

Abstract
Dopamine receptors regulate glutamatergic neurotransmission and Na+,K+-ATPase via protein kinase A (PKA) and dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32)-dependent signaling. Consequently, dopamine receptor activation may modulate neonatal hypoxic–ischemic (H–I) neuronal damage in the selectively vulnerable putamen enriched with dopaminergic receptors. Piglets subjected to two durations of hypoxia followed by asphyxic cardiac arrest were treated with a D1-like (SCH23390) or D2-like (sulpiride) receptor antagonist. At 4 days of recovery from less severe H–I, the remaining viable neurons in putamen were 60% of control, but nearly completely salvaged by pretreatment with SCH23390 or sulpiride. After more severe H–I in which only 18% of neurons were viable, partial neuroprotection was seen with SCH23390 pretreatment (50%) and posttreatment (39%) and with sulpiride pretreatment (35%), but not with sulpiride posttreatment (24%). Dopamine was significantly elevated in microdialysis samples from putamen during asphyxia and the first 15 mins of reoxygenation. Pretreatment with SCH23390 or sulpiride largely attenuated the increased nitrotyrosine and the decreased Na+,K+-ATPase activity that occurred at 3 h after severe H–I. Pretreatment with SCH23390, but not sulpiride, also attenuated H–I-induced increases in PKA-dependent phosphorylation of Thr34 on DARPP-32, Ser943 on the α subunit of Na+,K+-ATPase, and Ser897 of the N-methyl-D-aspartate (NMDA) receptor NR1 subunit. These findings indicate that D1 and D2 dopamine receptor activation contribute to neuronal death in newborn putamen after H–I in association with increased protein nitration and decreased Na+,K+-ATPase activity. Furthermore, mechanisms of D1 receptor toxicity may involve DARPP-32-dependent phosphorylation of NMDA receptor NR1 and Na+,K+-ATPase.
Keywords: cardiac arrest, DARPP-32, global cerebral ischemia, Na, K-ATPase, neonate, nitric oxide, nitrotyrosine, NMDA receptor, peroxynitrite
Introduction
The basal ganglia, thalamus, and peri-Rolandic cortex represent regions of selective vulnerability in neonatal hypoxic-ischemic (H–I) encephalopathy (Miller et al, 2005). Among the mechanisms responsible for this injury, N-methyl-D-aspartate (NMDA) receptor-mediated excitotoxicity, intracellular Ca2+ accumulation, and oxidative stress have received much attention. However, blockage of NMDA receptors can produce adverse effects. Therefore, it is of interest to investigate mechanisms that modulate NMDA receptor function without completely blocking the receptor.
Dopamine is a major neurotransmitter in striatum and is known to regulate the function of several proteins, including those important in ischemic injury, such as glutamate receptors and Na+,K+-ATPase. Dopamine acts on two classes of receptors: D1-like receptors (D1, D5) coupled to Gs proteins that activate adenylate cyclase and protein kinase A (PKA), and D2-like receptors (D2, D3, D4) coupled to Gi/o proteins that inhibit adenylate cyclase and PKA (Svenningsson et al, 2004). Many of the effects of D1 and D2 receptor activation depend on the phosphorylation state of multiple sites on the dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32). For example, phosphorylation of Thr34 on DARPP-32 by PKA after D1 receptor activation inhibits protein phosphatase-1 and thereby permits sustained phosphorylation of serine and threonine sites on other proteins targeted by PKA (Svenningsson et al, 2004). Stimulation of D1 receptors leads to phosphorylation of Ser897 on the NR1 subunit of NMDA receptors (Dudman et al, 2003; Dunah and Standaert, 2001) by a mechanism dependent on DARPP-32 (Snyder et al, 1998). Phosphorylation of NR1 by PKA can increase receptor sensitivity to glutamate and increase Ca2+ currents (Flores-Hernandez et al, 2002; Maldve et al, 2002), which could then increase the activity of nitric oxide (NO) synthase and other downstream cascades involved in excitotoxicity. Although D2 receptor activation exerts the opposite effects of D1 receptor activation on PKA and DARPP-32 Thr34 phosphorylation, D1 and D2 receptor activation reduces Na+,K+-ATPase activity in a synergistic rather than in an antagonist manner (Bertorello et al, 1990). Decreased Na+,K+-ATPase activity could have important consequences on ionic regulation in postischemic neurons. Together, these findings open the possibility that dopamine receptors may influence ischemic neuronal damage by modulating NMDA receptors, Na+,K+-ATPase, or other components involved in excitotoxicity. Whether D2 receptors exert opposing or similar actions to those exerted by D1 receptors during ischemia is uncertain.
The effects of D1 and D2 antagonists on neuronal injury and on phosphorylation of various target proteins in striatum with its enriched dopaminergic innervation have not been studied in detail in either newborn or adult animal models of cerebral ischemia. Some dopamine receptor agonists have been reported to protect hippocampal neurons (O’Neill et al, 1998). Increases in extracellular dopamine in newborn pig striatum are particularly sensitive to hypoxia (Pastuszko et al, 1993). In the present study, we used a well-characterized model of H–I in newborn piglets in which oxidative stress and neuronal death in selectively vulnerable putamen rapidly evolved during the early hours of reoxygenation, followed by neurodegeneration in primary sensorimotor cortex (Martin et al, 1997a, b, 2000). Within the striatum in this model, NMDA receptor phosphorylation at the PKA-sensitive site Ser897 on the NR1 subunit is increased (Guerguerian et al, 2002), NO-dependent protein nitration is increased (Martin et al, 2000), and Na+,K+-ATPase activity is decreased without detectable increases in tyrosine nitration (Golden et al, 2001, 2003). We examined the effects of the prototypic D1 receptor antagonist SCH23390 and D2 receptor antagonist sulpiride on neuronal damage in putamen after neonatal H–I and investigated potential molecular mechanisms that involve NMDA receptor phosphorylation, nitrotyrosine formation, and activity and phosphorylation of Na+,K+-ATPase.
Materials and methods
Animal Model of Hypoxia–Ischemia
The animal protocol was approved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions and modified from the method described previously (Martin et al, 1997a, b). In brief, male piglets at 4 to 7 days of age (2.5 to 3 kg) were anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneal), intubated, and ventilated mechanically with 30% O2. Because of evidence of sex differences in the mechanism of H–I-induced cell death in immature brain (Zhu et al, 2006), only males were selected to reduce the potential increase in variance that could arise from grouping males and females. After a femoral artery and vein were catheterized under aseptic conditions, 10 mL/h of 5% dextrose and 0.45% saline were infused intravenously during the period of anesthesia. Additional analgesia and neuromuscular blockage were provided with a bolus intravenous injection of fentanyl (40 μg/kg) and pancuronium (0.3 mg/kg) approximately 10 mins before initiating H–I. The injection did not alter arterial pressure. The antibiotic cefazolin (25 mg/kg) was administered daily. Rectal temperature was maintained at 38.5°C to 39°C with a heating lamp and warm water-perfused blanket.
At approximately 15 mins after completing the surgery, inspired O2 was decreased to either 10% O2 for 30 mins or to 9.8% O2 for 40 mins, followed by ventilation with 21% O2 for 5 mins and airway occlusion for 7 mins to produce cardiac arrest. Ventilation with 10% O2 for 30 mins was used to produce moderate injury, whereas ventilation with 9.8% O2 for 40 mins was used to produce severe injury. Ventilation with 21% O2 for 5 mins after hypoxia was necessary for successful cardiac resuscitation after subsequent asphyxia. Piglets were resuscitated by ventilation with 50% O2, manual chest compressions, and intravenous injection of epinephrine (0.1 mg/kg) until return of spontaneous circulation. Defibrillation was performed when needed. Piglets that did not have return of spontaneous circulation within 3 mins of resuscitation were excluded. After resuscitation, inspired O2 was gradually reduced to 30% to maintain arterial O2 oxyhemoglobin saturation greater than 95%, as guided by arterial blood analysis. Sodium bicarbonate was administered to correct base deficits > 5 mmol/L. Arterial blood pressure, blood gases, pH, and glucose were monitored until the piglets regained consciousness. Piglets were extubated when they began to regain consciousness, usually within 6 to 8 h. Sham control animals were anesthetized and subjected to the surgical preparation but not the H–I. Piglets recovered in an individual cage with a warming blanket and received an intravenous infusion of 5% dextrose and 0.45% saline for the first 12 to 24 h of recovery. They were fed formula milk from a syringe during the first day of recovery. By the second day, they usually were able to drink milk from a bowl and were housed with a littermate.
