shea J, Knierim K, Stebbins G, Gabrieli J. Memory encoding in Alzheimer’s disease: An fMRI study of explicit and implicit memory. Brain. 2005;128:773–787. doi: 10.1093/brain/awh400. [DOI] [PubMed] [Google Scholar]
- Gopinath K, Wierenga CE, Conway T, Crosson B, Briggs R. Differential BOLD hemodynamics in young and elderly: Emphasis on post-stimulus undershoot. Proceedings of the International Society for Magnetic Resonance in Medicine. 2006;14 [Google Scholar]
- Gould RL, Brown RG, Owen AM, Bullmore ET, Williams SCR, Howard RJ. Functional neuroanatomy of successful paired associate learning in Alzheimer’s disease. American Journal of Psychiatry. 2005;162:2049–2060. doi: 10.1176/appi.ajp.162.11.2049. [DOI] [PubMed] [Google Scholar]
- Greicius MD, Krasnow B, Reiss AL, Menon V. Functional connectivity in the resting brain: A network analysis of the default mode hypothesis. Proceedings of the National Academy of Science, USA. 2003;100:253–258. doi: 10.1073/pnas.0135058100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: Evidence from functional MRI. Proceedings of the National Academy of Sciences, USA. 2004;101:4637–4642. doi: 10.1073/pnas.0308627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grober E, Kawas C. Learning and retention in preclinical and early Alzheimer’s disease. Psychology and Aging. 1997;12:183–188. doi: 10.1037//0882-7974.12.1.183. [DOI] [PubMed] [Google Scholar]
- Gron G, Riepe MW. Neural basis for the cognitive continuum in episodic memory from health to Alzheimer disease. American Journal of Geriatric Psychiatry. 2004;12:648–652. doi: 10.1176/appi.ajgp.12.6.648. [DOI] [PubMed] [Google Scholar]
- Grossman M, Koenig P, Glosser G, DeVita C, Moore P, Rhee J, Detre J, Alsop D, Gee J. Neural basis for semantic memory difficulty in Alzheimer’s disease: An fMRI study. Brain. 2003;126:292–311. doi: 10.1093/brain/awg027. [DOI] [PubMed] [Google Scholar]
- Guye M, Parker GJ, Symms M, Boulby P, Wheeler-Kingshott CA, Salek-Haddadi A, Barker GJ, Duncan JS. Combined functional MRI and tractography to demonstrate the connectivity of the human primary motor cortex in vivo. NeuroImage. 2003;19:1349–13460. doi: 10.1016/s1053-8119(03)00165-4. [DOI] [PubMed] [Google Scholar]
- Hamalainen A, Pihlajamaki M, Tanila H, Hanninen T, Niskanen E, Tervo S, Karjalainen PA, Vanninen RL, Soininen H. Increased fMRI responses during encoding in mild cognitive impairment. Neurobiology of Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.08.008. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Han SD, Drake AI, Cessante LM, Jak AJ, Houston WS, Delis DC, Filoteo JV, Bondi MW. APOE and TBI in a U.S. military population: Evidence of a neuropsychological compensatory mechanism? Journal of Neurology, Neurosurgery & Psychiatry. 2007 doi: 10.1136/jnnp.2006.108183. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SD, Houston WS, Jak AJ, Eyler LT, Nagel BJ, Fleisher AS, Brown GG, Corey-Bloom J, Salmon DP, Thal LJ, Bondi MW. Verbal paired-associate learning by APOE genotype in non-demented older adults: fMRI evidence of a right hemisphere compensatory response. Neurobiology of Aging. 2006 doi: 10.1016/j.neurobiolaging.2005.12.013. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri AR, Goldberg TE, Mattay VS, Kolachana BS, Callicott JH, Egan MF, Weinberger DR. Brain-derived neurotrophic factor val66met polymorphism affects human memory-related hippocampal activity and predicts memory performance. Journal of Neuroscience. 2003;23:6690–6694. doi: 10.1523/JNEUROSCI.23-17-06690.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoge RD, Atkinson J, Gill B, Crelier GR, Marrett S, Pike GB. Linear coupling between cerebral blood flow and oxygen consumption in activated human cortex. Proceedings of the National Academy of Sciences, USA. 1999;96(16):9403–9408. doi: 10.1073/pnas.96.16.9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston WS, Delis DC, Lansing A, Cobell C, Jacobson M, Salmon DP, Bondi MW. Executive function asymmetry in older adults genetically at risk for Alzheimer’s disease: Verbal versus design fluency. Journal of the International Neuropsychological Society. 2005;11:863–870. doi: 10.1017/s1355617705051015. [DOI] [PubMed] [Google Scholar]
