

Abstract
The tyrosine phosphorylated protein Crk-associated substrate (CAS) has previously been shown to participate in the cellular processes regulating dynamic changes in the actin architecture and arterial constriction. In the present study, treatment of rat mesenteric arteries with phenylephrine (PE) led to the increase in CAS tyrosine phosphorylation and the association of CAS with the adapter protein CrkII. CAS phosphorylation was catalyzed by Abl in an in vitro study. To determine the role of Abl tyrosine kinase in arterial vessels, plasmids encoding Abl short hairpin RNA (shRNA) were transduced into mesenteric arteries by chemical loading plus liposomes. Abl silencing diminished increases in CAS phosphorylation upon PE stimulation. Previous studies have shown that assembly of the multiprotein compound containing CrkII, neuronal Wiskott - Aldrich Syndrome Protein (N-WASP) and the Arp2/3 (Actin Related Protein) complex triggers actin polymerization in smooth muscle as well as in nonmuscle cells. In this study, Abl silencing attenuated the assembly of the multiprotein compound in resistance arteries upon contractile stimulation. Furthermore, the increase in F/G-actin ratios (an index of actin assembly) and constriction upon contractile stimulation were reduced in Abl-deficient arterial segments compared to control arteries. However, myosin regulatory light chain phosphorylation (MRLCP) elicited by contractile activation was not inhibited in Abl-deficient arteries. These results suggest that Abl may play a pivotal role in mediating CAS phosphorylation, the assembly of the multiprotein complex, actin assembly, and constriction in resistance arteries. Abl does not participate in the regulation of myosin activation in arterial vessels during contractile stimulation.
Keywords: Tyrosine kinase, actin cytoskeleton, contraction, vascular smooth muscle, adapter protein
Introduction
Actin cytoskeleton remodeling has recently emerged as an important cellular process mediating smooth muscle contraction.1-7 A pool of globular actin (G-actin) is stimulated onto filamentous actin (F-actin) in a variety of smooth muscle cells and tissues in response to agonist stimulation. Blockage of actin polymerization by the inhibitors cytochalasin and latrunculin attenuates active force upon activation with contractile stimuli whereas myosin regulatory light chain phosphorylation (MRLCP) is not disrupted.5, 8-10 These studies suggest that actin dynamics and MRLCP are independently regulated, and that both dynamic changes in the actin cytoskeleton and myosin activation are required for force development during contractile stimulation of smooth muscle.4, 8-11 However, the mechanisms that regulate the actin cytoskeleton in smooth muscle are not completely understood.
The tyrosine phosphorylated protein Crk-associated substrate (CAS) has been implicated in the modulation of the actin cytoskeleton in smooth muscle cells as well as in nonmuscle cells including COS-7 cells and NIH3T3 cells.9, 12-14 Downregulation of CAS by antisense dramatically attenuates force development and actin polymerization in carotid arteries in response to contractile stimulation.9 CAS undergoes tyrosine phosphorylation in vascular smooth muscle cells and tissues upon stimulation with angiotensin II or serotonin 12, 15 as well as in non-muscle cells in response to growth factors and cell adhesion. 13, 16 CAS phosphorylation has been proposed to be a key event regulating the actin cytoskeleton; phosphorylated CAS may enhance its binding to the adapter protein CrkII, which may facilitate the formation of the multiprotein compound including CrkII, neuronal Wiskott - Aldrich Syndrome Protein (N-WASP) and the Arp2/3 (Actin Related Protein) complex and initiating actin polymerization and branching mediated by the Arp2/3 complex. 1-3, 17, 18
The upstream regulatory molecules of CAS in smooth muscle are not completely elucidated. The non-receptor tyrosine kinase Abl (Abelson tyrosine kinase, c-Abl) is ubiquitously expressed and has been implicated to function in a range of cellular processes including the regulation of the actin cytoskeleton that mediates cell migration and spreading, membrane ruffling, cell adhesion, neurite extension, and cell growth and survival in nonmuscle cells including NIH3T3 fibroblasts, mouse embryonic fibroblasts, neurons, epithelial cells, and COS-7 cells.19, 20 Abl is able to stimulate the formation of actin microspikes in fibroblasts spreading on fibronectin; this function of Abl is dependent on kinase activity and is not shared by c-Src tyrosine kinase.21 In addition, platelet-derived growth factor (PDGF) enhances the activity of Abl and increases plasma membrane ruffling. Mouse embryonic fibroblasts lacking Abl exhibit four- to five-fold fewer dorsal/circular membrane ruffles when stimulated by PDGF. Expression of wild type Abl in these cells rescues the effects of PDGF.22, 23
The objective of this study was to test the hypothesis that Abl may be a pivotal upstream molecule of CAS in the resistance artery, a well-recognized arterial smooth muscle model.6, 24 Our results demonstrate that silencing of Abl by small interfering RNA (siRNA) attenuates CAS phosphorylation, the formation of the protein complex containing CrkII and dynamic changes in the actin cytoskeleton without affecting myosin activation during stimulation with the α1-adrenoceptor agonist phenylephrine (PE).
Materials and Methods
Materials, tissue preparation, immunoblot analysis, in vitro kinase assay, cDNA constructs, chemical loading and organ culture, fluorescence microscopy, co-immunoprecipitation, analysis of F/G-actin ratios, analysis of myosin regulatory light chain phosphorylation (MRLCP) and statistical analysis are described in the Materials and Methods section of Online Data Supplement available at http://circres.ahajournals.org.