Neurobehavioral Assessment
Neurobehavioral assessments were performed at 1, 2, 3, and 4 days after H–I. Neurologic deficits were graded on a scale of 0 to 154 (normal score, 0; maximal deficit score, 154), as described previously (Agnew et al, 2003). The scale is based on levels of consciousness, brainstem function, sensory responses, motor function, spatial orientation, behavioral activity, and seizure activity.
Administration of Dopamine Receptors Antagonists
Sham or H–I piglets were injected intravenously with (1) 0.9% saline, (2) the dopamine D1-like receptor antagonist SCH23390 (0.5 mg/kg; Sigma, St Louis, MO, USA), or (3) the dopamine D2-like receptor antagonist sulpiride (30 mg/kg; Sigma) either at 20 mins before hypoxia for the pretreatment protocol or at 5 mins of recovery for the posttreatment protocol. These doses have been used by others to produce effects within the central nervous system after systemic administration (Bourne et al, 2001). Binding of SCH23390 in human putamen reaches a maximum at approximately 20 mins after injection and then decreases by approximately 40% over a subsequent 40-min period (Hirvonen et al, 2001). A maintenance dose of saline, SCH23390 (0.2 mg/kg/h), or sulpiride (3.3 mg/kg/h) was continuously infused from 5 mins through 3 h after resuscitation in both the pretreatment and posttreatment protocols.
Microdialysis
Changes in extracellular dopamine in putamen during H–I were assessed by analyzing microdialysates from eight piglets. Holes were drilled in the skull 8 mm anterior and 7.5 mm lateral from bregma. The tip of the microdialysis probes (CMA 10; CMA/Microdialysis, Stockholm, Sweden) with a 5-mm membrane length was inserted 16.5 mm below the dura. The probes were perfused with dye at the end of the experiment to ensure that the membrane was within putamen. The probes were perfused with artificial cerebrospinal fluid at a rate of 2.5 μL/min starting at least 45 mins after placement. The first sample collected over 15 mins was not used in the analysis. Microdialysates were collected at 15-min intervals over a 45-mins baseline period followed by H–I and 3 h of recovery with allowance for the time delay of the dead space of the probe. Dialysates were analyzed for dopamine concentration by high-pressure liquid chromatography with electrochemical detection. Isoproterenol was used as an internal standard. Dialysate concentrations were not adjusted for incomplete recovery of dopamine across the dialysis membrane.
Immunoblotting
At 3 h of recovery from H–I or an equivalent time in sham-operated piglets, additional pentobarbital was administered and the animal was perfused transcardially with ice-cold phosphate-buffered saline (PBS). Brains were removed rapidly, transected midsagittally and subdissected carefully to obtain samples selectively from putamen. Tissues were homogenized in ice-cold homogenization buffer (20 mmol/L Tris–HCl, pH 7.4, with 10% sucrose, 1 mmol/L ethylenediaminetetraacetate, 0.1 mmol/L phenylmethylsulfonyl fluoride, and protease inhibitor cocktail (Roche, Germany)) and centrifuged at 1000g for 10 mins. The supernatant was removed and centrifuged at 54,000g for 40 mins. The resulting membrane-enriched pellet was resuspended and washed with sucrose-free homogenization buffer and centrifuged at 54,000g for 40 mins twice. The final pellet was resuspended in homogenization buffer that contained 20% glycerol instead of sucrose. Protein concentrations were determined by bicinchoninic acid protein assay.
Samples (20 μg of protein) were separated by 4% to 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and electrotransferred onto nitrocellulose membranes. After being blocked with 5% nonfat milk in 0.01 mol/L PBS (pH 7.4) for 1 h, the membranes were probed overnight at 4°C with the following primary antibodies: anti-NMDAR1 (1:2000, BD Pharmingen, San Jose, CA, USA), anti-phospho-NR1 Ser896 or anti-phospho-NR1 Ser897 (1:2000, Upstate, Lake Placid, NY, USA), anti-DARPP-32 or anti-phospho-DARPP-32 Thr34 (1:1500, Cell Signaling Technology, Danvers, MA, USA), anti-Na+,K+-ATPase α1 (1:50,000, Sigma) or Na+,K+ ATPase α3 (1:50,000, Affinity Bioreagents, Golden, CO), anti-phospho Na+,K+-ATPase α Ser23 or Na+,K+-ATPase α Ser943 (1:2000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-nitrotyrosine (1:40,000, Upstate) or anti-synaptophysin (1:20,000, Chemicon, Temecula, CA, USA). Membranes were then washed and incubated with the corresponding secondary antibodies conjugated to horse-radish peroxidase (Amersham, Piscataway, NJ, USA). Immunoreactive bands were visualized with an enhanced chemiluminescence detection system (Amersham). Synaptophysin, a synaptic vesicle membrane protein, whose level does not change during the first 24 h after H–I (Martin et al, 2000), was used as a protein loading standard. To elucidate whether the antibodies reacted specifically with phosphorylated protein, 20 μg protein aliquots from animals in the H–I saline group were also treated with 400 U/mL of λ-protein phosphatase (Upstate) for 4 h before electrophoresis. After immunoblotting, the bands were scanned and analyzed with Quantityone software (Bio-Rad, Hercules, CA, USA). Values of optical density (OD) were normalized by the value of a sham animal treated with saline on each gel. Each immunoblot experiment was performed in quadruplicate using tissue from four piglets per treatment group.
Immunohistochemistry
The anesthetized piglets were perfused transcardially with ice-cold PBS and 4% paraformaldehyde. Brains were removed and bisected midsagittally, and each hemisphere cut into 1 cm slabs. The left forebrain was paraffin embedded for histology with hematoxylin and eosin (H&E) staining. The right forebrain was cryoprotected in 20% glycerol-PBS for 24 h, frozen, and cut into serial 40-μm sections through putamen. Immunohistochemistry was completed on free-floating sections to detect D1 or D2 receptors of piglet putamen. Endogenous peroxidase was quenched with 30-min incubation of 0.3% H2O2 in methanol. Sections were blocked in normal serum and incubated with rabbit anti-D1R (1:300, Chemicon) or anti-D2R (1:300, Chemicon) for 48 h at 4°C. After rinsing in PBS, sections were incubated with biotinylated anti-rabbit IgG (1:200, Vector Laboratories, Burlingame, CA, USA) and VECTASTAIN Elite ABC reagent (Vector). Immunostaining was developed with diaminobenzidine as a chromogen (Vector). Negative controls were treated without primary antibodies and showed no positive signals. The positive signals were detected with Inquiry quantitative autoradiography software (version 3.08b, Loat Associates, Westminster, MD, USA) and expressed as optical density per mm2 (OD/mm2) or positive cells per mm2 (cells/mm2).
Na+, K+-ATPase Biochemical Assay
The biochemical activity of Na+,K+-ATPase in samples of putamen was determined by measuring ouabain-sensitive hydrolysis of ATP as described previously (Golden et al, 2001). Samples from each animal were analyzed in triplicate.
Data and Statistical Analysis
Profile counting was used to estimate ischemic neuronal damage in mid-coronal sections of putamen on H&E-stained paraffin sections. In sections that were matched for level, the number of ischemic and nonischemic neuronal profiles was counted in an observer-blinded fashion in seven nonoverlapping, microscopic fields at ×1000 power. In each microscopic field, the seven values in each animal were averaged to obtain a single value of viable neurons per mm2 for each piglet to be used in the statistical analysis.
All values are expressed as mean±s.d. Statistical analysis among groups was performed with one-way analysis of variance (ANOVA) followed by the Student–Newman–Keuls multiple range test, with P < 0.05 considered statistically different. The neurologic deficit score was analyzed by two-way ANOVA with repeated measures.
