

Abstract
Respiratory syncytial virus (RSV) is the most common cause of bronchiolitis in infants and children worldwide. We wished to determine whether intracheal administration of β-agonists improved alveolar fluid clearance (AFC) across the distal respiratory epithelium of RSV infected mice. Following intranasal infection with RSV strain A2, AFC was measured in anesthetized, ventilated BALB/c mice by instillation of 5% BSA into the dependent lung. We found that direct activation of protein kinase A by forskolin or 8-bromo-cAMP increased AFC at day 2 after infection with RSV. In contrast, short- and long-acting β-agonists had no effect at either day 2 or day 4. Insensitivity to β-agonists was not a result of elevated plasma catecholamines, or lung epithelial cell β-adrenergic receptor degradation. Instead, RSV infected mice had significantly higher levels of phosphorylated PKCζ in the membrane fractions of their lung epithelial cells. In addition, insensitivity to β-agonists was mediated in a paracrine fashion by KC (the murine homolog of CXCL8) and reversed by inhibition of either PKCζ or G protein-coupled receptor kinase 2 (GRK2). These results indicate that insufficient response to β-agonists in RSV may be caused, at least in part, by impaired β-adrenergic receptor signaling, as a consequence of GRK2-mediated uncoupling of β-adrenergic receptors from adenylyl cyclase.
Keywords: Paramyxovirus, Protein kinase C, G protein-coupled receptor kinase 2, CXCL8
Introduction
Respiratory syncytial virus (RSV) is the most common cause of lower respiratory tract disease in infants and children worldwide (44), is a frequent initiator of acute asthma exacerbations in young children, and has a disease impact comparable to that of nonpandemic influenza A in the elderly (8). Approximately 2–3% of all cases of RSV bronchiolitis result in severe hypoxia, or a need for parenteral fluid supplementation that necessitates hospitalization (44). β-agonists are frequently used to treat RSV bronchiolitis, primarily because of their perceived ability to relax airway smooth muscle and cause bronchodilation, with the ultimate aim of alleviating hypoxemia. However, it is not clear that these drugs are clinically effective: meta-analyses have shown little or no overall benefit, irrespective of viral bronchiolitis severity (15,20). Their lack of efficacy has not been explained, although it has often been ascribed to difficulties associated with drug delivery to the small airways of young infants, particularly in the presence of bronchoconstriction and inflammatory exudates or airway obstruction (28). β-agonists increase total body oxygen consumption, thereby increasing oxygen demands in infants hospitalized for respiratory compromise, and can potentially exacerbate ventilation-perfusion mismatch by inducing vasodilation without bronchodilation (12). Indeed, because of an aggregate preponderance of potential harm over therapeutic benefit, the American Academy of Pediatrics recently recommended that bronchodilators should not be used routinely in the management of bronchiolitis.
The process of alveolar fluid clearance (AFC) is crucial to efficient gas exchange in the lung (reviewed in (24,25)) and patients with acute lung injury with intact AFC have lower morbidity and mortality than those with compromised AFC (45). More than 90% of pulmonary β-adrenergic receptors (β-AR) are actually expressed on alveolar epithelial cells, rather than in bronchial epithelium or smooth muscle (3), and β-agonists have been shown to improve AFC in animal models of lung injury in which AFC is impaired (reviewed in (33)). β-agonist prophylaxis has also been shown to be of value in reducing the incidence of high altitude pulmonary edema (itself a consequence of impaired AFC secondary to hypoxia at high altitude) in susceptible mountaineers (42), and intravenous salbutamol treatment can reduce extravascular lung water in patients with acute lung injury (35).
Previously, we demonstrated that infection of BALB/c mice with RSV significantly impairs AFC at early time points after infection (by 43% from mock-infected values at day 2). This decrease in AFC, which is mediated by de novo synthesized UTP acting on P2Y purinergic receptors, was temporally associated with hypoxemia in RSV-infected mice (5). Our studies also showed that RSV-mediated nucleotide release, AFC inhibition, and the associated hypoxemia, could be prevented by pretreatment of mice with the de novo pyrimidine synthesis inhibitor leflunomide (5). These findings suggest that bronchoalveolar edema, occurring as a consequence of reduced active Na+ transport by the respiratory epithelium, may be an unrecognized component of RSV disease that plays a role in development of hypoxemia, either by impairing alveolar gas exchange or by contributing to obstruction of small airways. As described above, results of previous studies indicate that the AFC deficit caused by RSV infection in this model should be corrected by β-agonists (33). We were therefore able to use our model as a functional in vivo assay to directly determine whether or not intra-alveolar instillation of short and long term acting β-agonists can increase AFC after RSV infection. Having determined that β-agonists failed to increase AFC in RSV infected mice, we designed a series of physiological and biochemical studies to identify the potential cellular mechanisms underlying this β-agonist insensitivity.
Materials and Methods
Reagents
8-bromo-cAMP (Sigma-Aldrich, St. Louis, MO, USA), β-agonists (Sigma-Aldrich), propranolol (Sigma-Aldrich), 14–22 amide (EMD Biosciences, La Jolla, CA, USA), adenosine deaminase (Sigma-Aldrich), metRANTES (R & D Systems, Minneapolis, MN, USA), anti-KC mAb (MAB453, R & D Systems), anti-KC pAb (AF-453-NA, R & D Systems), anti-CXCR2 mAb (MAB2164, R & D Systems), anti-CXCR4 pAb (TP503, Torrey Pines Biolabs, Houston, TX, USA), and rat IgG2A (MAB006, R & D Systems) were reconstituted in normal saline. Forskolin (Sigma-Aldrich), amiloride (Sigma-Aldrich), GRK2 inhibitor (EMD Biosciences), and GF109203X (EMD Biosciences) were reconstituted in DMSO. Indomethacin (Sigma-Aldrich) was reconstituted in ethanol. Fresh terbutaline stocks were prepared weekly.
Preparation of viral inocula and infection of mice
Preparation of viral stocks and intranasal infection of eight to twelve week-old pathogen-free BALB/c mice of either sex with endotoxin- and mycoplasma-free RSV strain A2 (106 PFU in 100μl) were performed as previously described (5). All mouse procedures were approved by the UAB Institutional Animal Care and Use Committee.
Alveolar fluid clearance measurements
AFC was measured as previously described All reagents were added to the AFC instillate from stock solutions directly prior to instillation, in a minimal volume of solvent (1–10 μl/ml). Previous studies have demonstrated that measured declines in AFC are not a consequence of instillate dilution by intrapulmonary edema fluid (14,18).
Measurement of plasma catecholamines
EDTA plasma was collected from mice, euthanized following administration of an identical anesthetic regimen to that used in AFC procedures. Epinephrine and norepinephrine levels were measured using the CatCombi ELISA (RDI, Concord, MA).
Alveolar cell isolation and cytoplasmic and membrane fraction preparation
Alveolar cells were isolated from BALB/c mice using an adaptation of the method of Warshamana et al. (46). Cell cytoplasm and membrane fractions were prepared as follows. Briefly, control and RSV-infected cells were lysed in 500 μl of lysis buffer (50 mM Tris-HCl, pH 7.5, 2 mM EDTA, 2 mM EGTA, and 0.2 mM Na3VO4) supplemented with 1X protease inhibitor cocktail (BD Pharmingen, San Diego, CA), then centrifuged at 16,000g for 20 mins at 4°C to separate cytosolic and membrane fractions. The membrane pellet was then lysed in the above buffer plus 1% Triton X-100, 0.5% Nonidet P-40, and 150mM NaCl, and cleared by centrifugation at 16,000g for 10 mins. The supernatant containing membrane proteins was then carefully removed. Protein concentrations in all preparations were measured by the BCA method using BSA as a standard. All protein samples were stored at −80°C prior to use.