Results
Increases in CAS phosphorylation and the association of CAS with CrkII are dose- and time-dependent in mesenteric arteries upon PE stimulation
Our previous studies suggest that CAS participates in the cellular processes that mediate vascular smooth muscle contraction.8, 9 Recent report has shown that serotonin stimulation elicits the tyrosine phosphorylation (Tyr-410) of CAS in rat aorta (elastic artery).12 To evaluate whether CAS phosphorylation occurs in resistance arteries, rat mesenteric rings were treated with different concentrations of PE for different time points, or they were not stimulated. CAS tyrosine phosphorylation was assessed by immunoblot analysis using phospho-CAS (Tyr-410) antibody. Stimulation with PE (5 min) resulted in the dose-dependent increase in CAS phosphorylation in mesenteric arteries (Fig. 1A). CAS phosphorylation was increased as early as 1 min after stimulation with PE (10 μmol/L) and maintained for the 30-min period (Fig. 1B).
Figure 1. Activation with phenylephrine (PE) results in increases in CAS phosphorylation, the association of CAS with CrkII and Abl phosphorylation in resistance arteries.



Unstimulated or PE-stimulated rat mesenteric arteries were frozen for biochemical analysis. (A) Representative blots show the dose-dependent enhancement of CAS phosphorylation (Tyr-410) upon PE stimulation (5 min). The level of CAS phosphorylation elicited by PE is normalized to the value of unstimulated arteries (n = 4-5). (B) CAS phosphorylation upon PE stimulation (10 μmol/L) is time-dependent (n = 4). (C) Representative blots illustrate the increased amount of CAS co-immunoprecipitated with CrkII upon PE stimulation (5 min). CAS/CrkII ratios stimulated by PE are expressed as magnitude of the level of unstimulated arteries (n = 4). (D) The time-dependent increase in CAS/CrkII coupling in response to PE (10 μmol/L) stimulation (n = 4-5). (E) Representative blots illustrate the effects of PE stimulation (5 min) on Abl phosphorylation (Tyr-412). PE-stimulated Abl phosphorylation is normalized to the level of unstimulated tissues (n = 4-6). (F) The time-dependent enhancement of Abl phosphorylation upon PE (10 μmol/L) stimulation (n = 4). Values represent mean ± SE.
CAS phosphorylation enhances its ability to interact with the SH2-containing adapter protein CrkII in nonmuscle cells in response to integrin activation.25, 26 To determine whether contractile stimulation leads to the increase in the association of CAS with CrkII, blots of CrkII immunoprecipitates from unstimulated and PE-stimulated arteries were detected for CAS and CrkII. Activation with PE led to the increase in the amount of CAS co-immunoprecipitated with CrkII; CAS/CrkII ratios upon PE stimulation were obviously augmented in a dose- and time-dependent manner (Fig. 1, C and D).
Activation with PE induces Abl phosphorylation in resistance arteries
We also evaluated Abl phosphorylation in response to contractile stimulation In nonmuscle cells, Abl has been implicated in regulating actin cytoskeleton remodeling.19, 20 Immunoblot analysis was used to assess Abl phosphorylation (Tyr-412) in mesenteric arteries treated with PE. The phosphorylation level of Abl in resistance arteries was augmented upon contractile activation, which was dose- and time-dependent (Fig. 1, E and F). The increase in Abl phosphorylation in response to PE stimulation was slightly earlier than the enhancement of CAS phosphorylation and the interaction of CAS with CrkII.
CAS phosphorylation is catalyzed by Abl in vitro
Src has been implicated in the regulation of CAS phosphorylation in vascular smooth muscle tissues and non-muscle cells.12, 25 Abl phosphorylation is attenuated by a Src inhibitor in cultured smooth muscle cells.27 Thus, Abl kinase may be able to catalyze CAS phosphorylation in smooth muscle.
To assess whether Abl mediates CAS phosphorylation directly, Abl (enzyme) was immunoprecipitated from rat resistance arteries followed by the addition of purified CAS (substrate). In vitro kinase assay was initiated by the addition of ATP, and CAS phosphorylation was evaluated by immunoblot analysis. CAS tyrosine phosphorylation increased by 2.5-fold in the presence of Abl (Fig. 2).
Figure 2. Abl catalyzes CAS phosphorylation in vitro.