Results
Dopamine Concentration in Microdialysates
The time course of extracellular dopamine concentration in putamen during H–I was monitored in microdialysis samples collected from eight piglets (Figure 1). During hypoxia, the concentration of dopamine increased in half of the piglets, but the increase for the group as a whole was not statistically significant. However, dopamine increased substantially during asphyxia (P < 0.005) and the first 15 mins of recovery (P < 0.001; repeated measures ANOVA and paired t-test) compared with baseline and with those during hypoxia.
Figure 1.
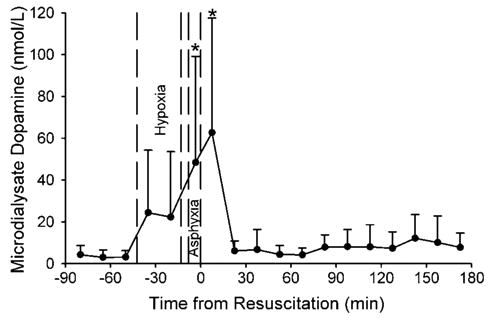
Concentration of dopamine (±s.d.) in microdialysis outflow from putamen of eight piglets during baseline, hypoxia, asphyxia, and 3 h of recovery. *P < 0.05 from baseline.
Reduced Neuronal Damage by SCH23390 and Sulpiride in Putamen
In this model of H–I, neuronal cell death in striatum occurs within the first 24 h of recovery (Martin et al, 2000). Sham groups treated with saline, SCH23390, or sulpiride had similar striatal neuronal density. With 30 mins of hypoxia plus 7 mins of asphyxia, the number of viable neurons in putamen at 4 days of recovery was 63%±26% (n = 9) of the values in combined sham-operated animals. Viable neurons were better preserved in H–I groups pretreated with SCH23390 (89%±17%; n = 7) or sulpiride (93%± 13%; n = 8). The values in the drug-treated H–I groups were not significantly different from the values in combined sham-operated piglets (100%± 18%; n = 17).
To test the efficacy of dopamine antagonists with a more severe model of injury, the duration of hypoxia was extended from 30 mins to 40 mins and the inspired O2 was slightly decreased from 10% to 9.8%. Hematoxylin and eosin staining revealed normal cytoarchitecture and little neuronal damage in putamen of sham-operated animals treated with saline (n = 4), SCH23390 (n = 4), or sulpiride (n = 4) at 4 days after surgery (Figures 2A, 2B, 2C and 2G). Hypoxia–ischemia induced selective neuronal damage in putamen. Most putaminal neurons exhibited ischemic morphology that consisted of cytoplasmic microvacuolation, eosinophilia, and nuclear pyknosis, or eventual cell homogenization (Figure 2D). Only 18% of putamen neurons in the saline H–I group (n = 15) were viable compared with the sham saline group (Figure 2G). Pretreatment with SCH23390 or sulpiride significantly alleviated neuronal damage in putamen (Figures 2E, 2F, and 2G). The number of viable neurons in the SCH23390 pretreatment group (n = 8) was 50% of that in the sham saline group, whereas that in the sulpiride pretreatment group (n = 9) was 35% of that in the sham saline group (Figure 2G). Moreover, treatment with SCH23390 (n = 7) starting 5 mins after resuscitation also significantly increased the number of viable neurons to 39% of the shamsaline value. However, improvement to 24% viable neurons with posttreatment of sulpiride (n = 6) was not significant (Figure 2G).
Figure 2.
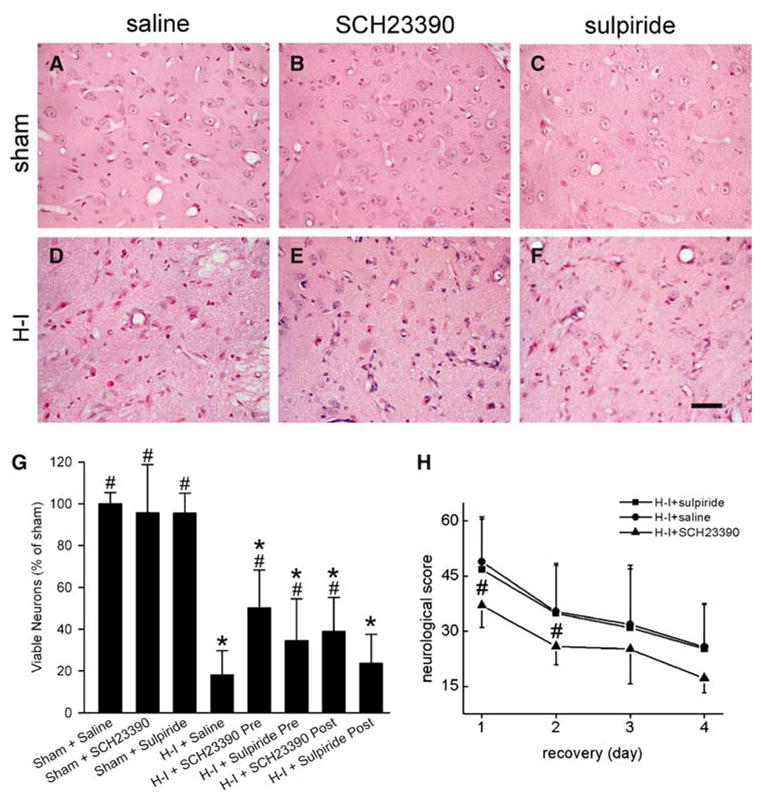
Effects of SCH23390, a D1 receptor antagonist, and sulpiride, a D2 receptor antagonist, on neuronal damage at 4 days of recovery and neurologic deficits in groups subjected to 40 mins of hypoxia and 7 mins of asphyxia (H–I). Hypoxic–ischemic or sham piglets were injected intravenous with saline, SCH23390, or sulpiride, either at 20 mins before hypoxia for pretreatment protocol, or at 5 mins of recovery for posttreatment protocol, followed by continuous infusion until 3 h of recovery. (A–F) Representative photographs of H&E-stained sections showing that pretreatment with SCH23390 or sulpiride partially alleviated ischemic neuronal damage in putamen of H–I piglets. Bar = 50 μm. (G) Quantitative results for viable putamen neurons expressed as a percent of the mean value in the sham group. (H) Neurologic score during the 4-day recovery. Data represent means±s.d. (n = 4 to 15 per group). *P < 0.05 versus sham group treated with saline; #P < 0.05 versus H–I group treated with saline; ANOVA followed by the Student–Newman–Keuls test.
Neuronal injury in primary sensorimotor cortex was more variable than in striatum, and pretreatment with antagonists did not have a significant effect. The percent of cortical neurons with ischemic cytopathology was 48%±44% in the saline H–I group, 48%±6% in the SCH23390 H–I group, and 22%±43% in the sulpiride H–I group.
Neurobehavioral deficits were greatest on the first day of recovery from H–I. Most H–I piglets treated with saline showed stupor or clouded-like consciousness, low muscle tone, no light and/or no auditory reflexes, and, sometimes, no response to pain stimulation on the first day of recovery. Two-way repeated measures ANOVA indicated an overall effect of treatment (P < 0.001) and time (P < 0.001). On individual days, pretreatment with SCH23390 significantly improved neurobehavioral recovery of H–I piglets at 24 and 48 h of recovery, whereas sulpiride had no effect (Figure 2H). Sham animals treated with saline, SCH23390, or sulpiride did not show obvious neurologic deficit after recovery from anesthesia.
Pretreatment with dopamine receptor antagonists could potentially alter cardiovascular physiologic parameters at baseline and the cardiovascular response to H–I. During the 40-min period of hypoxia, mean arterial blood pressure (MABP) was maintained near the baseline values and did not differ among groups (Table 1). During asphyxia, severe bradycardia and hypotension occurred. By 7 mins of airway occlusion, MABP was 19±8 mm Hg in the saline group, 15±6 mm Hg in the SCH23390 group, and 21±7 mm Hg in the sulpiride group. Successful resuscitation with ventilation, chest compression, and epinephrine administration led to a transient overshoot and a recovery to baseline levels of MABP. The percent of piglets without return of spontaneous circulation within 3 mins of cardiopulmonary resuscitation was 21% with saline pretreatment, 13% with SCH23390 pretreatment, and 37% with sulpiride pretreatment. In the saline H–I group, mean arterial PO2 decreased to 21 mm Hg during hypoxia, recovered to 76 mm Hg during the 5 mins of reoxygenation used to improve subsequent cardiac resuscitation from asphyxia, and declined to 13 mm Hg by 5 mins of airway occlusion. Similar changes were produced in drug-treated groups. The corresponding changes in arterial oxyhemoglobin saturation were 26% at the end of hypoxia, 88% during ventilation with 21% O2, and 5% during airway occlusion. Metabolic acidosis was present by the end of hypoxia without a change in arterial PCO2. During airway occlusion, metabolic and respiratory acidosis was severe, with no differences among groups. Hyperglycemia occurred in all groups during H–I. Rectal temperature was maintained near the normal temperature of 38.5°C for swine. Physiologic values in the groups with SCH23390 and sulpiride posttreatment (not shown) were similar to those of the other H–I groups. Physiologic values in the sham groups were similar to the baseline values of the H–I groups and were unchanged throughout the observation period.