Western blotting protocol
Alveolar cell membranes and cytoplasmic proteins were separated by SDS-PAGE and western blots performed using a standard protocol. Blots were probed with rabbit antibodies to PKCζ (sc-216, Santa Cruz Biotechnology, Santa Cruz, CA), then stripped and re-probed for phospho-PKCζ (sc-12894-R, Santa Cruz Biotechnology), and β-actin (Cell Signaling Technology, Danvers, MA). Bound primary antibodies were detected with HRP-conjugated goat anti-rabbit secondary antibodies and enhanced chemiluminescence (GE Healthcare Life Sciences, Piscataway, NJ), followed by exposure to X-ray film. Band intensity was measured using AlphaEaseFC on a FluorChem imager (Alpha Innotech, San Leandro, CA)and normalized to β-actin levels.
Peripheral lung total cell membrane protein preparation
Mice were euthanized, using an identical anesthetic regimen to that for AFC studies, and a thoracotomy was performed. The right ventricle was cannulated and the pulmonary circulation flushed with ice-cold PBS until no visible evidence of blood remained. The peripheral lung tissue from 15–20 mice was pooled for each group (uninfected, day 2, or day 6) and then homogenized in Hb buffer. Total cell membrane protein was isolated and cell membrane fractions were stored at −80°C until used. The assay was repeated three times for each time point.
β2-AR saturation binding assay
Saturation binding was performed as previously described (21). Briefly, 100 μg of whole peripheral lung cell membrane proteins were incubated with incremental concentrations (1–50nM) of the β2-AR-specific inverse agonist [3H]-ICI-118,551 at 37°C for 1 hour with gentle shaking. The reaction was terminated by rapid vacuum filtration, using a cell harvester (Molecular Devices Micro 96, Sunnyvale, CA), through presoaked Printed Filtermat B filters (Wallac, Tuku, Finland), which were washed 5 times and then counted with a micro-beta counter. Non-specific binding was determined by incubating 100 μg of protein with 200 μM unlabeled ICI-118,551 prior to addition of 50 nM [3H]-ICI-118,551 and harvesting. Specific binding was determined by subtracting non-specific binding from total binding. Triplicate measurements were made for each concentration of [3H]-ICI-118,551 tested. Scatchard analysis was performed using Prism statistical software (Graphpad, San Diego, CA).
β2-AR competition binding assay
Competition binding assays were performed as previously described (21). Total peripheral lung cell membranes were prepared as described above for β2-AR saturation binding assays, except two additional centrifugations were performed to ensure the removal of endogenous GTP. 100 μg membranes were incubated with 10 nM of [3H]-ICI-118,551 and 24 incremental concentrations of the β2-AR-specific agonist procaterol (10−10 to 2 × 10−3 M) for 1 h at 37°C. All reactions were performed in duplicate. Competition data were evaluated with one- and two-binding site models by an iterative least-squares technique.
Statistical Analyses
Descriptive statistics were calculated using Instat software (GraphPad). Gaussian data distribution was verified by the method of Kolmogorov and Smirnov. Differences between group means were analyzed by ANOVA, with Tukey-Kramer multiple comparison post-tests. Student’s t test was used to compare group means for data in Figure 2b only. P<0.05 was considered statistically significant. All data are presented as mean ± SEM.
Fig. 2. Effect of β-adrenergic agonists on alveolar fluid clearance in RSV-infected mice.
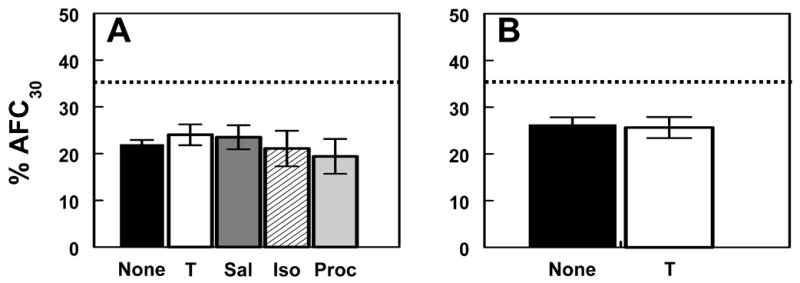
(a) Effect of 100 μM terbutaline (T; n=17), 100 μM salmeterol (Sal; n=14), 100 μM isoproterenol (Iso; n=8), and 100 μM procaterol (Proc; n=7) on AFC at day 2 (no treatment, None; n=10). (b) Effect of 100 μM terbutaline (T; n=9) on AFC at day 4 (no treatment, None; n=16). Dotted line indicates mean AFC rate in untreated, mock-infected animals (n=11).
Results
Effect of protein kinase A activation on AFC
Previously, we demonstrated that AFC is significantly inhibited at day 2 and day 4 after infection of BALB/c mice with RSV (5). For our current studies of β-agonist effects on impaired AFC after RSV infection, we concentrated predominantly on the day 2 time point, when AFC is most suppressed. The mean AFC rate over 30 minutes in RSV-infected mice is 21% at day 2 and 27% at day 4. Thus AFC is reduced by 43% and 26%, respectively, at these time points, from the mean rate in mock-infected animals (36%, which is identical to the rate in uninfected animals (5). There was no difference between day 2 AFC values derived from 3 separate infections using the viral inoculum preparation used in all the experiments described in the current study (21.8 ± 2.3%, n=10) and those we reported previously using 2 historically distinct viral preparations (22 ± 1%, n=25 (5)). Likewise, day 4 AFC values derived using the current inoculum (26.3 ± 1.6%, n=16) were comparable to those we previously reported with a distinct virus preparation (27.4 ± 2.1%, n=9 (5)).
Stimulatory effects of β-agonists on AFC have been shown to be mediated via activation of adenylyl cyclase (AC), which generates cAMP and thereby stimulates cAMP-dependent protein kinase (protein kinase A; PKA – reviewed in (19)). AC can be activated directly using the bacterial toxin forskolin. Data shown here indicate that, when added to the AFC instillate, forskolin (50 μM) increased mean AFC by 53% in RSV-infected mice at day 2, thereby restoring AFC to its level in mock-infected mice (Fig. 1a). The PKA activator 8-bromo-cAMP (50 μM) had a comparable effect on AFC at day 2. The stimulatory effect of forskolin on AFC was significantly inhibited (by 27 %) by concomitant addition of the epithelial Na+ channel (ENaC) inhibitor amiloride (1.5 mM) or the PKA inhibitor 14–22 amide (100 μM) to the instillate (Fig. 1b). Forskolin only increased amiloride-insensitive AFC by 8% at day 2, suggesting that the majority of its stimulatory effect on AFC was due to activation of ENaC.
Fig. 1. Effect of protein kinase A activation on alveolar fluid clearance in RSV-infected mice.
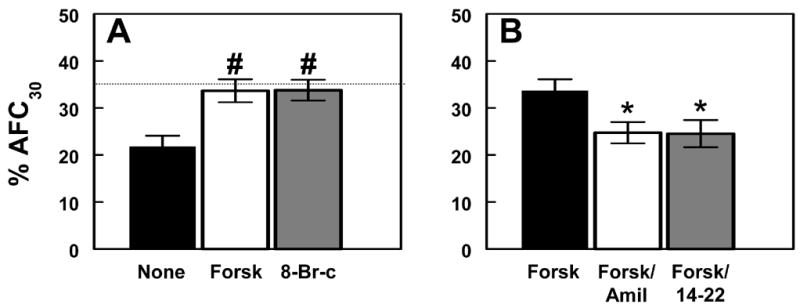
(a) Effect of 50 μM forskolin (Forsk; n=10), and 50 μM 8-Br-cAMP (8-Br-c; n=9) on AFC at day 2 (no treatment, None; n=10). (b) Effect of 1.5 mM amiloride (Forsk/Amil; n=8), and 100 μM 14–22 amide (Forsk/14–22; n=10) on forskolin-stimulated AFC at day 2 (forskolin-stimulated, Forsk; n=10). Dotted line indicates mean AFC rate in untreated, mock-infected animals (n=11). # P<0.005, compared with untreated, infected mice at day 2. * P<0.05, compared with forskolin-treated, RSV-infected mice at day 2.