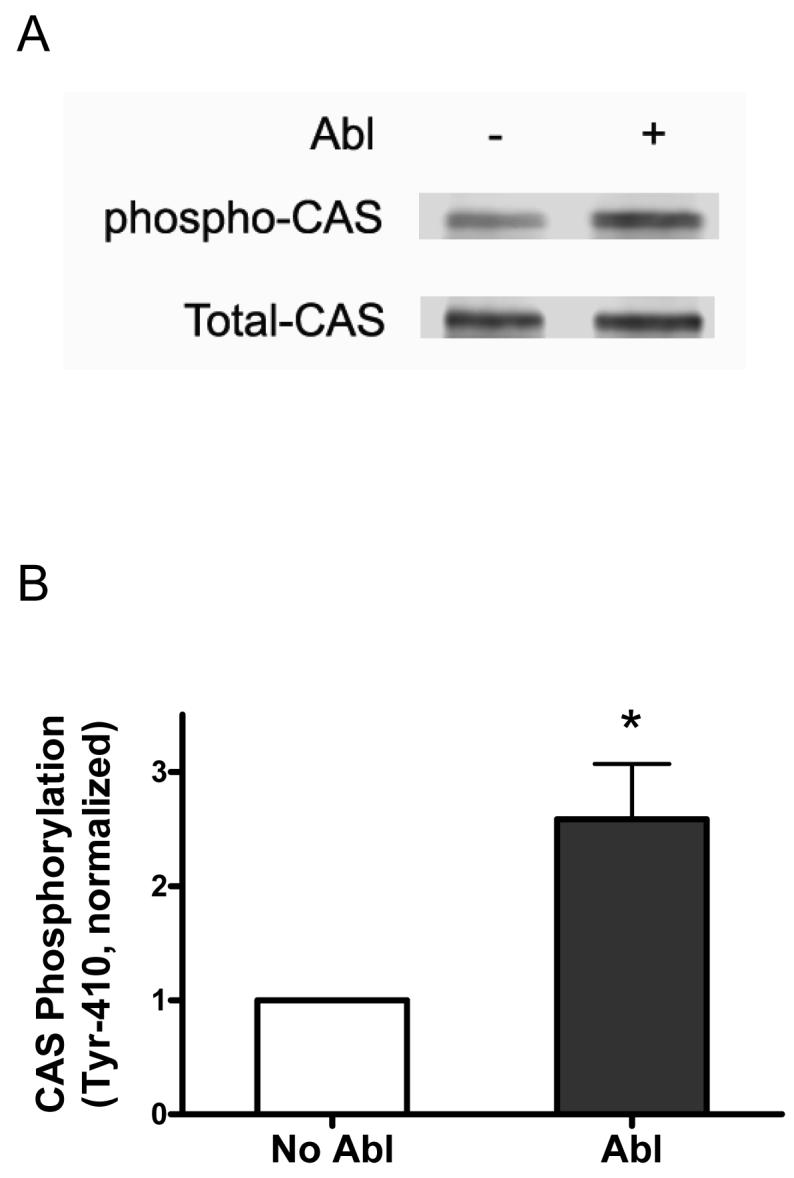
(A) The immunoblots illustrate the phosphorylation of CAS (Tyr-410) by Abl. Abl-mediated CAS phosphorylation 30 min after the initiation of reaction was determined as described under “Materials and Methods.” (B) The phosphorylation level after Abl treatment (30 min) is normalized to the level of CAS phosphorylation in the absence of Abl. *Significantly higher phosphorylation levels in the presence of Abl compared to the level without Abl treatment (n = 3, P < 0.05).
Abl silencing attenuates CAS phosphorylation upon contractile stimulation
To elucidate the role of Abl in regulating CAS in smooth muscle, we used plasmid-based short hairpin RNA (shRNA) to silence Abl in resistance arteries and then determine the effects of Abl silencing on CAS phosphorylation. Green fluorescence protein (GFP)-plasmids encoding Abl-shRNA or luciferase-shRNA (control RNA) were introduced into mesenteric arteries using a modified method of chemical loading (also known as reversible permeabilization).1, 2, 28 Protein expression in these tissues was assessed by immunoblot analysis. Fluorescence microscopy was used to assess whether GFP fluorescence (an indication of transfection) is present in the smooth muscle layer of the arterial wall.
As shown in Fig. 3A, the level of Abl protein was reduced in arteries producing Abl shRNA compared to untreated tissues or arteries generating luciferase shRNA. However, smooth muscle specific α-actin content was not different in these tissues. Ratios of Abl/actin were significantly lower in Abl-deficient tissues than in untreated arteries or tissues producing luciferase shRNA (Fig. 3B). Moreover, GFP fluorescence was widely detected in the medial smooth muscle layer of arteries treated with plasmids, indicating efficient transfection in the tissues (Fig. 3C).
Figure 3. Silencing of Abl gene in mesenteric segments by short hairpin RNA (shRNA).

(A) Blots of protein extracts from untreated arteries (UT) or arteries that had been cultured for 2 days with plasmids encoding luciferase shRNA (Luc) or Abl shRNA (Abl) were probed with antibodies against Abl and α-actin. The level of Abl was lower in arteries producing Abl shRNA than in untreated arteries or segments generating Luc shRNA. Similar amounts of actin were detected in all three samples. (B) Abl/actin ratios in arteries producing Luc shRNA or Abl shRNA are normalized to ratios obtained in tissues not treated with plasmids (n = 5). Values are mean ± SE. *Significantly lower protein ratios in segments producing Abl shRNA compared to untreated arteries or arteries generating Luc shRNA (P < 0.01). (C) Representative images showing green fluorescence protein (GFP) signals observed in the medial smooth muscle layer of the arterial walls treated with plasmids, indicating effective transfection in the smooth muscle layer of arteries. Weak autofluorescence (panel a) was observed from the untreated mesenteric artery (panel a'). GFP signal (panel b) was detected from the artery transfected with plasmids encoding luciferase (Luc) shRNA (panel b'); the fluorescence (panel c), the artery transfected with plasmids for Abl shRNA (panel c'). PC, phase contrast images. A, adventitia; M, media. Intima was removed during preparation. Bar, 15 μm.
CAS phosphorylation in Abl-depleted arteries and control tissues was evaluated by immunoblot analysis. Basal CAS phosphorylation (Tyr-410) was similar in untreated arteries, or in segments producing luceferase shRNA or Abl shRNA. However, CAS phosphorylation upon stimulation with PE (10 μmol/L, 5 min) was significantly lower in arterial segments generating Abl shRNA compared to untreated tissues or arteries producing luciferase shRNA (Fig. 4A) (n = 5, P < 0.01).
Figure 4. CAS phosphorylation in response to contractile stimulation is diminished in Abl-deficient arteries.