Table 1.
Physiologic parameters during and after hypoxia–ischemia
| Baseline | Hypoxia | 21% O2 | Asphyxia | Post resuscitation
|
|||
|---|---|---|---|---|---|---|---|
| 37 mins | 5 mins | 5 mins | 1 h | 2 h | 3 h | ||
| MABP (mm Hg) | |||||||
| Saline | 73±9 | 74±20 | 86±13 | 29±8 | 70±9 | 68±10 | 68±9 |
| SCH23390 | 73±9 | 76±20 | 97±18 | 21±11 | 75±7 | 65±14 | 67±10 |
| Sulpiride | 73±12 | 61±10 | 93±11 | 30±8 | 69±5 | 65±7 | 66±8 |
| Arterial PO2 (mm Hg) | |||||||
| Saline | 117±14 | 22±2 | 76±17 | 13±5 | 111±13 | 112±14 | 118±13 |
| SCH23390 | 115±15 | 21±2 | 72±20 | 14±9 | 121±20 | 106±15 | 108±15 |
| Sulpiride | 127±17 | 21±2 | 71±17 | 10±6 | 131±30 | 120±20 | 125±23 |
| Arterial PCO2 (mm Hg) | |||||||
| Saline | 39±3 | 38±5 | 37±10 | 84±15 | 38±5 | 40±5 | 37±5 |
| SCH23390 | 37±2 | 40±5 | 38±5 | 79±12 | 41±7 | 37±6 | 37±4 |
| Sulpiride | 40±4 | 43±5 | 40±7 | 84±11 | 41±4 | 40±5 | 41±3 |
| Arterial pH | |||||||
| Saline | 7.42±0.03 | 7.21±0.11 | 7.17±0.13 | 6.86±0.09 | 7.36±0.08 | 7.44±0.06 | 7.48±0.03 |
| SCH23390 | 7.40±0.04 | 7.12±0.12 | 7.08±0.12 | 6.84±0.11 | 7.26±0.14 | 7.42±0.10 | 7.48±0.07 |
| Sulpiride | 7.40±0.02 | 7.20±0.07 | 7.17±0.07 | 6.89±0.08 | 7.36±0.08 | 7.45±0.06 | 7.47±0.04 |
| Arterial glucose (mmol/L) | |||||||
| Saline | 2.6±0.9 | 9.3±4.2 | 7.3±3.2 | 9.8±5.9 | 5.4±3.4 | 4.4±2.4 | 4.3±2.3 |
| SCH23390 | 2.8±0.7 | 9.6±3.6 | 7.7±3.8 | 7.5±5.1 | 5.9±4.3 | 6.1±4.6 | 5.6±3.2 |
| Sulpiride | 2.6±0.9 | 9.0±5.1 | 7.6±5.0 | 9.9±4.9 | 5.7±3.1 | 4.4±2.9 | 3.7±1.0 |
| Rectal temperature (°C) | |||||||
| Saline | 38.4±0.4 | 38.8±0.2 | 38.9±0.3 | 38.9±0.2 | 38.8±0.6 | 38.6±0.5 | 38.6±0.5 |
| SCH23390 | 38.5±0.2 | 38.6±0.2 | 38.7±0.2 | 38.8±0.2 | 38.9±0.3 | 39.0±0.4 | 38.9±0.3 |
| Sulpiride | 38.1±0.3 | 38.6±0.3 | 38.6±0.3 | 38.7±0.2 | 38.8±0.5 | 38.5±0.6 | 38.4±0.3 |
D1 and D2 Immunohistochemistry at 4 Days of Recovery
On immunoblots of piglet putamen, antibodies against D1 and D2 receptors yielded single bands at 50 kDa, which were similar to the bands obtained from rat striatum (Figure 3I). Immunohistochemistry for D1 and D2 receptors in putamen of sham piglets revealed diffuse staining throughout the neuropil, with little immunoreactivity in cell bodies (Figures 3A and 3E). However, staining in the neuropil became faint 4 days after H–I and was largely restricted to cell bodies (Figures 3B and 3F). The change in the pattern of staining was quantified in two ways: by counting immunopositive cell bodies and by OD measurements of stained sections of putamen. Immunopositive cell bodies for D1 or D2 receptors were scarce in sham animals treated with saline, SCH23390, or sulpiride, but increased markedly in the H–I saline group (Figures 3B and 3F). The overall OD was decreased in the H–I saline group compared with the sham groups (Figures 3L and 3M), presumably reflecting decreased staining in the neuropil (Figures 3B and 3F).
Figure 3.
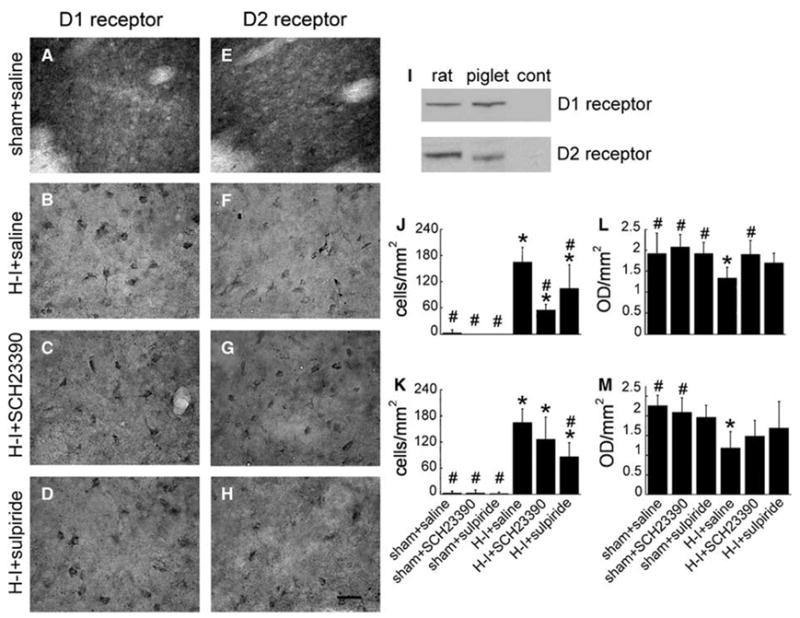
Representative sections of immunohistochemical staining for D1 receptors (A–D) and D2 receptors (E–H) in putamen of piglets 4 days after recovery from sham surgery or hypoxia–ischemia (H–I) with pretreatment of saline, SCH23390, or sulpiride. Scale bar = 20 μm. Reactivity of specific antibodies against D1 or D2 receptors with cytoplasmic membrane-enriched fractions of striatum (20 μg protein/lane) is shown (I) with noninjured rat brain, piglet brain, and control (no protein loaded, cont) by SDS-PAGE. Diffuse staining of neuropil was present in sham piglets, whereas discrete cell body staining was evident in H–I piglets and quantified for D1 (J) and D2 (K) staining (±s.d.). Overall optical density (OD) showed a decrease in H–I piglets treated with saline for D1 (L) and D2 (M). *P < 0.05 from sham + saline group; #P < 0.05 from H–I + saline group; one-way ANOVA followed by the Student–Newman–Keuls test.