- Huang W, Qui C, von Strauss E, Winblad B, Fratiglioni L. APOE genotype, family history of dementia, and Alzheimer disease risk: A 6-year follow-up study. Archives of Neurology. 2004;61:1930–1934. doi: 10.1001/archneur.61.12.1930. [DOI] [PubMed] [Google Scholar]
- Hyder F. Neuroimaging with calibrated fMRI. Stroke. 2004;35(11 Suppl 1):2635–2641. doi: 10.1161/01.STR.0000143324.31408.db. [DOI] [PubMed] [Google Scholar]
- Jacobs DM, Sano M, Dooneief G, Marder K, Bell KL, Stern Y. Neuropsychological detection and characterization of preclinical Alzheimer’s disease. Neurology. 1995;45:957–962. doi: 10.1212/wnl.45.5.957. [DOI] [PubMed] [Google Scholar]
- Jacobson MW, Delis DC, Lansing A, Houston WS, Olsen R, Wetter S, Bondi MW, Salmon DP. Asymmetries in global and local processing in elderly with the APOE ε4 allele. Neuropsychology. 2005a;19:822–829. doi: 10.1037/0894-4105.19.6.822. [DOI] [PubMed] [Google Scholar]
- Jacobson M, Delis DC, Bondi MW, Salmon DP. Asymmetry in auditory and spatial attention span in normal elderly genetically at risk for Alzheimer’s disease. Journal of Clinical and Experimental Neuropsychology. 2005b;27:240–253. doi: 10.1080/13803390490515441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M, Delis DC, Bondi MW, Salmon DP. Do neuropsychological tests detect preclinical Alzheimer’s disease: Individual-test versus cognitive-discrepancy score analysis. Neuropsychology. 2002;16:132–139. doi: 10.1037//0894-4105.16.2.132. [DOI] [PubMed] [Google Scholar]
- Johnson SC, Baxter LC, Susskind-Wilder L, Connor DJ, Sabbagh MN, Caselli RJ. Hippocampal adaptation to face repetition in healthy elderly and mild cognitive impairment. Neuropsychologia. 2004;42:980–989. doi: 10.1016/j.neuropsychologia.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Johnson SC, Saykin AJ, Baxter LC, Flashman LA, Santulli RB, McAllister TW, Mamourian AC. The relationship between fMRI activation and cerebral atrophy: Comparison of normal aging and Alzheimer disease. Neuroimage. 2000;11:179–187. doi: 10.1006/nimg.1999.0530. [DOI] [PubMed] [Google Scholar]
- Johnson SC, Schmitz TW, Trivedi MA, Ries ML, Torgerson B, Carlsson C, Asthana S, Hermann B, Sager MA. The influence of Alzheimer disease family history and APOE e4 on mesial temporal lobe activation. Journal of Neuroscience. 2006;26:6069–6076. doi: 10.1523/JNEUROSCI.0959-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorm AF, Mather KA, Butterworth P, Anstey KJ, Christensen H, Easteal S. APOE genotype and cognitive functioning in a large age-stratified population sample. Neuropsychology. 2007;21:1–8. doi: 10.1037/0894-4105.21.1.1. [DOI] [PubMed] [Google Scholar]
- Kantarci K, Petersen RC, Boeve BF, Knopman DS, Weigand SD, O’Brien PC, Shiung MM, Smith GE, Ivnik RJ, Tangalos EG, Jack CR. DWI predicts future progression to Alzheimer disease in amnestic mild cognitive impairment. Neurology. 2005;64:902–904. doi: 10.1212/01.WNL.0000153076.46126.E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Knopman D, Liu HY. Dissociation of regional activation in mild AD during visual encoding —a functional MRI study. Neurology. 2001;57:812–816. doi: 10.1212/wnl.57.5.812. [DOI] [PubMed] [Google Scholar]
- Kawamura J, Terayma Y, Takashima S, Obara K, Pavol MA, Meyer JS, Mortel KF, Weathers S. Leuko-araiosis and cerebral perfusion in normal aging. Experimental Aging Research. 1993;19:225–240. doi: 10.1080/03610739308253935. [DOI] [PubMed] [Google Scholar]