(A) Blots of extracts from resistance arteries that had been treated with plasmids encoding Luc shRNA or Abl shRNA, or untreated arteries were detected with use of phospho-CAS (Tyr-410) antibody, stripped, and reprobed with (total) CAS antibody. CAS phosphorylation upon stimulation with PE was depressed in Abl-deficient arteries (n = 5). Values are normalized to the phosphorylation level in corresponding unstimulated arteries. (B) Representative blots illustrate the effects of KCl stimulation (5 min) on Abl phosphorylation (Tyr-412). KCl-stimulated Abl phosphorylation is normalized to the level of unstimulated tissues (n = 5). (C) Representative blots show the increase in CAS phosphorylation (Tyr-410) upon KCl stimulation (5 min). The level of CAS phosphorylation elicited by KCl is normalized to the value of unstimulated arteries (n = 5). (D) Representative blots illustrate the effects of Abl silencing on KCl-induced CAS phosphorylation. Values are normalized to the phosphorylation level in corresponding unstimulated arteries (n = 5). Values are mean ± SE. *Significantly lower levels upon PE or KCl stimulation compared to corresponding untreated (control) arteries or tissues treated with Luc shRNA (P < 0.01).
We also observed that stimulation with KCl resulted in the increase in phosphorylation of Abl and CAS (Fig. 4, B and C). Therefore, we determined whether Abl silencing affects KCl-induced phosphorylation of CAS in resistance arteries. CAS phosphorylation in response to stimulation with KCl (80 mM, 5 min) was significantly reduced in segments producing Abl shRNA compared to untreated arteries or tissues generating luciferase shRNA (Fig. 4D) (n = 5, P < 0.01).
Abl silencing inhibits the association of CAS with CrkII and formation of the protein complex containing CrkII during PE stimulation
We evaluated the effects of Abl silencing on CAS/CrkII coupling and formation of the complex containing CrkII. Untreated arteries or arteries that had been treated with plasmids encoding luciferase shRNA, or Abl shRNA were stimulated with PE, or left unstimulated. Blots of CAS or CrkII immunoprecipitates from these arteries were probed with antibodies against CAS, CrkII, N-WASP or Arp2.
In untreated arteries and arteries producing luciferase shRNA, the amount of CrkII in CAS immunoprecipitates was increased during PE stimulation. In contrast, the CrkII amount co-immunoprecipitated with CAS in response to PE stimulation was attenuated in tissues producing Abl shRNA (Fig. 5A). Similarly, the amounts of N-WASP and Arp2 co-immunoprecipitated with CrkII were increased upon contractile stimulation in untreated arteries and arteries producing luciferase shRNA; however, the increase in the amounts of N-WASP and Arp2 in CrkII immunoprecipitates elicited by PE stimulation was reduced in tissues generating Abl shRNA (Fig. 5B). Treatment with shRNAs did not affect CAS/CrkII coupling and the complex formation in unstimulated arteries (Fig. 5, A and B). Increases in protein ratios of CrkII/CAS, N-WASP/CrkII, and Arp2/CrkII in response to PE activation were significantly reduced in arteries producing Abl shRNA compared to untreated tissues and tissues generating luciferase shRNA (Fig. 5C, n = 3, P < 0.05).
Figure 5. Association of CAS with CrkII and formation of protein complex containing CrkII are attenuated in Abl-deficient arteries upon PE stimulation.

Untreated arteries or arteries that had been treated with plasmids encoding Luc shRNA, or Abl shRNA were stimulated with PE, or left unstimulated. (A) Blots of CAS immunoprecipitates from these arteries were probed with antibodies against CAS and CrkII. The immunoblots illustrate the effects of Abl silencing on CAS/CrkII coupling. (B) Blots of CrkII immunoprecipitates from these arteries were probed with antibodies against CrkII, N-WASP, and Arp2. Abl silencing inhibits the complex formation. (C) Individual protein ratios are normalized to protein ratios in corresponding unstimulated arteries. *Significantly lower protein ratios in response to PE stimulation compared to corresponding PE-induced protein ratios in untreated arteries or arteries producing Luc shRNA (n = 3, P < 0.05). Values are mean ± SE.
Increases in F/G-actin ratios elicited by PE are attenuated in Abl-depleted arteries
Contractile stimulation increases ratios of F/G-actin in smooth muscle tissues and cells.1-3, 8, 9, 29 Abl has been implicated in regulating the actin cytoskeleton in nonmuscle cells such as fibroblasts and COS cells.19, 20 We evaluated the effects of Abl downregulation on F/G-actin ratios in resistance arteries upon contractile stimulation. Untreated arteries or arteries that had been treated with plasmids encoding luciferase shRNA or Abl shRNA were stimulated with PE or they were not stimulated. Ratios of F/G-actin in these tissues were evaluated using a fractionation assay.1, 3, 8, 9, 29
Stimulation with PE led to the increase in F/G-actin ratios in untreated arteries and segments producing luciferase shRNA. However, the increase in F/G-actin ratios upon PE stimulation was dramatically depressed in Abl-deficient resistance arteries. Treatment with shRNAs did not affect the F/G-actin ratios in unstimulated arteries (Fig. 6).
Figure 6. Abl silencing inhibits the increase in F/G-actin ratios upon PE activation.

Arteries producing Luc shRNA or Abl shRNA were stimulated with PE, or left unstimulated. F/G-actin ratios were then assessed. (A) The representative blot illustrating the effects of Abl silencing on the amounts of G-actin and F-actin. (B) *Significantly higher F/G-actin ratios upon PE stimulation compared to corresponding unstimulated values (P < 0.01). **PE-stimulated F/G-actin ratios in tissues producing Abl shRNA are significantly lower than those in untreated tissues or arteries generating Luc shRNA (P < 0.05). Values are mean ± SE (n = 5).