Pretreatment with SCH23390 decreased the number of D1 immunopositive cell bodies and increased the overall OD for D1 immunoreactivity (Figures 3J and 3L). Pretreatment with sulpiride decreased the number of D2 immunopositive cell bodies (Figure 3K). Although the overall OD for D2 immunoreactivity after sulpiride treatment was not significantly different from that of the sham saline group, the OD was also not different from the H–I saline group value (Figure 3M). Treatment with SCH23390 had no significant effect on the number of D2-immunopositive cell bodies (Figure 3K) or on the overall OD for D2 immunoreactivity (Figure 3M), compared with the H–I saline group, although the OD was not different from the sham saline group. Sulpiride had an intermediate attenuating effect on the number of D1-immunopositive cell bodies (Figure 3J) and on the overall OD for D1 immunoreactivity (Figure 3L).
Reduced Phosphorylation of DARPP-32 at Thr34 by SCH23390 in H–I Piglets
To investigate possible involvement of the cAMP-PKA-DARPP-32 pathway in neuronal injury after H–I, immunoblotting was used to examine the level of phosphorylated DARPP-32 at Thr34 and total DARPP-32 protein in putamen of sham and H–I piglets at 3 h of recovery. SCH23390 and sulpiride did not change the basal level of phosphorylated DARPP-32 at Thr34 in sham animals. However, the level of phosphorylated Thr34 on DARPP-32 (1.68± 0.53, ratio of sham) was significantly increased after H–I (Figures 4A and 4C). This increase was markedly reduced by SCH23390 (1.12±0.17), but not by sulpiride (1.47±0.24). In contrast to Thr34 phosphorylation, the level of total DARPP-32 protein was unchanged 3 h after H–I and by treatment with SCH23390 or sulpiride (Figures 4A and 4B).
Figure 4.
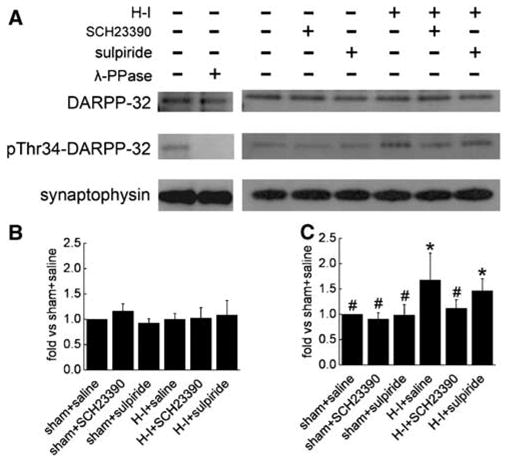
Western blot analysis of phosphorylated DARPP-32 at Thr34 and total DARPP-32 in lysates from piglet putamen 3 h after hypoxia–ischemia (H–I). In total, 20 μg of protein was loaded onto gels and antibodies against phosphorylated DARPP-32 at Thr34 (1:1500) and total DARPP-32 (1:1500) were used to detect the protein at 32 kDa. Synaptophysin was used as a loading control. (A) Representative blots showing the expression of phosphorylated DARPP-32 at Thr34 and total DARPP-32. Lane 2 was treated for 4 h with 400 U/mL λ-phosphatase (λ-PPase). Bar graphs (mean±s.d.) summarize the expression of total DARPP-32 (B) and phosphorylated Thr34 (C) normalized to the sham + saline value from four independent gels. *P < 0.05 versus sham + saline groups; #P < 0.05 versus H–I group treated with saline; one-way ANOVA followed by the Student–Newman–Keuls test.
Decreased Phosphorylation of NMDA NR1 Ser897 by SCH23390
Previous work in piglets demonstrated phosphorylation of NMDA NR1 serine residues after H–I (Guerguerian et al, 2002). To determine if NMDA NR1 colocalizes in cells with D1 and D2 receptors at this stage of development in piglets, double-immunofluorescent experiments were performed. Most D1 or D2 receptor-positive cells were found to costain with NR1 in putamen of H–I piglet at 3 h of recovery (data not shown). Immunoblots showed that H–I increased phosphorylation of NR1 at both the PKA-dependent site (Ser897) and the PKC-dependent site (Ser896) in putamen at 3 h of recovery (Figure 5). The increase in Ser897 phosphorylation (2.14±1.03, ratio of sham) was significantly inhibited by SCH23390 (0.88±0.48) but not by sulpiride (1.98±1.06; Figure 5C). SCH23390 and sulpiride did not change the basal level of phosphorylated NR1 Ser897 in sham animals. In contrast to the effects on phosphorylated NR1 Ser897, the H–I-induced increase in PKC-dependent phosphorylation of NR1 Ser896 was not inhibited by SCH23390 or sulpiride (Figure 5B). SCH23390 and sulpiride also had no effect on the basal Ser896 phosphorylation in sham animals. Consistent with earlier work (Guerguerian et al, 2002), the levels of total NR1 protein remained unchanged 3 h after H–I (Figure 5D). The level of NR1 protein was also unaffected by SCH23390 or sulpiride treatment.
Figure 5.
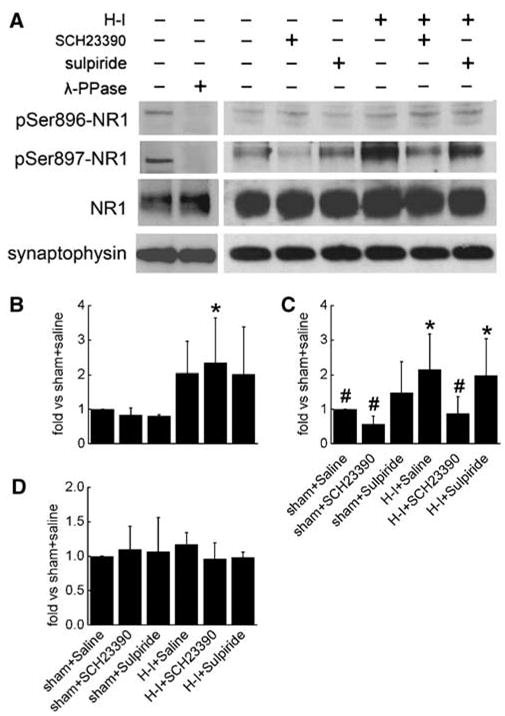
Western blot analysis of phosphorylated NMDA NR1 at Ser896 and Ser897 and total NR1 in lysates from piglet putamen 3 h after hypoxia–ischemia (H–I). In total, 20 μg of protein was loaded onto gels, and antibodies against phosphorylated NR1 at Ser896 and Ser897 (1:2,000) and total NR1 (1:2,000) were used to detect the protein at 120 kDa. Synaptophysin was used as a loading control. (A) Representative blots showing the expression of phosphorylated NR1 at Ser896 and Ser897 and total NR1. Lane 2 was treated for 4 h with 400 U/mL λ-phosphatase (λ-PPase). Bar graph summarizing the expression of phosphorylated NR1 at Ser896 (B) and Ser897 (C) and total NR1 (D). Data (means±s.d.) were normalized to the sham + saline value for four independent gels. *P < 0.05 versus sham groups treated with saline; #P < 0.05 versus H–I group treated with saline; one-way ANOVA followed by the Student–Newman–Keuls test.
SCH23390 and Sulpiride Decrease Nitrotyrosine Immunoreactivity
Administration of SCH23390 or sulpiride did not alter the basal level of 3-nitrotyrosine immunoreactivity integrated over multiple protein bands in sham animals (Figure 6). With H–I, the total amount of 3-nitrotyrosine increased in the saline group (2.84±1.06), and this increase was reduced by the administration of SCH23390 (1.39±1.75) and sulpiride (0.87±1.00).
Figure 6.
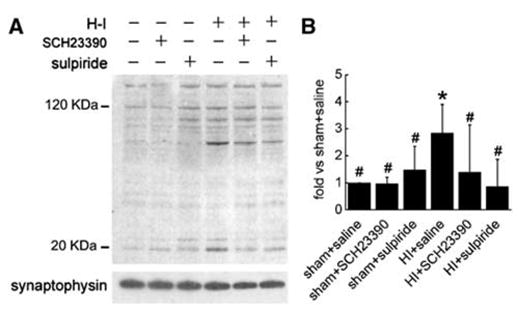
(A) Western blot showing the effects of pretreatment with SCH23390 or sulpiride on nitrotyrosine immunoreactivity on multiple protein bands in putamen of sham or hypoxia–ischemia (H–I) piglets at 3 h of recovery. In all, 20 μg of protein was loaded onto gels, and antibodies against 3-nitrotyrosine (1:40,000) were used to detect proteins with nitrotyrosine. Synaptophysin was used as a loading control. (B) Optical density was integrated over all protein band values (means± s.d.) and was normalized to the sham + saline value from four independent gels. *P < 0.05 versus sham groups treated with saline; #P < 0.05 versus H–I group treated with saline; one-way ANOVA followed by the Student–Newman–Keuls test.