- Kawas C, Katzman R. The epidemiology of dementia and Alzheimer disease. In: Terry RD, Katzman R, Bick KL, Sisodia SS, editors. Alzheimer disease. 2. New York: Raven Press; 1999. [Google Scholar]
- Knopman DS, DeKosky ST, Cummings JL, Chui H, Corey-Bloom J, Relkin N, Small GW, Miller B, Stevens JC. Practice parameter: Diagnosis of dementia (an evidence-based review) Neurology. 2001;56:1143–1153. doi: 10.1212/wnl.56.9.1143. [DOI] [PubMed] [Google Scholar]
- Lange KL, Bondi MW, Galasko DG, Delis DC, Salmon DP, Thal LJ. Decline in verbal memory during preclinical Alzheimer’s disease: Examination of the effect of Apolipoprotein E genotype. Journal of the International Neuropsychological Society. 2002;8:943–955. doi: 10.1017/s1355617702870096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rue A, Matsuyama SS, McPherson S, Sherman J, Jarvik LF. Cognitive performance in relatives of patients with probable Alzheimer’s disease: An age at onset effect? Journal of Clinical and Experimental Neuropsychology. 1992;14:533–538. doi: 10.1080/01688639208402842. [DOI] [PubMed] [Google Scholar]
- Levy JA, Bergeson J, Putnam K, Rosen V, Cohen R, Lalonde F, Mirza N, Linker G, Sunderland T. Context-specific memory and apolipoprotein E (ApoE) e4: Cognitive evidence from the NIMH prospective study of risk for Alzheimer’s disease. Journal of the International Neuropsychological Society. 2004;10:362–370. doi: 10.1017/S1355617704103044. [DOI] [PubMed] [Google Scholar]
- Li SC, Lindenberger U. Cross-level unification: A computational exploration of the link between deterioration of neurotransmitter systems dedifferentiation of cognitive abilities in old age (pp. 103–146) In: Nilsson LG, Markowitsch HJ, editors. Cognitive neuroscience of memory. Seattle, WA: Hogrefe & Huber; 1999. [Google Scholar]
- Lind J, Ingvar M, Persson J, Sleegers K, Van Broeckhoven C, Adolfsson R, Nilsson LG, Nyberg L. Parietal cortex activation predicts memory decline in apolipoprotein E e4 carriers. NeuroReport. 2006a;17:1683–1686. doi: 10.1097/01.wnr.0000239954.60695.c6. [DOI] [PubMed] [Google Scholar]
- Lind J, Persson J, Ingvar M, Larsson A, Cruts M, Van Broeckhoven C, Adolfsson R, Backman L, Nilsson LG, Petersson KM, Nyberg L. Reduced functional brain activity response in cognitively intact apolipoprotein E e4 carriers. Brain. 2006b;129:1240–1248. doi: 10.1093/brain/awl054. [DOI] [PubMed] [Google Scholar]
- Lustig C, Snyder AZ, Ghakta M, O’Brien KC, McAvoy M, Raichle ME, Morris JC, Buckner RL. Functional deactivations: Change with age and dementia of the Alzheimer type. Proceedings of the National Academy of Sciences, USA. 2003;100:14505–14509. doi: 10.1073/pnas.2235925100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machulda MM, Ward HA, Borowski B, Gunter JL, Cha RH, O’Brien PC, Petersen RC, Boeve BF, Knopman D, Tang-Wai DF, Ivnik RJ, Smith GE, Tangalos EG, Jack CR. Comparison of memory fMRI response among normal, MCI, and Alzheimer’s patients. Neurology. 2003;61:500–506. doi: 10.1212/01.wnl.0000079052.01016.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden DJ, Spaniol J, Whiting WL, Bucor B, Provenzale JM, Cabeza R, White LE, Huttel SA. Adult age differences in the functional neuroanatomy of visual attention: A combined fMRI and DTI study. Neurobiology of Aging. 2006;28:457–476. doi: 10.1016/j.neurobiolaging.2006.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus H, Cullinane M. Severely impaired cerebrovascular reactivity predicts stroke and TIA risk in patients with carotid artery stenosis and occlusion. Brain. 2001;124:457–467. doi: 10.1093/brain/124.3.457. [DOI] [PubMed] [Google Scholar]