Effect of β-adrenergic agonists on AFC
Unlike forskolin, the β2-AR agonist terbutaline (1, 10, or 100 μM) had no significant effect upon AFC at day 2 (Fig. 2a). 1 mM terbutaline did stimulate AFC at this time point (increasing AFC by 42% to 30.6 ± 0.67 % over 30 minutes, n=13), but AFC still remained significantly lower (P<0.05) than in mock-infected animals (34.8 ± 1.5, n=11 (5)). 100 μM salmeterol (a highly potent, long-acting β2-AR agonist), isoproterenol (a nonspecific β-AR agonist), and procaterol (a short-acting β2-AR agonist) likewise had no stimulatory effect upon AFC at day 2 after RSV infection (Fig. 2a). Responsiveness to 100 μM terbutaline was also absent at day 4 after infection (Fig. 2b).
These data demonstrate that PKA is functional in RSV-infected mice, and that direct activation of PKA by forskolin or 8-bromo-cAMP restores AFC to normal levels, primarily by stimulating Na+ reabsorption via amiloride-sensitive ENaC-like channels in the bronchoalveolar epithelium. Based on this finding, and on previous reports using comparable β-agonist doses (10,36), upstream activation of β-AR by β-agonists should therefore result in PKA activation and hence the same increase in AFC. However, β-agonists had no effect on AFC in RSV-infected mice, except at very high doses. Our subsequent studies therefore focused upon determining the underlying cause of this insensitivity to β-agonists following RSV infection, and ultimately demonstrate that this phenomenon is not a simple dose effect, since lower concentrations of β-agonists can stimulate AFC, provided that the intracellular milieu is appropriately modulated.
Role of endogenous catecholamines in β-agonist insensitivity in RSV-infected mice
β2-AR can become desensitized as a result of prolonged exposure to exogenous β-agonists or high levels of endogenous catecholamines (9,32). We therefore examined the possibility that RSV infection might induce sufficient physiologic stress to trigger activation of the adrenal medulla and thereby chronically elevate systemic endogenous catecholamine (norepinephrine and epinephrine) levels. This might be sufficient to induce β2-AR internalization or sequestration, with loss of receptors from the cell surface. However, as shown in Figure 3a, RSV infection had no effect upon plasma catecholamine levels, which remained at control levels at day 2 (Fig. 3a). To support these findings, we examined the effect of β-agonists on AFC in RSV-infected adrenalectomized BALB/c mice, in which the majority of the catecholamine response is absent (although central pathways remain intact). Normal, adrenalectomized mice showed a reduced rate of AFC compared to control animals (Fig. 3b), an effect which has been reported previously (7). Infection of adrenalectomized mice with RSV for 2 days resulted in a reduction in AFC to a final level identical to that seen in intact mice after infection (21%). However, like intact RSV-infected animals, adrenalectomized, RSV-infected mice showed no increase in AFC in response to 100 μM terbutaline at day 2. Taken together, these findings indicate that β-AR desensitization at day 2 after RSV infection is not due to receptor sequestration or degradationmediated by endogenous catecholamines.
Fig. 3. Role of endogenous catecholamines in β-agonist insensitivity in RSV-infected mice.
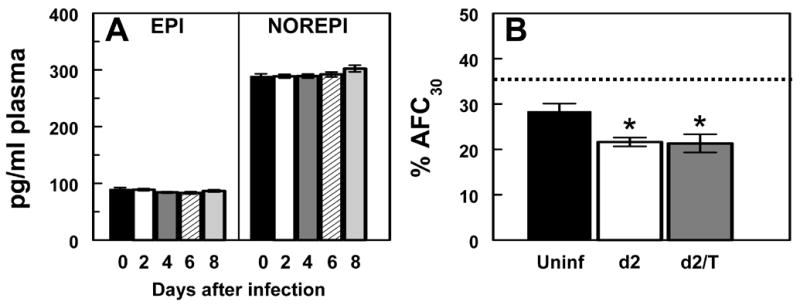
(a) Effect of RSV infection on plasma epinephrine (EPI) and norepinephrine (NOREPI) levels at day 0 to day 8 after infection (n=12–16 per group). (b) Effect of RSV infection for 2 days (d2; n=8), and RSV infection for 2 days + 100 μM terbutaline (d2/T; n=10) on AFC in adrenalectomized mice (uninfected, Uninf; n=9). Dotted line indicates mean AFC rate in untreated, mock-infected intact mice (n=11). * P<0.05, compared with uninfected, adrenalectomized mice.
Effect of RSV infection on membrane-bound β2-adrenergic receptor characteristics
To further investigate effects of RSV on β-AR function, we quantified membrane-bound β2-AR levels in homogenates of peripheral lung tissue using a saturation binding method with the highly β2-AR-specific inverse agonist ICI 118.551 (Fig. 4a, data plot from 2 pooled experiments). Interestingly, infection with RSV for 2 days actually resulted in a 32% increase in β2-AR receptor density (Bmax, Fig. 4b). However, the kd (the concentration at which 50% of receptors are bound) was 4-fold greater at day 2, as compared to uninfected mice (Fig. 4c). The precise binding site(s) of ICI 118,551 to the β2AR are not known, thus the Kd may not be an accurate index of receptor affinity for ligand. Mice infected with RSV for 6 days had normal β2-AR receptor density and ligand affinity. AFC rate is normal at this timepoint (5).
Fig. 4. Effect of RSV infection on β2-adrenergic receptor expression in the peripheral lung.
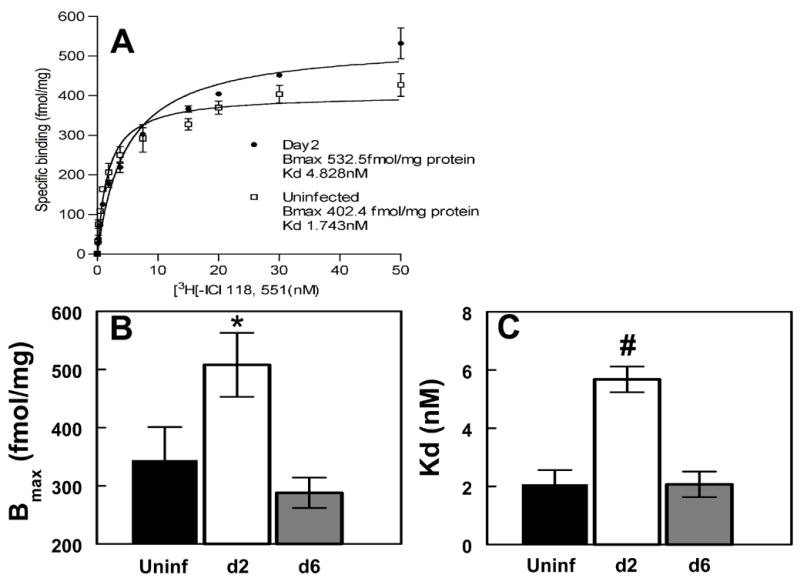
(a) Representative Scatchard plots for β2-AR density and affinity in peripheral lung homogenates from mice infected with RSV for 2 days, compared with uninfected mice (mean data from 2 experiments). (b) Effect of RSV infection for 2 and 6 days on β2-AR density in mouse peripheral lung homogenates (Uninf, uninfected). (c) Effect of RSV infection for 2 and 6 days on β2-AR ligand affinity in mouse peripheral lung homogenates (Uninf, uninfected). n=3 replicate assays per group (15–20 mice per group per replicate). * P<0.05, # P<0.005, compared with untreated mice (by paired ANOVA).