Effects of SCH23390 and Sulpiride on H–I-induced phosphorylation of the Na+,K+-ATPase α Subunit
To elucidate whether the neuroprotective role of SCH23390 or sulpiride is associated with alterations in Na+,K+-ATPase, both enzymatic activity and protein expression were measured. Consistent with previous work (Golden et al, 2001), activity of Na+, K+-ATPase was markedly decreased at 3 h of recovery after H–I in putamen (Figure 7A). Administration of SCH23390 or sulpiride had no significant effect on activity in sham animals. However, administration of either SCH23390 or sulpiride completely restored Na+,K+-ATPase activity 3 h after H–I.
Figure 7.
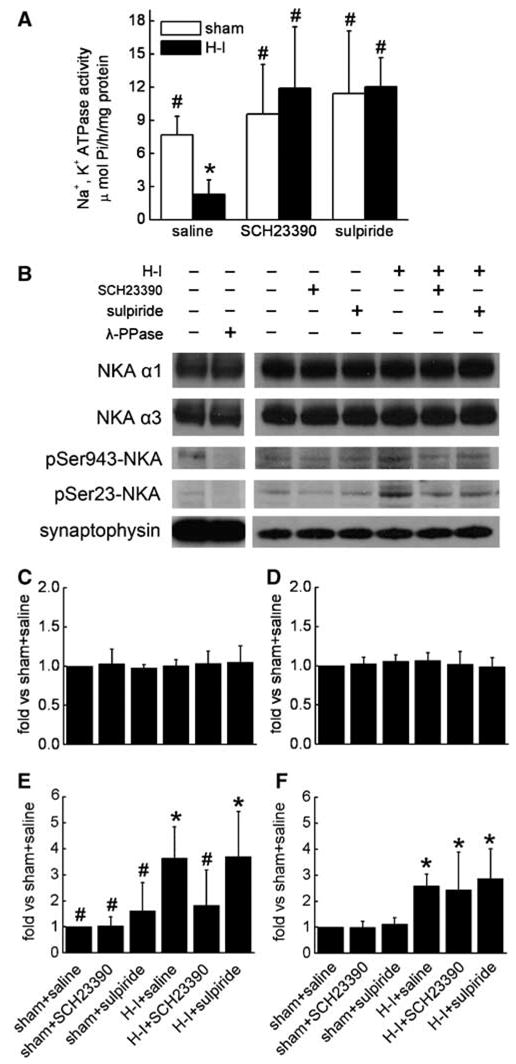
(A) Effects of pretreatment with SCH23390 or sulpiride on Na+,K+-ATPase activity in putamen of sham or hypoxia–ischemia (H–I) piglets at 3 h of recovery (n = 4 to 7 per group). (B) Representative Western blots of Na+,K+-ATPase (NKA) subunits α1 and α3 and phosphorylation of the combined α subunits at Ser943 and Ser23 in lysates from piglet putamen 3 h after hypoxia–ischemia (H–I). In total, 20 μg of protein was loaded onto gels, and antibodies against NKA α1 and α3 (1:50,000) and phosphorylated NKA at Ser943 and Ser23 (1:2000) were used to detect the protein at 110 kDa. Synaptophysin was used as a loading control. Lane 2 was treated for 4 h with 400 U/mL λ-phosphatase (λ-PPase). Bar graphs (means±s.d.) summarize the expression of NKA α1 (C) and α3 (D) subunits and phosphorylated NKA at Ser943 (E) and Ser23 (F), normalized to the sham + saline value, from four independent gels. *P < 0.05 versus sham groups treated with saline; #P < 0.05 versus H–I group treated with saline; one-way ANOVA followed by the Student–Newman–Keuls test.
Also consistent with previous findings (Golden et al, 2001), the decrease in Na+,K+-ATPase activity after H–I was not associated with a change in protein expression of the neuron-specific catalytic subunit α3 (Figures 7B and 7D) or the more widespread α1 isoform (Figures 7B and 7C) present in most non-neuronal cells (Blanco, 2005). Moreover, their expression remained unchanged with administration of SCH23390 or sulpiride in both sham and H–I groups (Figure 7). The α subunit of Na+,K+-ATPase is also a target of phosphorylation by PKA (Blanco, 2005). To understand whether SCH23390 or sulpiride had any effect on the PKA-dependent phosphorylation of Na+,K+-ATPase, we examined the level of phosphorylated Na+,K+-ATPase α at Ser943 at 3 h of recovery. SCH23390 and sulpiride did not change the basal level of Ser943 phosphorylation in sham animals (Figures 7B and 7E). However, H–I significantly increased Ser943 phosphorylation (3.64±1.21, ratio of sham). This increase was markedly reduced by SCH23390 (1.82± 1.36), but not by sulpiride (3.69±1.74). In contrast, PKC-dependent phosphorylation of Ser23 on Na+, K+-ATPase α (Figures 7B and 7F), which was also increased by H–I (2.58±0.45), was not inhibited by SCH23390 (2.43±1.45) or sulpiride (2.86±1.16).
Discussion
The major findings of this study are: (1) pretreatment with the D1 receptor antagonist SCH23390 or the D2 receptor antagonist sulpiride provided near complete neuroprotection in putamen in a model of mild H–I injury in neonatal piglets and partial neuroprotection in a more severe model; (2) D1 and D2 antagonists individually reduced protein nitration and restored Na+,K+-ATPase activity early after H–I; (3) the D1 antagonist selectively reduced phosphorylation of PKA-dependent sites on DARPP-32, the NMDA NR1 subunit, and the Na+, K+-ATPase α subunit early after H–I; and (4) posttreatment with the D1 receptor antagonist in the severe model also provided partial neuroprotection in putamen.
Increases in striatal extracellular dopamine have been reported in immature piglets during hypocapnic and hypercapnic hypoxia (Huang et al, 1995; Pastuszko et al, 1993). Our results with microdialysis confirm that a large increase in extracellular dopamine occurs in a survival model during the period of asphyxia and early reoxygenation. The new findings that either a D1 or D2 antagonist provides complete neuroprotection in putamen from a mild insult and partial neuroprotection from a severe insult show for the first time an important role for these receptors in modulating injury from neonatal H–I injury. This protective effect with systemic drug administration was not attributed to effects on the severity of hypoxia or on MABP. Neuroprotection in putamen with SCH23390 was associated with improved recovery of neurologic deficits over the first 2 days. However, significance was lost by 72 h because deficits spontaneously diminished over time in the control group. In addition, deficits were not improved with sulpiride pretreatment despite neuroprotection in putamen. Perhaps neurobehavior tests that are more sensitive for assaying striatal function would better detect long-term protection by D1 and D2 receptor antagonists than deficit scores of gross sensorimotor function. Moreover, variability in the neurologic deficit score may depend on synaptic deficits, white matter injury, and cortical injury. Cortical injury evolves later and is more variable than injury in putamen.
Loss of D2 immunoreactivity has been described in infants with H–I injury in basal ganglia (Meng et al, 1998). Traditionally, the D1 receptor family (D1 and D5) has been thought to be expressed on medium spiny neurons that primarily project to substantia nigra pars reticulata and the internal segment of the globus pallidus, whereas the D2 receptor family (D2–D4) has been thought to be expressed on medium spiny neurons that primarily project to the external segment of the globus pallidus (Gerfen 2000; Hersch et al, 1995). However, a significant portion of striatal neurons have some coexpression of both receptor families (Aizman et al, 2000; Surmeier et al, 1996). At 4 days after H–I, the pattern of D1 and D2 immunoreactivity changed from diffuse staining of the neuropil to discrete staining of cell bodies. This change in the pattern of immunostaining may reflect dendritic alterations in the remaining surviving neurons. The partial but selective reversal of the D1 immunoreactive pattern with the D1 antagonist and the partial but selective reversal of the D2 immunoreactive pattern with the D2 antagonist imply that the D1 and D2 antagonists are primarily protecting distinct populations of neurons. Combined pretreatment with SCH23390 and sulpiride was attempted to determine if the drugs provided an additive protective effect; however, cardiac resuscitation proved difficult and the effect of combined pretreatment could not be tested. Although the doses used in this study are comparable with those used by others, greater neuroprotection at higher doses of antagonists cannot be excluded.