- Masdeu JC, Zubieta JL, Arbizu J. Neuroimaging as a marker of the onset and progression of Alzheimer’s disease. Journal of the Neurological Sciences. 2005;236:55–64. doi: 10.1016/j.jns.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Matsuda H. Cerebral blood flow and metabolic abnormalities in Alzheimer’s disease. Annals of Nuclear Medicine. 2001;15:85–92. doi: 10.1007/BF02988596. [DOI] [PubMed] [Google Scholar]
- Mayeux R, Sano M. Treatment of Alzheimer’s disease. New England Journal of Medicine. 1999;341:1670–1679. doi: 10.1056/NEJM199911253412207. [DOI] [PubMed] [Google Scholar]
- Medina D, de T-M, Urresta F, Gabrieli JDE, Moseley M, Fleischman D, Bennett DA, Leurgans S, Turner DA, Stebbins GT. White matter changes in mild cognitive impairment and AD: A diffusion tensor imaging study. Neurobiology of Aging. 2006;27:663–672. doi: 10.1016/j.neurobiolaging.2005.03.026. [DOI] [PubMed] [Google Scholar]
- Mondadori CRA, Buchmann A, Mustovic H, Schmidt CF, Boesiger P, Nitsch RM, Hock C, Streffer J, Henke K. Enhanced brain activity may precede the diagnosis of Alzheimer’s disease by 30 years. Brain. 2006a;129:2908–2922. doi: 10.1093/brain/awl266. [DOI] [PubMed] [Google Scholar]
- Mondadori CRA, de Quervain DJ-F, Buchmann A, Mustovic H, Wollmer MA, Schmidt CF, Boesiger P, Hock C, Nitsch RM, Papassotiropoulos A, Henke K. Cerebral Cortex. 2006b. Better memory and neural efficiency in young apolipoprotein E e4 carriers. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Mosconi L, Brys M, Glodzik-Sobanska L, De Santi S, Rusinek H, de Leon MJ. Experimental Gerontology. Early detection of Alzheimer’s disease using neuroimaging. in press. [DOI] [PubMed] [Google Scholar]
- Muller MJ, Greverus D, Weibrich C, Dellani PR, Scheurich A, Stoeter P, Fellgiebel A. Diagnostic utility of hippocampal size and mean diffusivity in amnestic MCI. Neurobiology of Aging. 2007;28:398–403. doi: 10.1016/j.neurobiolaging.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Pariente J, Cole S, Henson R, Clare L, Kennedy A, Rossor M, Cipoloti L, Puel M, Demonet JF, Chollet F, Frackowiak RSJ. Alzheimer’s patients engage an alternative network during a memory task. Annals of Neurology. 2005;58:870–879. doi: 10.1002/ana.20653. [DOI] [PubMed] [Google Scholar]
- Peck KK, Wierenga CE, Bacon Moore A, Maher L, Gopinath K, Gaiefsky M, Briggs RW, Crosson B. Comparison of baseline conditions to investigate syntactic production using functional magnetic resonance imaging. NeuroImage. 2004;23:104–110. doi: 10.1016/j.neuroimage.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Persson J, Lind J, Larsson A, Ingvar M, Cruts M, Van Broeckhoven C, Adolfsson R, Nilsson LG, Nyberg L. Altered brain white matter integrity in healthy carriers of the APOE epsilon4 allele: a risk for AD? Neurology. 2006;66:1029–1033. doi: 10.1212/01.wnl.0000204180.25361.48. [DOI] [PubMed] [Google Scholar]
- Petersen RC. Mild cognitive impairment: Where are we? Alzheimer Disease Association Dis. 2006;19:166–169. doi: 10.1097/01.wad.0000179417.95584.90. [DOI] [PubMed] [Google Scholar]
- Petersen RC, Morris JC. Mild cognitive impairment as a clinical entity and treatment target. Archives of Neurology. 2005;62:1160–1166. doi: 10.1001/archneur.62.7.1160. [DOI] [PubMed] [Google Scholar]
- Petersen RC, Smith G, Ivnik RJ, Tangalos EG, Schaid DJ, Thibodeau SN, Kokmen E, Waring SC, Kurland LT. APOE status as a predictor of the development of Alzheimer’s disease in memory-impaired individuals. Journal of the American Medical Association. 1995;273:1274–1278. [PubMed] [Google Scholar]
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: Clinical characterization and outcome. Archives of Neurology. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- Petrella JR, Coleman RE, Doraiswamy PM. Neuroimaging and early diagnosis of Alzheimer disease: A look to the future. Radiology. 2003;226:315–336. doi: 10.1148/radiol.2262011600. [DOI] [PubMed] [Google Scholar]
- Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proceedings of the National Academy of Sciences, USA. 2001;98:676–682. doi: 10.1073/pnas.98.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajah MN, D’Esposito M. Region-specific changes in prefrontal function with age: a review of PET and fMRI studies on working and episodic memory. Brain. 2005;128(Pt 9):1964–1983. doi: 10.1093/brain/awh608. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the e4 allele for apolipoprotein E. The New England Journal of Medicine. 1996;334:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proceedings of the National Academy of Sciences, USA. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Correlations between apolipoprotein E e4 gene dose and brain-imaging measurements of regional hypometabolism. Proceedings of the National Academy of Sciences, USA. 2005;102:8299–8302. doi: 10.1073/pnas.0500579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Uecker A, Caselli RJ, Lewis S, Bandy D, de Leon MJ, De Santi S, Convit A, Osborne D, Weaver A, Thibodeau SN. Hippocampal volumes in cognitively normal persons at genetic risk for Alzheimer’s disease. Annals of Neurology. 1998;44:288–291. doi: 10.1002/ana.410440226. [DOI] [PubMed] [Google Scholar]
- Restom K, Perthen JE, Ances BM, Liu TT. Proceedings of the International Society for Magnetic Resonance in Medicine. 2007. Calibrated BOLD in the medial temporal lobe during a memory encoding task. (abstract) [Google Scholar]
- Roberts W, Dember WN, Brodwick M. Alternation and exploration in rats with hippocampal lesions. Journal of Computational and Physiological Psychology. 1962;55:695. doi: 10.1037/h0045168. [DOI] [PubMed] [Google Scholar]
- Rombouts SARB, Barkhof F, Goekoop R, Stam CJ, Scheltens P. Altered resting state networks in mild cognitive impairment and mild Alzheimer’s disease: An fMRI study. Human Brain Mapping. 2005a;26:231–239. doi: 10.1002/hbm.20160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombouts SARB, Barkhof F, Veltman DJ, Machielsen WCM, Witter MP, Bierlaagh MA, Lazeron RHC, Valk J, Scheltens P. Functional MR imaging in Alzheimer’s disease during memory encoding. American Journal of Neuroradiology. 2000;21:1869–1875. [PMC free article] [PubMed] [Google Scholar]
- Rombouts SARB, Goekoop R, Stam CJ, Barkhof F, Scheltens P. Delayed rather than decreased BOLD response as a marker for early Alzheimer’s disease. Neuroimage. 2005b;26:1078–1085. doi: 10.1016/j.neuroimage.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Rose SE, McMahon KL, Janke AL, O’Dowd B, de Zubicaray G, Strudwick MW, Chalk JB. MRI diffusion indices and neuropsychological performance in amnestic mild cogntive impairment. Journal of Neurology Neurosurgery and Psychiatry. 2006;77:1122–1128. doi: 10.1136/jnnp.2005.074336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager MA, Hermann B, La Rue A. Middle-aged children of persons with Alzheimer’s disease: APOE genotypes and cognitive function in the Wisconsin Registry for Alzheimer’s Prevention. Journal of Geriatric Psychiatry and Neurology. 2005;18:245–249. doi: 10.1177/0891988705281882. [DOI] [PubMed] [Google Scholar]