Effect of protein kinase inhibition on β-agonist sensitivity in RSV-infected mice
The results of our Scatchard analyses were incompatible with β2-AR desensitization due to receptor internalization or degradation. However, β2-AR can also become desensitized as a result of serine phosphorylation by G protein-coupled receptor kinase 2 (GRK2; reviewed in (19).) This process, which is rapid and readily reversible, uncouples β2-AR from its signal transduction pathway, so that β-agonist binding no longer results in AC activation. Using AFC as a functional readout, we therefore examined the effects of GRK2 inhibition on β-agonist sensitivity after RSV infection. Co-instillation of 100 μM terbutaline with a specific cell-permeable GRK2inhibitor (100 μM, EMD Biosciences) resulted in restoration of terbutaline sensitivity and increased AFC to its level in uninfected mice (Table 1). In the absence of terbutaline, the GRK2 inhibitor had no effect on AFC at day 2 after infection. Thus, GRK2 blockade prevented desensitization of β2-AR to β-agonists. The increase in AFC induced by terbutaline following GRK2 inhibition could be blocked by 14–22 amide or amiloride, confirming that it results fromactivation of PKA and upregulation of Na+ transport via amiloride-sensitive ENaC pathways. Inhibition of GRK2 also restored terbutaline sensitivity at day 4 after infection (increasing AFC over 30 minutes from 25.6 ± 1.7 %, n=9, to 33.2 ± 1.2 %, n=8).
Table 1.
Effect of G protein-coupled receptor kinase 2 inhibition on terbutaline-sensitive alveolar fluid clearance at day 2 after RSV infection.
| Treatment | Conc. (μM) | n* | % AFC30† | ΔTERBUT (%)‡ |
|---|---|---|---|---|
| None | - | 10 | 21.8 ± 2.3 | - |
| Terbutaline | 100 | 17 | 24 ± 2.3 | 10 |
| GRK2 inhibitor (GRK2i) | 100 | 10 | 23.4. ± 2.1 | - |
| GRK2i + Terbutaline | 100 + 100 | 8 | 32.9 ± 2.2 §# | 50 |
| GRK2i + Terbutaline + Amiloride | 100 + 100 + 1500 | 7 | 19. 6 ± 1.9 | −11 |
| GRK2i + Terbutaline + 14–22 Amide | 100 + 100 + 100 | 6 | 19.9 ± 2.1 | −9 |
Number of mice in which AFC was evaluated
Mean % AFC ± SEM
% change in mean AFC with terbutaline
% AFC30 in mock-infected mice is 34.8 ± 1.5 (n=11) (from ref. 4)
P<0.05 versus untreated AFC at day 2
Protein kinase C (PKC), which is known to be activated during RSV infection (30), canserine phosphorylate GRK2, promoting its translocation from the cytosol to the plasma membrane and increasing its activity (34). Addition of the general PKC inhibitor bisindolylmaleimide 1 (GF109203X, 100 μM), or a cell-permeable inhibitor of the PKCζ isoform (myristoylated PKCζ inhibitory peptide, 2 μg/ml), to the AFC instillate again resulted in a significant increase in AFC in response to concomitant terbutaline stimulation at day 2 (Table 2). In contrast, an inhibitor of the PKCα/β1 isoforms (Gö6976, 10 μM), or a cell-impermeable inhibitor of the PKCζ isoform (unmyristoylated PKCζ inhibitory peptide, 2 μg/ml), had no effect on terbutaline sensitivity. In the absence of terbutaline, none of these PKC inhibitors had any effect on AFC in RSV-infected mice at day 2.
Table 2.
Effect of protein kinase C inhibition on terbutaline-sensitive alveolar fluid clearance at day 2 after RSV infection.
| Treatment | Conc. | n* | % AFC30† | ΔTERBUT (%)‡ |
|---|---|---|---|---|
| None | - | 10 | 21.8 ± 2.3 | - |
| Terbutaline | 100μM | 17 | 24 ± 2.3 | 10 |
| PKC inhibitor (GF109203X) | 100μM | 6 | 19.7 ± 1.9 | - |
| GF109203X + Terbutaline | 100μM + 100μM | 7 | 33.7 ± 2.1 §# | 54 |
| PKCα/β1 inhibitor (Gö6976) | 10μM | 9 | 19.8 ± 2.5 | - |
| Gö6976 + Terbutaline | 10μM + 100μM | 11 | 23.8 ± 2.5 | 8 |
| Myristoylated PKCζ inhibitor (mPKCζi) | 2μg/ml | 8 | 23.2 ± 2 | - |
| mPKCζi + Terbutaline | 2μg/ml + 100μM | 8 | 33.3 ± 1.1** | 52 |
| Unmyristoylated PKCζ inhibitor + Terb. | 2μg/ml + 100μM | 6 | 23.8 ± 1.3 | 9 |
Number of mice in which AFC was evaluated
Mean % AFC ± SEM
% change in mean AFC with terbutaline
% AFC30 in mock-infected mice is 34.8 ± 1.5 (n=11) (from ref. 4)
P<0.05,
P<0.005 versus untreated AFC at day 2
Effect of RSV infection on PKCζ phosphorylation in mouse lung alveolar cells
To confirm that infection with RSV results in activation of PKCζ, mouse lung alveolar cells from 3 uninfected mice and 3 infected animals were analyzed by Western blotting using specific antibodies. When normalized to β-actin levels, RSV infection reduced total PKCζ levels in both the cytosolic (not shown) and membrane fraction from alveolar cells. In contrast, infection with RSV caused a significant increase in activated Thr-410 phospho-PKCζ levels in the membrane fraction at day 2 (Fig. 5a and 5b). Thus, the ratio of phosphoPKCζ:PKCζ in the membrane fraction of alveolar cells from RSV-infected mice increased by about 300% at day 2, compared to uninfected mice. While activation of several PKC isoforms, including PKCζ, has been demonstrated previously in vitro following RSV infection,(1,30,41) this is the first demonstration of PKCζ activation in RSV-infected mouse lung ex vivo.
Fig. 5. Effect of RSV infection on PKCζ phosphorylation in mouse lung alveolar cells.
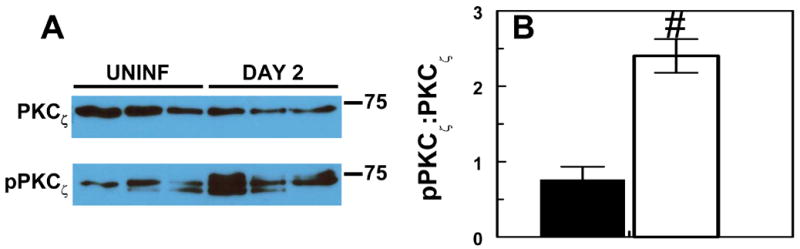
(a) Representative Western blot, showing total and phospho-PKCζ (both 73 kDa approx.) in the membrane fraction in uninfected and RSV-infected mice at day 2. (b) Effect of RSV infection for 2 days on the mean ratio of phospho-PKCζ:total PKCζ (measured by densitometry) in the membrane fraction from alveolar cells (n=3 per group). # P<0.005, compared with uninfected mice.
Effect of RSV infection on β2-adrenergic receptor phosphorylation
The above findings suggest that activated GRK2 desensitizes β2-AR either directly by receptor phosphorylation, or indirectly by phosphorylation or sequestration of a stimulatory G protein α subunit (Gsα), either of which will prevent activation of AC in response to β-agonists. In our next series of experiments, we therefore performed competition binding assays (with ICI 118.551 and procaterol) to quantify the fraction of high- and low-affinity receptors in pooled homogenates of peripheral lung tissue. Comparison of curve fit analysis from 2 separate sets of experiments for both RSV and uninfected controls was most consistent with a two-site binding site model (Fig. 6). However, no significant differences were detected in the fraction of low-affinity (phosphorylated) β2-AR between uninfected mice (logEC50 -7.59 ± 0.22, n=2) and RSV-infected mice at day 2 (logEC50 −7.97 ± 0.16, n=2). Likewise, infection with RSV for 2 days had no effect on the fraction of high-affinity (unphosphorylated) β2-AR detected by this technique (logEC50 −5.93 ± 0.06 at day 2, versus −5.79 ± 0.13 in uninfected mice). These data suggest that β2-AR insensitivity is not a result of increased phosphorylation of membrane bound receptors.
Fig. 6. Effect of RSV infection on β2-adrenergic receptor phosphorylation.