Many of the modulatory effects of dopamine receptor activation are mediated by phosphorylation of DARPP-32. Activation of D1 receptors or adenosine A2A receptors increases PKA-dependent phosphorylation of Thr34 on DARPP-32, which leads to inhibition of PP-1, whereas activation of D2 receptors can produce opposite effects. The increased Thr34 phosphorylation 3 h after H–I and the attenuation by SCH23390 are consistent with D1 receptor activation exerting a major influence on DARPP-32. Because activation of D2 receptors decreases PKA and Thr34 phosphorylation (Nishi et al, 1997), sulpiride augmentation of Thr34 phosphorylation induced after H–I could have been anticipated. The lack of a significant effect of sulpiride increases the possibility that stimulation of A2A receptors, which are coexpressed in D2 neurons, produces Thr34 phosphorylation that is not overcome by D2 receptor inhibition at the 3-h time of recovery.
Phosphorylation is an important mechanism for the regulation of NMDA receptor function. Agonists for D1 but not D2 receptors increase overall phosphorylation of the NMDA NR1 subunit (Snyder et al, 1998). The present results showing that SCH23390 but not sulpiride attenuates both Thr34 DARPP-32 and Ser897 NR1 phosphorylation indicate that dopamine promotes Ser897 phosphorylation after H–I by a D1 receptor/PKA/DARPP-32 cascade, although DARPP-32-independent mechanisms might also be operative. It is not precisely known how phosphorylation at Ser897 modulates the NMDA receptor activity. Several studies suggest that phosphorylation of NR1 by PKA enhances NMDA currents (Flores-Hernandez et al, 2002; Maldve et al, 2002) and Ca2+ influx (Dudman et al, 2003; Skeberdis et al, 2006) and promotes forward trafficking of the receptor from the endoplasmic reticulum to the synapse (Scott et al, 2003). These findings implicate phosphorylation of NR1 in augmentation of downstream cascades that result from NMDA receptor activation after H–I.
Enhanced Ca2+ influx may stimulate neuronal nitric oxide synthase bound to the NMDA receptor complex and produce NO, which, in the presence of excessive superoxide, generates highly toxic peroxynitrite. Although other chemical species can nitrate tyrosine, peroxynitrite is the major source of nitrotyrosine formation in vivo. At 3 h after H–I, we observed a marked increase in 3-nitrotyrosine on various protein bands in confirmation with previous findings in this model (Martin et al, 2000). The present results show that this nitration was attenuated by either SCH23390 or sulpiride. The concurrent decrease in Ser897 NR1 phosphorylation and protein nitration with SCH23390 is consistent with a link between D1 receptor-dependent NMDA NR1 phosphorylation and the excessive NO production that leads to protein nitration. However, the lack of effect of sulpiride on the increase in Ser897 NR1 phosphorylation indicates that D2 receptors contribute to increased protein nitration by a mechanism independent of PKA-dependent phosphorylation of this site. In this regard, it is of interest that coexpression of D1 and D2 receptors in the same complex permits a dopamine-induced increase in intracellular Ca2+ that is dependent on phospholipase C rather than on PKA or PKC (Lee et al, 2004). Increased Ca2+ by coactivation of D1 and D2 receptors during H–I may be adequate for stimulating NO and superoxide production in quantities sufficient for peroxynitrite formation and protein nitration.
A considerable portion of brain energy metabolism is used to support Na+,K+-ATPase activity for maintaining ion gradients, osmotic balance, cell volume regulation, resting membrane potential, the excitability of neurons, and the Na+-coupled reuptake of glutamate across the plasma membrane. In addition, Na+,K+-ATPase may modulate NMDA receptor activity by regulating intracellular Na+ (Yu and Salter 1998). The present results confirm previous findings of a marked decrease in Na+,K+-ATPase activity in this model of H–I in piglet putamen (Golden et al, 2001). Although exposure of cortical membranes to high concentrations of peroxynitrite can produce tyrosine nitration of Na+, K+-ATPase (Golden et al, 2003) and decrease its activity (Qayyum et al, 2001), increased tyrosine nitration of the Na+,K+-ATPase α3 subunit could not be detected in this model of H–I (Golden et al, 2003). Thus, we investigated dopaminergic mechanisms dependent on phosphorylation as a means for Na+, K+-ATPase inactivation.
Phosphorylation of Na+,K+-ATPase α subunit by PKA and PKC decreases activity in a cooperative manner (Blanco 2005; Cheng et al, 1997; Therien and Blostein 2000). Both D1 and D2 receptor activation inhibit Na+,K+-ATPase activity by mechanisms that require DARPP-32 despite the fact that D2 activation does not increase Thr34 phosphorylation on DARPP-32 (Nishi et al, 1999b). Thus, the inhibitory effect of D2 receptors on Na+,K+-ATPase activity depends on another mechanism. Our in vivo results generally support those obtained in vitro. The decrease in Na+,K+-ATPase activity at 3 h after resuscitation was associated with increased phosphorylation of DARPP-32 at Thr34, and of Na+, K+-ATPase α at the PKA-sensitive Ser943 site and the PKC-sensitive Ser23 site. SCH23390 restored Na+, K+-ATPase activity in association with inhibiting the increase in DARPP-32 Thr34 phosphorylation and Na+, K+-ATPase α Ser943 phosphorylation, whereas sulpiride restored Na+,K+-ATPase activity without inhibiting phosphorylation of either of these sites. These findings suggest that the suppressive action of D1 receptor activation on Na+,K+-ATPase during H–I is mediated by PKA-dependent phosphorylation of Ser943, along with DARPP-32 inhibition of PP-1. Although PKA does phosphorylate Ser943 (Fisone et al, 1994), increased phosphorylation of the α3/α2 subunit unexpectedly could not be demonstrated by dopamine in isolated rat neostriatal cells (Nishi et al, 1999a). Thus, it is possible that the effect of SCH23390 on Ser943 phosphorylation observed in the present experiment is related to an indirect effect of D1 receptor antagonism on the cell signaling process after the H–I insult. Alternatively, age differences, species differences, or an interaction of the different cell types in vivo may permit D1 receptor activation to directly produce Ser943 phosphorylation in our experiments. The observation that SCH23390 and sulpiride individually restored activity to control levels implies that both D1 and D2 receptor activation are required for suppressing Na+, K+-ATPase activity in vivo and supports the concept of a synergistic effect observed in acutely dissociated striatal neurons (Bertorello et al, 1990). However, the mechanism by which sulpiride restores Na+,K+-ATPase activity remains uncertain.
Pretreatment with SCH23390 was somewhat more effective than sulpiride in providing neuroprotection, possibly because of D1 receptor-specific regulation of NMDA receptors and other target proteins. When treatment was delayed until 5 mins after resuscitation, partial neuroprotection was sustained with SCH23390, but protection by sulpiride was no longer significant. These results imply that the adverse effects of D1 receptor activation continue into the reoxygenation period, but that the net adverse effects of D2 receptor activation are set into motion primarily during the period of asphyxia. Microdialysis data indicated that extracellular dopamine remained significantly elevated during the first 15 mins of reoxygenation. Because the microdialysis technique measures overall extracellular concentration, synaptic concentrations of dopamine may remain elevated for more prolonged periods of reoxygenation. Another consideration is that reoxygenation with 100% O2 has been noted to delay the recovery of dopamine in microdialysates, compared with reoxygenation with 21% O2, and to augment a secondary increase at 2 to 3 h of reoxygenation (Huang et al, 1995). We used 50% O2 for the initial resuscitation and gradually decreased the inspired O2 to 30% to offset the early effect of acidosis on oxyhemoglobin saturation during the first 30 mins of reoxygenation. A secondary increase in dopamine was observed in a few of the piglets at 2 to 3 h, but the effect was not significant. Posttreatment with SCH23390 and sulpiride may have had a larger impact if higher O2 concentrations had been used during reoxygenation.