- Salmon DP, Bondi MW. Annual Review of Psychology. 2008. Neuropsychology of aging and dementia. [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, St George-Hyslop PH, Perick-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD. Association of apolipoprotein E allele e4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- Saykin AJ, Flashman LA, Frutiger SA, Johnson SC, Mamourian AC, Moritz CH, O’Jile JR, Riordan HJ, Santulli RB, Smith CA, Weaver JB. Neuroanatomic substrates of semantic memory impairment in Alzheimer’s disease: Patterns of functional MRI activation. Journal of the International Neuropsychological Society. 1999;5:377–392. doi: 10.1017/s135561779955501x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slooter AJC, Breteler MMB, Ott A, Van Broeckhoven C, Van Duijn CM. APOE genotyping in differential diagnosis of Alzheimer’s disease. The Lancet. 1996;348:334. doi: 10.1016/s0140-6736(05)64501-1. [DOI] [PubMed] [Google Scholar]
- Small GW, Ercoli LM, Silverman DH, Huang SC, Komo S, Bookheimer SY, Lavretsky H, Miller K, Siddarth P, Rasgon NL, Mazziotta JC, Sexena S, Wu HM, Mega MS, Cummings JL, Saunders AM, Pericak-Vance MA, Roses AD, Barrio JR, Phelps ME. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proceedings of the National Academy of Science, USA. 2000a;97:6037–6042. doi: 10.1073/pnas.090106797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Nava AS, Perera GM, Delapaz R, Stern Y. Evaluating the function of hippocampal subregions with high-resolution MRI in Alzheimer’s disease and aging. Microscopy Research and Technique. 2000b;51:101–108. doi: 10.1002/1097-0029(20001001)51:1<101::AID-JEMT11>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, Lavretsky H, Burggren AC, Cole GM, Vinters HV, Thompson PM, Huang SC, Satyamurthy N, Phelps ME, Barrio JR. PET of brain amyloid and tau in mild cognitive impairment. New England Journal of Medicine. 2006;21:2652–2663. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- Small SA, Perera GM, DeLaPaz R, Mayeux R, Stern Y. Differential regional dysfunction of the hippocampal formation among elderly with memory decline and Alzheimer’s disease. Annals of Neurology. 1999;45:466–472. doi: 10.1002/1531-8249(199904)45:4<466::aid-ana8>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Smith CD, Andersen AH, Kryscio RJ, Schmitt FA, Kindy MS, Blonder LX, Avison MJ. Altered brain activation in cognitively intact individuals at high risk for Alzheimer’s disease. Neurology. 1999;53:1391–1396. doi: 10.1212/wnl.53.7.1391. [DOI] [PubMed] [Google Scholar]
- Smith CD, Andersen AH, Kryscio RJ, Schmitt FA, Kindy MS, Blonder LX, Avison MJ. Women at risk for AD show increased parietal activation during a fluency task. Neurology. 2002;58:1197–1202. doi: 10.1212/wnl.58.8.1197. [DOI] [PubMed] [Google Scholar]
- Smith CD, Kryscio RJ, Schmitt FA, Lovell MA, Blonder LX, Rayens WS, Andersen AH. Longitudinal functional alterations in asymptomatic women at risk for Alzheimer’s disease. Journal of Neuroimaging. 2005;15:271–277. doi: 10.1177/1051228405277340. [DOI] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Johansen-Berg H, Rueckert D, Nichols TE, Mackay CE, Watkins KE, Ciccarelli O, Cader MZ, Matthews PM, Behrens TE. Tract-based spatial statistics: Voxelwise analysis of multi-subject diffusion data. Neuroimage. 2006;31:1487–1505. doi: 10.1016/j.neuroimage.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Sperling RA, Bates JF, Chua EF, Cocchiarella AJ, Rentz DM, Rosen BR, Schacter DL, Albert MS. fMRI studies of associative encoding in young and elderly controls and mild Alzheimer’s disease. Journal of Neurology, Neurosurgery, and Psychiatry. 2003;74:44–50. doi: 10.1136/jnnp.74.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR. Memory and the hippocampus: A synthesis from findings with rats, monkeys, and humans. Psychological Review. 1992;99:195–231. doi: 10.1037/0033-295x.99.2.195. [DOI] [PubMed] [Google Scholar]