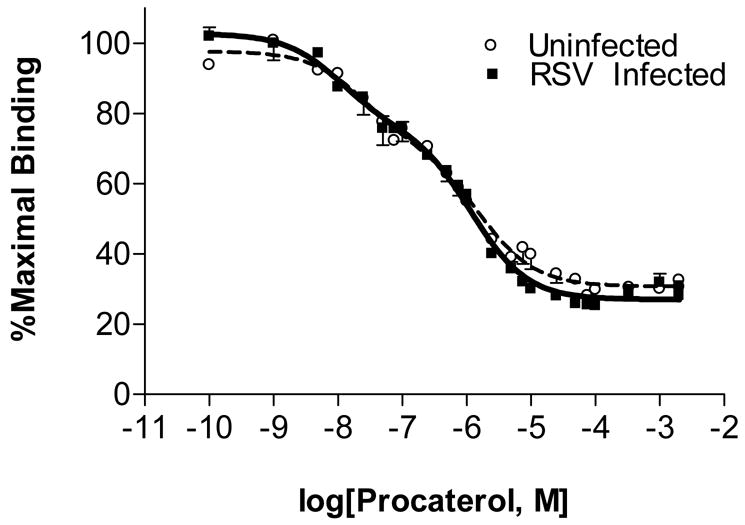
Results of competition binding assays on total peripheral lung cell membranes from uninfected mice and mice infected with RSV for 2 days, using [3H]-ICI-118,551 and the β2-AR-specific agonist procaterol. n=2 replicate assays per group (20 mice per group per replicate).
Effect of KC inhibition on β-agonist sensitivity in RSV-infected mice
Since RSV only infects a relatively small proportion of respiratory epithelial cells (23), generalized desensitization of β2-AR is unlikely to occur as a result of a direct effect of viral gene products on infected cells, and is more likely to be mediated by a soluble factor. Because infection with RSV does not elevate circulating catecholamine levels and thereby induce homologous β2-AR desensitization, we investigated the possibility that, by binding to its receptors on epithelial cells, another soluble mediator induces heterologous β2-AR desensitization following RSV infection. We have shown previously that, quantitatively, the predominant soluble mediator in the lung at days 2 and 4 after infection is the CXC chemokine KC (keratinocyte cytokine, the murine homolog of CXCL8/IL-8), which is present at log-fold higher concentrations in the BAL than other cytokines such as TNF-α and IL-1β (BAL KC concentration 913 ± 36 pg/ml at day 2 vs. 80 ± 21 pg/ml in mock-infected mice (4)). BAL KC levels also return to normal at days 6 and 8, when AFC has normalized. Mice express only functional CXCR2 receptors for KC, while humans also possess a CXCR1 receptor subtype which can bind CXCL8 (29). CXCR2 is coupled to inhibitory Gα protein subunits and can activate PKC (38). We therefore investigated the possibility that crosstalk signals from activation of CXCR2 by KC might account for β-agonist insensitivity at day 2 following RSV infection, and found that addition to the AFC instillate of a neutralizing monoclonal antibody to KC did indeed result in restoration of terbutaline sensitivity at day 2 (Table 3). This antibody had no effect on AFC in the absence of terbutaline, and an isotype-matched irrelevant antibody had no effect on AFC in either the presence or absence of terbutaline. A polyclonal neutralizing anti-KC antibody had a similar effect on terbutaline sensitivity at day 2. Likewise, addition to the AFC instillate of a neutralizing monoclonal antibody to CXCR2, which alone had no effect on AFC, also resulted in partial restoration of terbutaline sensitivity at day 2, while a neutralizing polyclonal antibody to the chemokine receptor CXCR4 (species matched to that used against KC) had no such effect. These findings suggest that β2-AR desensitization may occur as a consequence of crosstalk from CXCR2 chemokine receptors, activated by KC.
Table 3.
Effect of CXCL8 (KC) neutralization on terbutaline-sensitive alveolar fluid clearance at day 2 after RSV infection.
| Treatment | Conc. | n* | % AFC30† | ΔTERBUT (%)‡ |
|---|---|---|---|---|
| None | - | 10 | 21.8 ± 2.3 | - |
| Terbutaline | 100μM | 17 | 24 ± 2.3 | 10 |
| Rat IgG2A | 1μg/ml | 7 | 23.8 ± 1.1 | - |
| Rat IgG2A + Terbutaline | 1μg/ml + 100μM | 8 | 22.4 ± 1.8 | -6 |
| Anti-KC mAb§ | 1μg/ml | 13 | 21 ± 1.3 | - |
| Anti-KC mAb + Terbutaline | 1μg/ml + 100μM | 20 | 32 ± 0.94 a** | 53 |
| Anti-KC pAbb | 1μg/ml | 7 | 21.2 ± 2 | - |
| Anti-KC pAb + Terbutaline | 1μg/ml + 100μM | 13 | 30.8 ± 1.9# | 45 |
| Anti-CXCR2 mAb§ | 1μg/ml | 13 | 22.3 ± 2.1 | - |
| Anti-CXCR2 mAb + Terbutaline | 1μg/ml + 100μM | 9 | 28.7 ± 0.8# | 29 |
| Anti-CXCR4 pAbb | 1μg/ml | 8 | 23. 7 ± 3.1 | - |
| Anti-CXCR4 pAb + Terbutaline | 1μg/ml + 100μM | 10 | 24.5 ± 2.9 | 4 |
Number of mice in which AFC was evaluated
Mean % AFC ± SEM
% change in mean AFC with terbutaline
Rat IgG2A isotype
% AFC30 in mock-infected mice is 34.8 ± 1.5 (n=11) (from ref. 4)
Rabbit anti-rat polyclonal antibody
P<0.05,
P<0.005 versus untreated AFC at day 2
Discussion
Previously, we demonstrated that infection of BALB/c mice with RSV results in reduced AFC at days 2–4 after infection (5). We have restricted our current studies predominantly to a comparison of the effects of forskolin and β-agonists on AFC at day 2 after RSV infection – the time point at which AFC is most impaired in our model and therefore, at least in terms of its ion transport function, the epithelium is functionally most “injured”. While it has not yet been formally demonstrated that impaired AFC is a component of human RSV disease, a significant subset of hospitalized infants with RSV that require mechanical ventilation actually exhibit restrictive lung disease that fulfils clinical criteria for classification as ARDS (12,13). Impaired AFC is therefore likely to be present at least in this group of infants (45). Moreover, the AFC response to β-agonists provides a useful in vivo readout of epithelial β-AR (and associated signaling apparatus) function, irrespective of the overall contribution of impaired AFC to the pathogenesis of RSV bronchiolitis.
Initially, we found that forskolin stimulates AFC at day 2 after RSV infection, when AFC is most impaired, and returns AFC to its level in normal mice. This increase in AFC, which is comparable to that reported by other investigators using β-agonists in injured lung models (reviewed in (33)), can be completely inhibited by 14–22 amide or amiloride, indicating that forskolin, by activating PKA, is stimulating transepithelial Na+ transport predominantly through ENaC-like channels in RSV-infected mice (although we cannot exclude effects upon the Na+/K+ ATPase). These channels are the rate-limiting step in epithelial Na+ reabsorption, which is the main driving force for AFC. In contrast, low doses of either short- or long-acting, β2-specific, or nonspecific, β-agonists that had been shown in other studies to stimulate AFC (39), did not increase AFC at day 2 and day 4 post RSV infection. Although we did find some improvement in AFC at day 2 with 1 mM terbutaline, such a high dose is unlikely to be of either physiologic or clinical relevance, and would be highly likely to provoke significant side-effects such as tachycardia and arrhythmias (26,40).
Our data suggests that β-agonist insensitivity in RSV-infected mice does not result from receptor internalization or degradation due to chronic elevation of endogenous catecholamines, and is not characterized by any dramatic shift in β2-AR binding kinetics. Rather, it appears to result from β2-AR desensitization by GRK2, which on activation by PKCζ uncouples β2-AR from their normal AC stimulation pathway. In turn, PKCζ activation may result from ligation of CXCR2 by KC, released in response to RSV infection (Fig. 7). Moreover, β-agonist insensitivity is relatively prolonged, since it persists until day 4 after infection, and can be accounted for by the same mechanism throughout this period.