Constriction in response to contractile stimulation is depressed in Abl-deficient arteries
We assessed the effects of Abl silencing on arterial constriction in response to contractile stimulation. The contractile responses of rat mesenteric arteries to PE were determined, after which plasmids encoding luciferase shRNA (Luc shRNA) or Abl shRNA were transduced into these arteries, and were incubated for 2 days. Constriction of these arteries were then determined. Dose-response curves upon stimulation with PE were compared between three groups. In untreated segments or in arteries treated with plasmids producing luciferase shRNA, contractile responses to PE were dose-dependent; maximal constriction was 70% of the corresponding maximal value before preincubation. However, the dose-response curve in Abl deficient tissues was significantly right shifted. Maximal contraction was reduced to 30% of the preincubation value (Fig. 7A).
Figure 7. Constriction is diminished in Abl-deficient arteries in response to activation with PE or KCl.

(A) Compared to untreated tissues or arteries producing luciferase shRNA (Luc RNA), PE-induced dose-response curve in arteries producing Abl shRNA was right shifted. Maximal contraction was also significantly reduced (n = 4-6). (B) The dose-response curve in response to KCl stimulation was right shifted with the reduced maximal response compared to untreated tissues or arteries producing Luc RNA. Constriction in response to PE or KCl is normalized to corresponding maximal levels in each artery before incubation (n = 5). Values are mean ± SE. Asterisk indicates significantly lower response as compared to untreated tissues or tissues generating Luc shRNA (P < 0.05).
We also assessed the effects of Abl silencing on KCl-induced vasoconstriction. KCl-induced dose-response curve in Abl deficient segments was right shifted with a reduced maximal response compared to untreated arteries or segments producing luciferase shRNA (Fig. 7B). There were no significant differences in tension among the three experimental groups before contractile stimulation.
Abl silencing does not inhibit the increase in MRLCP upon contractile stimulation
MRLCP has been thought to initiate crossbridge cycling and force generation in smooth muscle. 30, 31 To determine whether the depressed contractile response in Abl-depleted arteries stem from the inhibition of MRLCP, a subset of mesenteric segments that had been treated with no plasmids, or plasmids encoding luciferase shRNA or Abl shRNA were stimulated with 10−5 mol/L PE for 5 min, or they were not stimulated. These arterial vessels were then frozen for the determination of MRLCP.
In spite of reduced constriction, PE stimulation was able to lead to a significant increase in MRLCP in Abl deficient arteries. The average increases in MRLCP in segments not treated with shRNA, or luciferase shRNA-treated and Abl shRNA-treated tissues were not significantly different 5 min after PE stimulation (Fig. 8).
Figure 8. Effects of Abl silencing on myosin regulatory light chain phosphorylation (MRLCP).

Increases in MRLCP upon PE stimulation are similar in arteries that have been treated with plasmids encoding Luc shRNA or Abl shRNA, or no plasmids (P > 0.05). Values shown are mean ± SE (n = 5).
Discussion
CAS is a 130-kDa tyrosine phosphorylated protein participating in the cellular processes that transduce external signals onto the actin cytoskeleton in various cell types including smooth muscle cells. In this report, Abl is able to catalyze CAS phosphorylation in the in vitro study. Moreover, Abl silencing attenuates CAS phosphorylation, the formation of the multiprotein complex containing CrkII, actin polymerization, and arterial constriction during contractile activation. These results suggest that Abl plays an essential role in regulating CAS-mediated signaling and constriction in resistance arteries.
CAS is required for actin filament assembly and active force development in arterial smooth muscle.9 In the present report, activation with the α adrenoceptor agonist led to the enhancement of CAS phosphorylation and the association of CAS with CrkII in resistance arteries. These observations are supported by previous studies on elastic artery and in cultured cells.12, 25 More importantly, our results showed that Abl underwent phosphorylation at Tyr-412 in resistance arteries upon contractile stimulation, implying its role in modulating arterial constriction. Tyr-412 is located at the activation loop of Abl kinase domain. When unstimulated, the activation loop of the Abl kinase domain folds into the active site, thereby preventing binding of both the substrate and ATP. Phosphorylation at Tyr-412 induces conformation changes; the activation loop no longer blocks the active site, which leads the increase in kinase activity.32
In the present study, arterial segments were transfected with plasmids encoding Abl shRNA using the method of chemical loading plus liposomes.1, 2, 28, 33, 34 Plasmid-based shRNA has been shown to inhibit gene expression in mammalian cells; shRNA can be cleaved by intracellular RNase III (dicers) to generate siRNA with 21-22 bases, which initiates the formation of RNA-induced silencing complex (RISC) and induces gene silencing.35 Abl silencing by shRNA depressed the expression of Abl and attenuated CAS tyrosine phosphorylation in response to contractile activation (Figs. 3 and 4). Moreover, our in vitro study suggests a direct role for Abl in catalyzing CAS tyrosine phosphorylation. Because Src may participate in the regulation of CAS phosphorylation in vascular smooth muscle tissues and non-muscle cells,12, 36 and Abl phosphorylation is attenuated by a Src inhibitor in cultured vascular smooth muscle cells,27 it is possible that contractile stimulation may activate Abl tyrosine kinase via Src; the activated Abl may directly catalyze CAS phosphorylation in resistance arteries.