- Stefanovic B, Warnking JM, Rylander KM, Pike GB. The effect of global cerebral vasodilation on focal activation hemodynamics. Neuroimage. 2005 doi: 10.1016/j.neuroimage.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Stoub TR, de T-M, Stebbins GT, Leurgans S, Bennett DA, Shah RC. Hippocampal disconnection contributes to memory dysfunction individuals at risk for Alzheimer’s disease. Proceedings of the National Academy of Sciences, USA. 2006;103:10041–10045. doi: 10.1073/pnas.0603414103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunderland T, Hampal H, Takeda M, Putnam KT, Cohen RM. Biomarkers in the diagnosis of Alzheimer’s disease: Are we ready? Journal of Geriatric Psychiatry and Neurology. 2006;19:172–179. doi: 10.1177/0891988706291088. [DOI] [PubMed] [Google Scholar]
- Takada H, Nagata K, Hirata Y, Satoh Y, Watahiki Y, Sugawara J, Yokoyama E, Kondoh Y, Shishido F, Inugami A, et al. Age-related decline of cerebral oxygen metabolism in normal population detected with positron emission tomography. Neurological Research. 1992;14(2 Suppl):128–131. doi: 10.1080/01616412.1992.11740031. [DOI] [PubMed] [Google Scholar]
- Takahashi E, Ohki K, Kim D. Diffusion tensor studies dissociated two fronto-temporal pathways in the human memory system. NeuroImage. 2007;34:827–838. doi: 10.1016/j.neuroimage.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariot PN, Federoff HJ. Current treatment for Alzheimer disease and future prospects. Alzheimer Disease and Associated Disorders. 2003;17(4 Suppl):105–113. doi: 10.1097/00002093-200307004-00005. [DOI] [PubMed] [Google Scholar]
- Tekes A, Mohamed MA, Browner NM, Calhoun VD, Yousem DM. Effect of age on visuomotor functional MR imaging. Academy of Radiology. 2005;12:739–745. doi: 10.1016/j.acra.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Hansen LA. The neuropathology of Alzheimer disease and the structural basis of its cognitive alterations. In: Terry RD, Katzman R, Bick KL, Sisodia SS, editors. Alzheimer disease. 2. Philadelphia: Lippincott Williams & Wilkins; 1999. [Google Scholar]
- Tiraboschi P, Hansen LA, Masliah E, Alford M, Thal LJ, Corey-Bloom J. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology. 2004;62:1977–1983. doi: 10.1212/01.wnl.0000128091.92139.0f. [DOI] [PubMed] [Google Scholar]
- Tohgi H, Takahashi S, Kato E, Homma A, Niina R, Sasaki K, Yonezawa H, Sasaki M. Reduced size of right hippocampus in 39- to 80-year-old normal subjects carrying the apolipoprotein E epsilon4 allele. Neuroscience Letters. 1997;236:21–24. doi: 10.1016/s0304-3940(97)00743-x. [DOI] [PubMed] [Google Scholar]
- Toosy AT, Ciccarelli O, Parker GJ, Wheeler-Kingshott CA, Miller DH, Thompson AJ. Characterizing function-structure relationships in the human visual system with functional MRI and diffusion tensor imaging. NeuroImage. 2004;21:1452–1463. doi: 10.1016/j.neuroimage.2003.11.022. [DOI] [PubMed] [Google Scholar]
- Trivedi MA, Schmitz TW, Ries M, Torgerson BM, Sager MA, Hermann BP, Asthana S, Johnson SC. Reduced hippocampal activation during episodic encoding in middle-aged individuals at genetic risk of Alzheimer’s disease: A cross-sectional study. BioMed Central. 2006;4:1–14. doi: 10.1186/1741-7015-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twamley EW, Ropacki S, Bondi MW. Neuropsychological and neuroimaging changes in preclinical Alzheimer’s disease. Journal of the International Neuropsychological Society. 2006;12:707–735. doi: 10.1017/S1355617706060863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uspenskaia O, Liebetrau M, Herms J, Danek A, Hamann GF. Aging is associated with increased collagen type IV accumulation in the basal lamina of human cerebral microvessels. BioMed Central Neuroscience. 2004;5:37. doi: 10.1186/1471-2202-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes-Sosa PA, Kotter R, Friston KJ. Introduction: Multimodal neuroimaging of brain connectivity. Phil Trans R Soc B. 2005;360:865–867. doi: 10.1098/rstb.2005.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Duijn CM, Clayton D, Chandra V, Fratiglioni L, Graves AB, Heyman A, Jorm AF, Kokmen E, Kondo K, Mortimer JA. Familial aggregation of Alzheimer’s disease and related disorders: A collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. International Journal of Epidemiology. 1991;20(Suppl 2):13–20. doi: 10.1093/ije/20.supplement_2.s21. [DOI] [PubMed] [Google Scholar]