Fig. 7. Proposed mechanism for heterologous desensitization of β2-AR by KC following RSV infection.
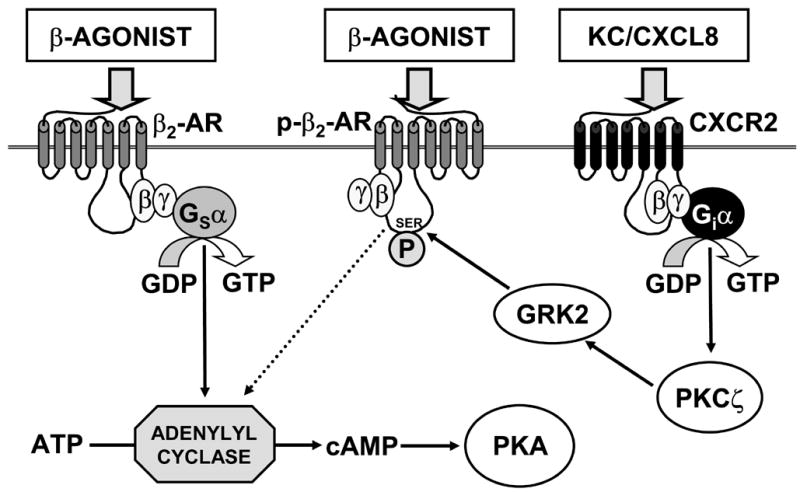
Normally, binding of β-agonist to β2-AR results in hydrolysis of GTP by the stimulatory α subunit of the receptor-associated heterotrimeric G protein (Gsα), which then dissociates and activates adenylyl cyclase. Adenylyl cyclase in turn catalyzes formation of cAMP from ATP. cAMP then activates cAMP-dependent protein kinase (protein kinase A, PKA), which stimulates AFC via effects on ENaC and the Na+/K+ ATPase (and possibly CFTR). However, following RSV infection, KC/CXCL-8 is produced. Upon binding to its receptors (CXCR2, and in humans also CXCR1), KC promotes hydrolysis of GTP by the inhibitory α subunit of the receptor-associated heterotrimeric G protein (Giα), which then activates PKCζ. In turn, PKCζ phosphorylates and activates G protein-coupled receptor kinase 2 (GRK2), which either serine phosphorylates β2-AR (p-β2-AR), uncoupling them from Gsα, or which can directly sequester Gsα (not shown). Upon binding of β-agonist, these phosphorylated receptors are unable to activate adenylyl cyclase and no AFC response is detected.
Several lines of evidence support our contention that β2-AR desensitization in RSV-infected mice occurs as a result of receptor uncoupling rather than internalization or degradationbecause of phosphorylation. Firstly, blockade of PKCζ, GRK2, or CXCL8 restores responsiveness to terbutaline comparable to that induced by forskolin, indicating that such blockade effectively recouples β2-AR to PKA. Secondly, RSV is known to trigger phosphorylation, sustained activation, and cytoplasm-to-membrane translocation of PKCζ (30,41), which is consistent with this mechanism. Thirdly, membrane β2-AR expression increases at day 2, which is incompatible with β-agonist desensitization as a result of either receptor internalization or degradation. Fourthly, terbutaline insensitivity in RSV-infected miceis rapidly reversible (inhibitors of GRK2, PKC and CXCL8 are only present during the 30-minute AFC procedure), indicating that β-agonist insensitivity cannot be a consequence of β2-AR degradation. Moreover, the ability to resensitize β2-AR to 100 μM terbutaline after GRK2, PKC, or CXCL8 inhibition demonstrates that terbutaline insensitivity after RSV infection cannot be a consequence of inhibition of β2-AR by the ketamine used in our AFC anesthetic regimen (43), or a simple dose effect artifact of the AFC procedure itself. In addition, the ability to increase AFC in response to both short-term treatment with either forskolin or β-agonists (when β2-AR phosphorylation is blocked) demonstrates that the AFC deficit is solely functional, and not a consequence of cell death, which is not a feature of RSV infection in polarized epithelia (48). Finally, the lack of effect of inhibitors of GRK2, PKC, and CXCL8 on AFC in the absence of terbutaline further confirms our findings from adrenalectomized mice that endogenous catecholamines play no role in β2-AR desensitization and indicate that desensitization is a specific effect of RSV, rather than a result of a physiologic catecholamine response to the stress of infection.
Our findings suggest that desensitization of epithelial β2-AR can occur by a paracrinemechanism, induced by activation of epithelial CXCR1/2 G protein-coupled receptors by the proinflammatory chemokine CXCL8 following RSV infection. While GRK2 generally preferentially phosphorylates agonist-occupied β2-AR (homologous desensitization), agonist-independent phosphorylation of β2-AR (heterologous desensitization) has been demonstrated in other systems. For example, the chemokine CCL17 can induce agonist-independent phosphorylation of β2-AR in human peripheral blood T cells (16). Thus, even though infection with RSV does not elevate circulating catecholamine levels, β2-AR desensitization can still occur in the absence of ligand, as a consequence of cross-activation of PKCζ and GRK2 by binding of CXCL8 to its receptors. CXCL8 mainly promotes neutrophil chemotaxis and survival (22), and an effect of this chemokine on β-agonist sensitivity has not been reported previously. However, we have found previously that KC is quantitatively the predominant proinflammatory mediator present in the mouse lung at days 2 and 4 (5), and an elevated plasma, nasal lavage or lung level of CXCL8 may be an indicator of increased disease severity in children with RSV (reviewed in (44)). Unfortunately, no studies of CXCL8 to date have stratified disease severity on the basis of persistence or absence of β-agonist sensitivity, so a relationship between the two in infants with RSV has not been demonstrated. Interestingly, however, BAL CXCL8 levels are elevated for a far longer period in infants hospitalized for severe bronchiolitis than are KC levels in lungs from RSV-infected mice (27), suggesting that, if mediated by the same mechanism, β2-AR desensitization might be much more prolonged in human subjects than in our mouse model. Moreover, it is possible that other CXCR1/2 ligands that are known to be induced by RSV infection, such as CXCL2 (MIP-2 (37)) and CXCL5 (ENA-78 (49)), may also contribute to β2-AR desensitization by the same mechanism.
The increased Kd noted in the saturation binding assays from day 2 infected mice (Figure 4c) are suggestive of decreased receptor affinity for ligand. However, this data must be interpreted in the context of the absence of data regarding where, and to how many, sites on the β2-AR ICI 118, 551 binds. Thus, this Kd may not be a useful index of β2-AR phosphorylation. To address this methodologic limitation we performed competition binding studies using a highly specific partial agonist (procaterol) and ICI 118,551 (Figure 6). These data are highly consistent with a receptor with two conformational states (phosphorylated and non-phosphorylated). As can be seen in Figure 6, this method shows no significant difference in the proportion of high-affinity, receptors between day 2 infected and sham infected mice (40.6% vs. 35.2%). It is possible that dilution factors related to the analysis of whole lung homogenates may have impeded our ability to detect differences in epithelial β2-AR phosphorylation levels following RSV infection. However, a more likely possibility is that β2-AR desensitization is not a direct consequence of β2-AR phosphorylation by GRK2, but results instead from sequestration of Gsα by activated GRK2, as has been described for the glutamate receptor (reviewed in (6)). To our knowledge, phosphorylation-independent regulation of β2-AR by GRK2 has not been reported previously.
A recent report demonstrated that in vitro infection of human airway smooth muscle cells with RSV resulted in insensitivity to β-agonists, which was associated with a reduction in β2-AR density (31). However, this study must be interpreted with some caution, since there is no evidence that RSV infects smooth muscle in vivo. Finally, it should be noted that influenza (17), rhinovirus (11), and parainfluenza virus (2) have been shown to desensitize β-AR in airway smooth muscle. To our knowledge, however, this remains the first report demonstrating loss of sensitivity to β-agonists by respiratory epithelium following viral infection.