CAS phosphorylation has been shown to occur in its substrate domain, which contains 15 YXXP tyrosine residues.13 Using site-directed mutagenesis combined with tryptic phosphopeptide mapping, Hanks and his colleagues have documented that Tyr-410 is one of the major phosphorylation sites on CAS; phosphorylation on the tyrosine residues creates docking sites for the SH2-containing signaling effector CrkII.13, 25 Previous studies have shown that CrkII is able to regulate the activity of N-WASP. When unstimulated, the C-terminal portion of N-WASP binds to its GTP-binding domain, masking its binding motif for the Arp2/3 complex and inhibiting the activity of N-WASP. When bound to CrkII, N-WASP undergoes conformational changes, exposing the binding motif for the Arp2/3 complex and initiating actin polymerization and branching mediated by the Arp2/3 complex.1-3, 17, 18
In this report, Abl silencing inhibited the association of CAS with CrkII, the formation of protein complex containing CrkII, N-WASP and the Arp2/3 complex, and actin filament assembly in arterial segments during contractile activation. Since Abl mediates CAS phosphorylation in the in vitro study and in arteries in response to contractile stimulation, we propose that Abl may regulate CAS tyrosine phosphorylation, which may sequentially facilitate the interaction of CAS with CrkII and the activation of N-WASP, promoting the Arp2/3 complex-mediated actin polymerization in arterial smooth muscle in response to contractile activation. In addition, Abl silencing did not attenuate increases in MRLCP during contractile stimulation, suggesting that Abl is not involved in the regulation of myosin activation in resistance arteries.
Actin polymerization may enhance force development by the following mechanisms. First, the actin filaments of smooth muscle cells connect to extracellular matrix via transmembranous β integrins and linker proteins such as vinculin and talin, facilitating force transmission between actin filaments to matrix.1, 2, 37, 38 Actin polymerization is initiated by the Arp2/3 complex in nonmuscle and smooth muscle cells, promoting nascent actin polymerization at cell cortex. Cortical actin assembly may reinforce the linkage of actin filaments to integrins strengthening the transduction of mechanical force.1-3, 17, 18 Second, actin assembly has been shown to increase the numbers of contractile units and the length of actin filaments, providing more and efficient contractile elements for force development 5, 39. Third, actin polymerization may also be a part of the reorganization process that allows for the rapid adjustment of cytoskeletal organization elicited by external stimulation. 1, 3, 40-43
In the present study, both PE stimulation and KCl depolarization induced the tyrosine phosphorylation of Abl and CAS. Furthermore, Abl silencing attenuated CAS phosphorylation and force development in response to activation with PE and KCl. These results suggest that Abl tyrosine kinase is an essential convergent molecule that regulates constriction in resistance arteries upon the activation with α-receptor and membrane depolarization. An interpretation for this is that KCl depolarization increases intracellular Ca2+ (well demonstrated), which may activate Abl and CAS via a Ca2+-dependent mechanism.31, 44 Therefore, Abl deficiency is able to disrupt the KCl-induced contraction. Because PE is also able to activate L-type channel and increase intracellular Ca2+, 44 it is possible that PE-mediated Abl/CAS activation and constriction may be mediated in part by Ca2+-dependent mechanisms. As some other protein kinases associated with smooth muscle contraction, Abl may also be activated by other signaling molecules such as small GTPases.31, 44
Summary
Although the role of CAS in regulating smooth muscle constriction has been recognized, the upstream regulator of CAS has not been well identified. In this study, we demonstrate a critical role of Abl in modulating CAS tyrosine phosphorylation and constriction in resistance arteries. Abl-mediated arterial constriction may be regulated by the formation of the multiprotein complex and actin filament assembly.
Supplementary Material
Acknowledgments
This work was supported by The National Heart, Lung, and Blood Institute Grant HL-75388 and an American Heart Association Scientist Development Grant (to D.D.T.).
Footnotes
Disclosures
None
References
- 1.Tang DD, Zhang W, Gunst SJ. The Adapter Protein CrkII Regulates Neuronal Wiskott-Aldrich Syndrome Protein, Actin Polymerization, and Tension Development during Contractile Stimulation of Smooth Muscle. J Biol Chem. 2005 June 17;280(24):23380–9. doi: 10.1074/jbc.M413390200. [DOI] [PubMed] [Google Scholar]
- 2.Tang DD, Gunst SJ. The small GTPase Cdc42 regulates actin polymerization and tension development during contractile stimulation of smooth muscle. J Biol Chem. 2004 December 10;279(50):51722–8. doi: 10.1074/jbc.M408351200. [DOI] [PubMed] [Google Scholar]
- 3.Zhang W, Wu Y, Du L, Tang DD, Gunst SJ. Activation of the Arp2/3 complex by N-WASp is required for actin polymerization and contraction in smooth muscle. Am J Physiol Cell Physiol. 2005 May;288(5):C1145–C1160. doi: 10.1152/ajpcell.00387.2004. [DOI] [PubMed] [Google Scholar]