- Vinogradova OS. Hippocampus as comparator: Role of the two input and two output systems of the hippocampus in selection and registration of information. Hippocampus. 2001;11:578–598. doi: 10.1002/hipo.1073. [DOI] [PubMed] [Google Scholar]
- Wang PJ, Saykin AJ, Flashman LA, Wishart HA, Rabin LA, Santulli RB, McHugh TL, MacDonald JW, Mamouriuan AC. Regionally specific atrophy of the corpus callosum in AD, MCIl and cognitive complaints. Neurobiology of Aging. 2006;27:1613–1617. doi: 10.1016/j.neurobiolaging.2005.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zang Z, He Y, Liang M, Zhang X, Tian L, Wu T, Jiang T, Li K. Changes in hippocampal connectivity in the early stages of Alzheimer’s disease: Evidence from resting state fMRI. Neuroimage. 2006;31:496–504. doi: 10.1016/j.neuroimage.2005.12.033. [DOI] [PubMed] [Google Scholar]
- Wetter S, Delis DC, Houston WS, Jacobson MW, Lansing A, Cobell K, Salmon DP, Bondi MW. Heterogeneity in verbal memory: A marker of preclinical Alzheimer’s disease? Aging, Neuropsychology, and Cognition. 2006;13:503–515. doi: 10.1080/138255890969492. [DOI] [PubMed] [Google Scholar]
- Wetter S, Delis DC, Houston WS, Jacobson MW, Lansing A, Cobell K, Salmon DP, Bondi MW. Deficits in inhibition and flexibility are associated with the APOE ε4 allele in nondemented older adults. Journal of Clinical and Experimental Neuropsychology. 2005;27:943–952. doi: 10.1080/13803390490919001. [DOI] [PubMed] [Google Scholar]
- Wierenga CE, Benjamin M, Gopinath K, Perlstein WM, Leonard CM, Rothi LJ, Conway T, Cato MA, Briggs R, Crosson B. Age-related changes in word retrieval: Role of bilateral frontal and subcortical networks. Neurobiology of Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.10.024. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Wishart HA, Saykin AJ, Rabin LA, Santulli RB, Flashman LA, Guerin SJ, Mamourian AC, Belloni DR, Rhodes CH, McAllister TW. Increased brain activation during working memory in cognitively intact adults with the APOE e4 allele. American Journal of Psychiatry. 2006;163:1603–1610. doi: 10.1176/ajp.2006.163.9.1603. [DOI] [PubMed] [Google Scholar]
- Wolf H, Jelic V, Gertz HJ, Nordberg A, Julin P, Wahlund LO. A critical discussion of the role of neuroimaging in mild cognitive impairment. Acta Neurologica Scandinavica. 2003;107(s179):52–76. doi: 10.1034/j.1600-0404.107.s179.10.x. [DOI] [PubMed] [Google Scholar]
- Woodard JL, Grafton ST, Votaw JR, Green RC, Dobraski ME, Hoffman JM. Compensatory recruitment of neural resources during overt rehearsal of word lists in Alzheimer’s disease. Neuropsychology. 1998;12:491–504. doi: 10.1037//0894-4105.12.4.491. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Kanno I, Uemura K, Shishido F, Inugami A, Ogawa T, Murakami M, Suzuki K. Reduction in regional cerebral metabolic rate of oxygen during human aging. Stroke. 1986;17:1220–1228. doi: 10.1161/01.str.17.6.1220. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Meyer JS, Sakai F, Yamaguchi F. Aging and cerebral vasodilator responses to hypercarbia: responses in normal aging and in persons with risk factors for stroke. Archives of Neurology. 1980;37:489–496. doi: 10.1001/archneur.1980.00500570037005. [DOI] [PubMed] [Google Scholar]
- Yip AG, McKee AC, Green RC, Wells J, Young H, Cupples LA, Farrer LA. APOE, vascular pathology, and the AD brain. Neurology. 2005;65:259–265. doi: 10.1212/01.wnl.0000168863.49053.4d. [DOI] [PubMed] [Google Scholar]