In conclusion, our data indicate that bronchoalveolar epithelial β2-AR are desensitized to β-agonists for a prolonged period following RSV infection, so that even with optimal drug delivery (which our AFC model permits) appropriate physiologic responses to β-agonists are blunted. Furthermore, desensitization appears to result from KC-mediated PKCζ activation and stimulation of GRK2, which uncouples β2-AR, thereby preventing appropriate activation of AC. While there are undoubtedly differences between the murine model of RSV infection and the human disease (including the presence of β2-AR polymorphisms in man which have recently been shown to affect responses to salmeterol (47)), and while other factors may also contribute to the lack of β-agonist effect, such direct in vivo evidence that β2-AR desensitization following RSV infection may account for the modest utility of β-agonists in therapy for bronchiolitis has not previously been reported.
Acknowledgments
The authors would like to acknowledge the excellent technical assistance of Glenda C. Davis. This work was funded by NIH grants HL31197 and HL51173 (SM), RR17626 (ICD) and HL066211 and HL071042 (PF).
Reference List
- 1.Bitko V, Barik S. Persistent activation of RelA by respiratory syncytial virus involves protein kinase C, underphosphorylated IkappaBbeta, and sequestration of protein phosphatase 2A by the viral phosphoprotein. J Virol. 1998;72:5610–5618. doi: 10.1128/jvi.72.7.5610-5618.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buckner CK, Clayton DE, in-Shoka AA, Busse WW, Dick EC, Shult P. Parainfluenza 3 infection blocks the ability of a beta adrenergic receptor agonist to inhibit antigen-induced contraction of guinea pig isolated airway smooth muscle. J Clin Invest. 1981;67:376–384. doi: 10.1172/JCI110045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carstairs R, Nimmo AJ, Barnes PJ. Autoradiographic visualization of beta-adrenoreceptor subtypes in human lung. Am Rev Respir Dis. 1985;132:541–547. doi: 10.1164/arrd.1985.132.3.541. [DOI] [PubMed] [Google Scholar]
- 4.Davis IC, Lazarowski ER, Hickman-Davis JM, Fortenberry JA, Chen FP, Zhao X, Sorscher E, Graves LM, Sullender WM, Matalon S. Leflunomide prevents alveolar fluid clearance inhibition by respiratory syncytial virus. Am J Respir Crit Care Med. 2006;173:673–682. doi: 10.1164/rccm.200508-1200OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis IC, Sullender WM, Hickman-Davis JM, Lindsey JR, Matalon S. Nucleotide-mediated inhibition of alveolar fluid clearance in BALB/c mice after respiratory syncytial virus infection. Am J Physiol Lung Cell Mol Physiol. 2004;286:L112–L120. doi: 10.1152/ajplung.00218.2003. [DOI] [PubMed] [Google Scholar]
- 6.Dhami GK, Ferguson SS. Regulation of metabotropic glutamate receptor signaling, desensitization and endocytosis. Pharmacol Ther. 2006;111:260–271. doi: 10.1016/j.pharmthera.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Factor P, Adir Y, Mutlu GM, Burhop J, Dumasius V. Effects of beta2-adrenergic receptor overexpression on alveolar epithelial active transport. J Allergy Clin Immunol. 2002;110:S242–S246. doi: 10.1067/mai.2002.129706. [DOI] [PubMed] [Google Scholar]
- 8.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med. 2005;352:1749–1759. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- 9.Finney PA, Donnelly LE, Belvisi MG, Chuang TT, Birrell M, Harris A, Mak JC, Scorer C, Barnes PJ, Adcock IM, Giembycz MA. Chronic systemic administration of salmeterol to rats promotes pulmonary beta(2)-adrenoceptor desensitization and down-regulation of G(s alpha) Br J Pharmacol. 2001;132:1261–1270. doi: 10.1038/sj.bjp.0703946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garat C, Carter EP, Matthay MA. New in situ mouse model to quantify alveolar epithelial fluid clearance. J Appl Physiol. 1998;84:1763–1767. doi: 10.1152/jappl.1998.84.5.1763. [DOI] [PubMed] [Google Scholar]
- 11.Grunstein MM, Hakonarson H, Whelan R, Yu Z, Grunstein JS, Chuang S. Rhinovirus elicits proasthmatic changes in airway responsiveness independently of viral infection. J Allergy Clin Immunol. 2001;108:997–1004. doi: 10.1067/mai.2001.120276. [DOI] [PubMed] [Google Scholar]
- 12.Hammer J, Numa A, Newth CJ. Albuterol responsiveness in infants with respiratory failure caused by respiratory syncytial virus infection. J Pediatr. 1995;127:485–490. doi: 10.1016/s0022-3476(95)70088-9. [DOI] [PubMed] [Google Scholar]
- 13.Hammer J, Numa A, Newth CJ. Acute respiratory distress syndrome caused by respiratory syncytial virus. Pediatr Pulmonol. 1997;23:176–183. doi: 10.1002/(sici)1099-0496(199703)23:3<176::aid-ppul2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 14.Hardiman KM, McNicholas-Bevensee CM, Fortenberry J, Myles CT, Malik B, Eaton DC, Matalon S. Regulation of amiloride-sensitive Na(+) transport by basal nitric oxide. Am J Respir Cell Mol Biol. 2004;30:720–728. doi: 10.1165/rcmb.2003-0325OC. [DOI] [PubMed] [Google Scholar]
- 15.Hartling L, Wiebe N, Russell K, Patel H, Klassen TP. A meta-analysis of randomized controlled trials evaluating the efficacy of epinephrine for the treatment of acute viral bronchiolitis. Arch Pediatr Adolesc Med. 2003;157:957–964. doi: 10.1001/archpedi.157.10.957. [DOI] [PubMed] [Google Scholar]
- 16.Heijink IH, Vellenga E, Oostendorp J, de Monchy JG, Postma DS, Kauffman HF. Exposure to TARC alters beta2-adrenergic receptor signaling in human peripheral blood T lymphocytes. Am J Physiol Lung Cell Mol Physiol. 2005;289:L53–L59. doi: 10.1152/ajplung.00357.2004. [DOI] [PubMed] [Google Scholar]
- 17.Henry PJ, Rigby PJ, Mackenzie JS, Goldie RG. Effect of respiratory tract viral infection on murine airway beta-adrenoceptor function, distribution and density. Br J Pharmacol. 1991;104:914–921. doi: 10.1111/j.1476-5381.1991.tb12526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hickman-Davis JM, Nicholas-Bevensee C, Davis IC, Ma HP, Davis GC, Bosworth CA, Matalon S. Reactive species mediate inhibition of alveolar type II sodium transport during mycoplasma infection. Am J Respir Crit Care Med. 2006;173:334–344. doi: 10.1164/rccm.200501-155OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson M. Molecular mechanisms of beta(2)-adrenergic receptor function, response, and regulation. J Allergy Clin Immunol. 2006;117:18–24. doi: 10.1016/j.jaci.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 20.Kellner JD, Ohlsson A, Gadomski AM, Wang EE. Efficacy of bronchodilator therapy in bronchiolitis. A meta-analysis. Arch Pediatr Adolesc Med. 1996;150:1166–1172. doi: 10.1001/archpedi.1996.02170360056009. [DOI] [PubMed] [Google Scholar]
- 21.Liggett SB, Caron MG, Lefkowitz RJ, Hnatowich M. Coupling of a mutated form of the human beta 2-adrenergic receptor to Gi and Gs. Requirement for multiple cytoplasmic domains in the coupling process. J Biol Chem. 1991;266:4816–4821. [PubMed] [Google Scholar]