- 4.Barany M, Barron JT, Gu L, Barany K. Exchange of the actin-bound nucleotide in intact arterial smooth muscle. J Biol Chem. 2001 December 21;276(51):48398–403. doi: 10.1074/jbc.M106227200. [DOI] [PubMed] [Google Scholar]
- 5.Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J. 2002 January;16(1):72–6. doi: 10.1096/cj.01-0104hyp. [DOI] [PubMed] [Google Scholar]
- 6.Chen X, Pavlish K, Zhang HY, Benoit JN. Effects of chronic portal hypertension on agonist-induced actin polymerization in small mesenteric arteries. Am J Physiol Heart Circ Physiol. 2006 May;290(5):H1915–H1921. doi: 10.1152/ajpheart.00643.2005. [DOI] [PubMed] [Google Scholar]
- 7.Hirshman CA, Zhu D, Pertel T, Panettieri RA, Emala CW. Isoproterenol induces actin depolymerization in human airway smooth muscle cells via activation of an Src kinase and GS. Am J Physiol Lung Cell Mol Physiol. 2005 May;288(5):L924–L931. doi: 10.1152/ajplung.00463.2004. [DOI] [PubMed] [Google Scholar]
- 8.Tang DD, Tan J. Downregulation of profilin with antisense oligodeoxynucleotides inhibits force development during stimulation of smooth muscle. Am J Physiol Heart Circ Physiol. 2003 June 12;285:H1528–H1536. doi: 10.1152/ajpheart.00188.2003. [DOI] [PubMed] [Google Scholar]
- 9.Tang DD, Tan J. Role of Crk-Associated Substrate in the Regulation of Vascular Smooth Muscle Contraction. Hypertension. 2003 July 28;42:858–63. doi: 10.1161/01.HYP.0000085333.76141.33. [DOI] [PubMed] [Google Scholar]
- 10.Mehta D, Gunst SJ. Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J Physiol. 1999;519(Pt 3):829–40. doi: 10.1111/j.1469-7793.1999.0829n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang DD, Wu MF, Opazo Saez AM, Gunst SJ. The focal adhesion protein paxillin regulates contraction in canine tracheal smooth muscle. J Physiol. 2002 July 15;542(Pt 2):501–13. doi: 10.1113/jphysiol.2002.021006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogden K, Thompson JM, Hickner Z, Huang T, Tang DD, Watts SW. A new signaling paradigm for serotonin: use of Crk-associated substrate in arterial contraction. Am J Physiol Heart Circ Physiol. 2006 December;291(6):H2857–H2863. doi: 10.1152/ajpheart.00229.2006. [DOI] [PubMed] [Google Scholar]
- 13.Shin NY, Dise RS, Schneider-Mergener J, Ritchie MD, Kilkenny DM, Hanks SK. Subsets of the major tyrosine phosphorylation sites in Crk-associated substrate (CAS) are sufficient to promote cell migration. J Biol Chem. 2004 September 10;279(37):38331–7. doi: 10.1074/jbc.M404675200. [DOI] [PubMed] [Google Scholar]
- 14.Cho SY, Klemke RL. Purification of pseudopodia from polarized cells reveals redistribution and activation of Rac through assembly of a CAS/Crk scaffold. J Cell Biol. 2002 February 18;156(4):725–36. doi: 10.1083/jcb.200111032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takahashi T, Kawahara Y, Taniguchi T, Yokoyama M. Tyrosine phosphorylation and association of p130Cas and c-Crk II by ANG II in vascular smooth muscle cells. Am J Physiol. 1998 April;274(4 Pt 2):H1059–H1065. doi: 10.1152/ajpheart.1998.274.4.H1059. [DOI] [PubMed] [Google Scholar]
- 16.Ojaniemi M, Vuori K. Epidermal growth factor modulates tyrosine phosphorylation of p130Cas. Involvement of phosphatidylinositol 3'-kinase and actin cytoskeleton. J Biol Chem. 1997;272(41):25993–8. doi: 10.1074/jbc.272.41.25993. [DOI] [PubMed] [Google Scholar]
- 17.Rohatgi R, Nollau P, Ho HY, Kirschner MW, Mayer BJ. Nck and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP-Arp2/3 pathway. J Biol Chem. 2001 July 13;276(28):26448–52. doi: 10.1074/jbc.M103856200. [DOI] [PubMed] [Google Scholar]
- 18.Pollard TD, Blanchoin L, Mullins RD. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct. 2000;29:545–76. doi: 10.1146/annurev.biophys.29.1.545. [DOI] [PubMed] [Google Scholar]
- 19.Hu H, Bliss JM, Wang Y, Colicelli J. RIN1 is an ABL tyrosine kinase activator and a regulator of epithelial-cell adhesion and migration. Curr Biol. 2005 May 10;15(9):815–23. doi: 10.1016/j.cub.2005.03.049. [DOI] [PubMed] [Google Scholar]
- 20.Woodring PJ, Hunter T, Wang JY. Regulation of F-actin-dependent processes by the Abl family of tyrosine kinases. J Cell Sci. 2003 July 1;116(Pt 13):2613–26. doi: 10.1242/jcs.00622. [DOI] [PubMed] [Google Scholar]
- 21.Woodring PJ, Litwack ED, O'Leary DD, Lucero GR, Wang JY, Hunter T. Modulation of the F-actin cytoskeleton by c-Abl tyrosine kinase in cell spreading and neurite extension. J Cell Biol. 2002 March 4;156(5):879–92. doi: 10.1083/jcb.200110014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furstoss O, Dorey K, Simon V, Barila D, Superti-Furga G, Roche S. c-Abl is an effector of Src for growth factor-induced c-myc expression and DNA synthesis. EMBO J. 2002 February 15;21(4):514–24. doi: 10.1093/emboj/21.4.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plattner R, Kadlec L, DeMali KA, Kazlauskas A, Pendergast AM. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999 September 15;13(18):2400–11. doi: 10.1101/gad.13.18.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watts SW. Serotonin-induced contraction in mesenteric resistance arteries: signaling and changes in deoxycorticosterone acetate-salt hypertension. Hypertension. 2002 March 1;39(3):825–9. doi: 10.1161/hy0302.104668. [DOI] [PubMed] [Google Scholar]