- 22.Mantovani A. The chemokine system: redundancy for robust outputs. Immunol Today. 1999;20:254–257. doi: 10.1016/s0167-5699(99)01469-3. [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Sobrido L, Gitiban N, Fernandez-Sesma A, Cros J, Mertz SE, Jewell NA, Hammond S, Flano E, Durbin RK, Garcia-Sastre A, Durbin JE. Protection against respiratory syncytial virus by a recombinant Newcastle disease virus vector. J Virol. 2006;80:1130–1139. doi: 10.1128/JVI.80.3.1130-1139.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matalon S, O’Brodovich H. Sodium channels in alveolar epithelial cells: molecular characterization, biophysical properties, and physiological significance. Annu Rev Physiol. 1999;61:627–661. doi: 10.1146/annurev.physiol.61.1.627. [DOI] [PubMed] [Google Scholar]
- 25.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82:569–600. doi: 10.1152/physrev.00003.2002. [DOI] [PubMed] [Google Scholar]
- 26.McAuley DF, Frank JA, Fang X, Matthay MA. Clinically relevant concentrations of beta2-adrenergic agonists stimulate maximal cyclic adenosine monophosphate-dependent airspace fluid clearance and decrease pulmonary edema in experimental acid-induced lung injury. Crit Care Med. 2004;32:1470–1476. doi: 10.1097/01.ccm.0000129489.34416.0e. [DOI] [PubMed] [Google Scholar]
- 27.McNamara PS, Ritson P, Selby A, Hart CA, Smyth RL. Bronchoalveolar lavage cellularity in infants with severe respiratory syncytial virus bronchiolitis. Arch Dis Child. 2003;88:922–926. doi: 10.1136/adc.88.10.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Modl M, Eber E, Malle-Scheid D, Weinhandl E, Zach MS. Does bronchodilator responsiveness in infants with bronchiolitis depend on age? J Pediatr. 2005;147:617–621. doi: 10.1016/j.jpeds.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Moepps B, Nuesseler E, Braun M, Gierschik P. A homolog of the human chemokine receptor CXCR1 is expressed in the mouse. Mol Immunol. 2006;43:897–914. doi: 10.1016/j.molimm.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 30.Monick M, Staber J, Thomas K, Hunninghake G. Respiratory syncytial virus infection results in activation of multiple protein kinase C isoforms leading to activation of mitogen-activated protein kinase. J Immunol. 2001;166:2681–2687. doi: 10.4049/jimmunol.166.4.2681. [DOI] [PubMed] [Google Scholar]
- 31.Moore PE, Cunningham G, Calder MM, DeMatteo AD, Jr, Peeples ME, Summar ML, Peebles RS., Jr Respiratory syncytial virus infection reduces beta2-adrenergic responses in human airway smooth muscle. Am J Respir Cell Mol Biol. 2006;35:559–564. doi: 10.1165/rcmb.2005-0282OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan EE, Stader SM, Hodnichak CM, Mavrich KE, Folkesson HG, Maron MB. Postreceptor defects in alveolar epithelial beta-adrenergic signaling after prolonged isoproterenol infusion. Am J Physiol Lung Cell Mol Physiol. 2003;285:L578–L583. doi: 10.1152/ajplung.00339.2002. [DOI] [PubMed] [Google Scholar]
- 33.Mutlu GM, Koch WJ, Factor P. Alveolar epithelial beta 2-adrenergic receptors: their role in regulation of alveolar active sodium transport. Am J Respir Crit Care Med. 2004;170:1270–1275. doi: 10.1164/rccm.200404-470CP. [DOI] [PubMed] [Google Scholar]
- 34.Penela P, Murga C, Ribas C, Tutor AS, Peregrin S, Mayor F., Jr Mechanisms of regulation of G protein-coupled receptor kinases (GRKs) and cardiovascular disease. Cardiovasc Res. 2006;69:46–56. doi: 10.1016/j.cardiores.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 35.Perkins GD, McAuley DF, Thickett DR, Gao F. The beta-agonist lung injury trial (BALTI): a randomized placebo-controlled clinical trial. Am J Respir Crit Care Med. 2006;173:281–287. doi: 10.1164/rccm.200508-1302OC. [DOI] [PubMed] [Google Scholar]
- 36.Planes C, Leyvraz C, Uchida T, Angelova MA, Vuagniaux G, Hummler E, Matthay M, Clerici C, Rossier B. In vitro and in vivo regulation of transepithelial lung alveolar sodium transport by serine proteases. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1099–L1109. doi: 10.1152/ajplung.00332.2004. [DOI] [PubMed] [Google Scholar]
- 37.Power UF, Huss T, Michaud V, Plotnicky-Gilquin H, Bonnefoy JY, Nguyen TN. Differential histopathology and chemokine gene expression in lung tissues following respiratory syncytial virus (RSV) challenge of formalin-inactivated RSV- or BBG2Na-immunized mice. J Virol. 2001;75:12421–12430. doi: 10.1128/JVI.75.24.12421-12430.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rose JJ, Foley JF, Murphy PM, Venkatesan S. On the mechanism and significance of ligand-induced internalization of human neutrophil chemokine receptors CXCR1 and CXCR2. J Biol Chem. 2004;279:24372–24386. doi: 10.1074/jbc.M401364200. [DOI] [PubMed] [Google Scholar]
- 39.Sakuma T, Gu X, Wang Z, Maeda S, Sugita M, Sagawa M, Osanai K, Toga H, Ware LB, Folkesson G, Matthay MA. Stimulation of alveolar epithelial fluid clearance in human lungs by exogenous epinephrine. Crit Care Med. 2006;34:676–681. doi: 10.1097/01.CCM.0000201403.70636.0F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakuma T, Hida M, Nambu Y, Osanai K, Toga H, Takahashi K, Ohya N, Inoue M, Watanabe Y. Beta1-adrenergic agonist is a potent stimulator of alveolar fluid clearance in hyperoxic rat lungs. Jpn J Pharmacol. 2001;85:161–166. doi: 10.1254/jjp.85.161. [DOI] [PubMed] [Google Scholar]
- 41.San-Juan-Vergara H, Peeples ME, Lockey RF, Mohapatra SS. Protein kinase C-alpha activity is required for respiratory syncytial virus fusion to human bronchial epithelial cells. J Virol. 2004;78:13717–13726. doi: 10.1128/JVI.78.24.13717-13726.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sartori C, Allemann Y, Duplain H, Lepori M, Egli M, Lipp E, Hutter D, Turini P, Hugli O, Cook S, Nicod P, Scherrer U. Salmeterol for the prevention of high-altitude pulmonary edema. N Engl J Med. 2002;346:1631–1636. doi: 10.1056/NEJMoa013183. [DOI] [PubMed] [Google Scholar]
- 43.Seeman P, Kapur S. Anesthetics inhibit high-affinity states of dopamine D2 and other G-linked receptors. Synapse. 2003;50:35–40. doi: 10.1002/syn.10221. [DOI] [PubMed] [Google Scholar]
- 44.Smyth RL, Openshaw PJ. Bronchiolitis. Lancet. 2006;368:312–322. doi: 10.1016/S0140-6736(06)69077-6. [DOI] [PubMed] [Google Scholar]
- 45.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 46.Warshamana GS, Corti M, Brody AR. TNF-alpha, PDGF, and TGF-beta(1) expression by primary mouse bronchiolar-alveolar epithelial and mesenchymal cells: tnf-alpha induces TGF-beta(1) Exp Mol Pathol. 2001;71:13–33. doi: 10.1006/exmp.2001.2376. [DOI] [PubMed] [Google Scholar]
- 47.Wechsler ME, Lehman E, Lazarus SC, Lemanske RF, Jr, Boushey HA, Deykin A, Fahy JV, Sorkness CA, Chinchilli VM, Craig TJ, DiMango E, Kraft M, Leone F, Martin RJ, Peters SP, Szefler SJ, Liu W, Israel E. beta-Adrenergic receptor polymorphisms and response to salmeterol. Am J Respir Crit Care Med. 2006;173:519–526. doi: 10.1164/rccm.200509-1519OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol. 2002;76:5654–5666. doi: 10.1128/JVI.76.11.5654-5666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Luxon BA, Casola A, Garofalo RP, Jamaluddin M, Brasier AR. Expression of respiratory syncytial virus-induced chemokine gene networks in lower airway epithelial cells revealed by cDNA microarrays. J Virol. 2001;75:9044–9058. doi: 10.1128/JVI.75.19.9044-9058.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]