- 25.Chodniewicz D, Klemke RL. Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim Biophys Acta. 2004 July 5;1692(23):63–76. doi: 10.1016/j.bbamcr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 26.Defilippi P, Di SP, Cabodi S. p130Cas: a versatile scaffold in signaling networks. Trends Cell Biol. 2006 May;16(5):257–63. doi: 10.1016/j.tcb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Ushio-Fukai M, Zuo L, Ikeda S, Tojo T, Patrushev NA, Alexander RW. cAbl tyrosine kinase mediates reactive oxygen species- and caveolin-dependent AT1 receptor signaling in vascular smooth muscle: role in vascular hypertrophy. Circ Res. 2005 October 14;97(8):829–36. doi: 10.1161/01.RES.0000185322.46009.F5. [DOI] [PubMed] [Google Scholar]
- 28.Marganski WA, Gangopadhyay SS, Je HD, Gallant C, Morgan KG. Targeting of a novel Ca+2/calmodulin-dependent protein kinase II is essential for extracellular signal-regulated kinase-mediated signaling in differentiated smooth muscle cells. Circ Res. 2005 September 16;97(6):541–9. doi: 10.1161/01.RES.0000182630.29093.0d. [DOI] [PubMed] [Google Scholar]
- 29.Meeks MK, Ripley ML, Jin Z, Rembold CM. Heat shock protein 20-mediated force suppression in forskolin-relaxed swine carotid artery. Am J Physiol Cell Physiol. 2005 March;288(3):C633–C639. doi: 10.1152/ajpcell.00269.2004. [DOI] [PubMed] [Google Scholar]
- 30.Tang DD, Gunst SJ. Depletion of focal adhesion kinase by antisense depresses contractile activation of smooth muscle. Am J Physiol Cell Physiol. 2001 April;280(4):C874–C883. doi: 10.1152/ajpcell.2001.280.4.C874. [DOI] [PubMed] [Google Scholar]
- 31.Somlyo AP, Somlyo AV. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol. 2000 January 15;522(Pt 2):177–85. doi: 10.1111/j.1469-7793.2000.t01-2-00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang JY. Controlling Abl: auto-inhibition and co-inhibition? Nat Cell Biol. 2004 January;6(1):3–7. doi: 10.1038/ncb0104-3. [DOI] [PubMed] [Google Scholar]
- 33.Wang R, Li Q, Tang DD. Role of vimentin in smooth muscle force development. Am J Physiol Cell Physiol. 2006 September;291(3):C483–C489. doi: 10.1152/ajpcell.00097.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang R, Li QF, Anfinogenova Y, Tang DD. Dissociation of Crk-associated substrate from the vimentin network is regulated by p21-activated kinase on ACh activation of airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2007 January;292(1):L240–L248. doi: 10.1152/ajplung.00199.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandy P, Ventura A, Jacks T. Mammalian RNAi: a practical guide. Biotechniques. 2005 August;39(2):215–24. doi: 10.2144/05392RV01. [DOI] [PubMed] [Google Scholar]
- 36.Ruest PJ, Shin NY, Polte TR, Zhang X, Hanks SK. Mechanisms of CAS substrate domain tyrosine phosphorylation by FAK and Src. Mol Cell Biol. 2001 November;21(22):7641–52. doi: 10.1128/MCB.21.22.7641-7652.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gunst SJ, Tang DD. The contractile apparatus and mechanical properties of airway smooth muscle. Eur Respir J. 2000 April;15(3):600–16. doi: 10.1034/j.1399-3003.2000.15.29.x. [DOI] [PubMed] [Google Scholar]
- 38.Gerthoffer WT, Gunst SJ. Invited review: focal adhesion and small heat shock proteins in the regulation of actin remodeling and contractility in smooth muscle. J Appl Physiol. 2001 August;91(2):963–72. doi: 10.1152/jappl.2001.91.2.963. [DOI] [PubMed] [Google Scholar]
- 39.Herrera AM, Martinez EC, Seow CY. Electron microscopic study of actin polymerization in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2004 June;286(6):L1161–L1168. doi: 10.1152/ajplung.00298.2003. [DOI] [PubMed] [Google Scholar]
- 40.Opazo SA, Zhang W, Wu Y, Turner CE, Tang DD, Gunst SJ. Tension development during contractile stimulation of smooth muscle requires recruitment of paxillin and vinculin to the membrane. Am J Physiol Cell Physiol. 2004 February;286(2):C433–C447. doi: 10.1152/ajpcell.00030.2003. [DOI] [PubMed] [Google Scholar]
- 41.Gerthoffer WT. Actin cytoskeletal dynamics in smooth muscle contraction. Can J Physiol Pharmacol. 2005 October;83(10):851–6. doi: 10.1139/y05-088. [DOI] [PubMed] [Google Scholar]
- 42.Li QF, Spinelli AM, Wang R, Anfinogenova Y, Singer HA, Tang DD. Critical Role of Vimentin Phosphorylation at Ser-56 by p21-activated Kinase in Vimentin Cytoskeleton Signaling. J Biol Chem. 2006 November 10;281(45):34716–24. doi: 10.1074/jbc.M607715200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang DD, Bai Y, Gunst SJ. Silencing of p21-activated kinase attenuates vimentin phosphorylation on Ser-56 and reorientation of the vimentin network during stimulation of smooth muscle cells by 5-hydroxytryptamine. Biochem J. 2005 June 15;388(Pt 3):773–83. doi: 10.1042/BJ20050065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horowitz A, Menice CB, Laporte R, Morgan KG. Mechanisms of smooth muscle contraction. Physiol Rev. 1996 October;76(4):967–1003. doi: 10.1152/physrev.1996.76.4.967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.