Clinically, H–I encephalopathy in term newborns presents with a wide range of severity of injury. Although two levels of H–I severity were tested, a protective effect of dopamine antagonists is likely to be diminished with more severe insults. Another limitation of the present study is that the time course of phosphorylation changes and the efficacy of delayed treatment were not determined. Nevertheless, the present study demonstrates that both D1 and D2 receptor activation play an important role in augmenting injury in the selectively vulnerable putamen in a model of neonatal H–I. Both D1 and D2 receptor activation contribute to the marked suppression of Na+,K+-ATPase activity and to peroxynitrite generation, as indicated by nitrotyrosine formation. The D1 receptor effects may be mediated by targeted phosphorylation of DARPP-32, NMDA NR1, and Na+,K+-ATPase α subunits. The precise mechanism by which D2 receptors exert their effects remains unknown, but the D2 receptor antagonist appears to protect a largely different population of neurons than the D1 receptor antagonist. Therapeutic use of dopaminergic receptor antagonists in neonatal H–I may provide a means for protecting striatum without the adverse effects of completely blocking NMDA receptors in the developing brain.
Acknowledgments
We thank Ellen Gordes for technical assistance and Tzipora Sofare for editorial assistance.
Footnotes
This work was supported by NIH Grant NS20020.
References
- Agnew DM, Koehler RC, Guerguerian AM, Shaffner DH, Traystman RJ, Martin LJ, Ichord RN. Hypothermia for 24 h after asphyxic cardiac arrest in piglets provides striatal neuroprotection that is sustained 10 days after rewarming. Pediatr Res. 2003;54:253–62. doi: 10.1203/01.PDR.0000072783.22373.FF. [DOI] [PubMed] [Google Scholar]
- Aizman O, Brismar H, Uhlen P, Zettergren E, Levey AI, Forssberg H, Greengard P, Aperia A. Anatomical and physiological evidence for D1 and D2 dopamine receptor colocalization in neostriatal neurons. Nat Neurosci. 2000;3:226–30. doi: 10.1038/72929. [DOI] [PubMed] [Google Scholar]
- Bertorello AM, Hopfield JF, Aperia A, Greengard P. Inhibition by dopamine of (Na(+)+K+)ATPase activity in neostriatal neurons through D1 and D2 dopamine receptor synergism. Nature. 1990;347:386–8. doi: 10.1038/347386a0. [DOI] [PubMed] [Google Scholar]
- Blanco G. Na,K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin Nephrol. 2005;25:292–303. doi: 10.1016/j.semnephrol.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Bourne JA, Fosbraey P, Halliday J. SCH 23390 affords protection against soman-evoked seizures in the freely moving guinea-pig: a concomitant neurochemical, electrophysiological and behavioural study. Neuropharmacology. 2001;40:279–88. doi: 10.1016/s0028-3908(00)00136-2. [DOI] [PubMed] [Google Scholar]
- Cheng XJ, Hoog JO, Nairn AC, Greengard P, Aperia A. Regulation of rat Na(+)-K(+)-ATPase activity by PKC is modulated by state of phosphorylation of Ser-943 by PKA. Am J Physiol. 1997;273:C1981–6. doi: 10.1152/ajpcell.1997.273.6.C1981. [DOI] [PubMed] [Google Scholar]
- Dudman JT, Eaton ME, Rajadhyaksha A, Macias W, Taher M, Barczak A, Kameyama K, Huganir R, Konradi C. Dopamine D1 receptors mediate CREB phosphorylation via phosphorylation of the NMDA receptor at Ser897-NR1. J Neurochem. 2003;87:922–34. doi: 10.1046/j.1471-4159.2003.02067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Standaert DG. Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J Neurosci. 2001;21:5546–58. doi: 10.1523/JNEUROSCI.21-15-05546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisone G, Cheng SX, Nairn AC, Czernik AJ, Hemmings HC, Jr, Hoog JO, Bertorello AM, Kaiser R, Bergman T, Jornvall H, et al. Identification of the phosphorylation site for cAMP-dependent protein kinase on Na+,K(+)-ATPase and effects of site-directed mutagenesis. J Biol Chem. 1994;269:9368–73. [PubMed] [Google Scholar]
- Flores-Hernandez J, Cepeda C, Hernandez-Echeagaray E, Calvert CR, Jokel ES, Fienberg AA, Greengard P, Levine MS. Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: role of D1 receptors and DARPP-32. J Neurophysiol. 2002;88:3010–20. doi: 10.1152/jn.00361.2002. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. Molecular effects of dopamine on striatal-projection pathways. Trends Neurosci. 2000;23:S64–70. doi: 10.1016/s1471-1931(00)00019-7. [DOI] [PubMed] [Google Scholar]
- Golden WC, Brambrink AM, Traystman RJ, Martin LJ. Failure to sustain recovery of Na,K-ATPase function is a possible mechanism for striatal neurode-generation in hypoxic–ischemic newborn piglets. Brain Res Mol Brain Res. 2001;88:94–102. doi: 10.1016/s0169-328x(01)00032-8. [DOI] [PubMed] [Google Scholar]
- Golden WC, Brambrink AM, Traystman RJ, Shaffner DH, Martin LJ. Nitration of the striatal Na,K-ATPase alpha3 isoform occurs in normal brain development but is not increased during hypoxia–ischemia in newborn piglets. Neurochem Res. 2003;28:1883–9. doi: 10.1023/a:1026132110850. [DOI] [PubMed] [Google Scholar]
- Guerguerian AM, Brambrink AM, Traystman RJ, Huganir RL, Martin LJ. Altered expression and phosphorylation of N-methyl-D-aspartate receptors in piglet striatum after hypoxia-ischemia. Brain Res Mol Brain Res. 2002;104:66–80. doi: 10.1016/s0169-328x(02)00285-1. [DOI] [PubMed] [Google Scholar]
- Hersch SM, Ciliax BJ, Gutekunst CA, Rees HD, Heilman CJ, Yung KK, Bolam JP, Ince E, Yi H, Levey AI. Electron microscopic analysis of D1 and D2 dopamine receptor proteins in the dorsal striatum and their synaptic relationships with motor corticostriatal afferents. J Neurosci. 1995;15:5222–37. doi: 10.1523/JNEUROSCI.15-07-05222.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirvonen J, Nagren K, Kajander J, Hietala J. Measurement of cortical dopamine d1 receptor binding with 11C[SCH23390]: a test–retest analysis. J Cereb Blood Flow Metab. 2001;21:1146–50. doi: 10.1097/00004647-200110000-00002. [DOI] [PubMed] [Google Scholar]
- Huang CC, Yonetani M, Lajevardi N, Delivoria-Papadopoulos M, Wilson DF, Pastuszko A. Comparison of postasphyxial resuscitation with 100% and 21% oxygen on cortical oxygen pressure and striatal dopamine metabolism in newborn piglets. J Neurochem. 1995;64:292–8. doi: 10.1046/j.1471-4159.1995.64010292.x. [DOI] [PubMed] [Google Scholar]
- Lee SP, So CH, Rashid AJ, Varghese G, Cheng R, Lanca AJ, O’Dowd BF, George SR. Dopamine D1 and D2 receptor co-activation generates a novel phospholipase C-mediated calcium signal. J Biol Chem. 2004;279:35671–8. doi: 10.1074/jbc.M401923200. [DOI] [PubMed] [Google Scholar]
- Maldve RE, Zhang TA, Ferrani-Kile K, Schreiber SS, Lippmann MJ, Snyder GL, Fienberg AA, Leslie SW, Gonzales RA, Morrisett RA. DARPP-32 and regulation of the ethanol sensitivity of NMDA receptors in the nucleus accumbens. Nat Neurosci. 2002;5:641–8. doi: 10.1038/nn877. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Brambrink A, Koehler RC, Traystman RJ. Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by hypoxia–ischemia. J Comp Neurol. 1997a;377:262–85. doi: 10.1002/(sici)1096-9861(19970113)377:2<262::aid-cne8>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Brambrink AM, Lehmann C, Portera-Cailliau C, Koehler R, Rothstein J, Traystman RJ. Hypoxia–ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann Neurol. 1997b;42:335–48. doi: 10.1002/ana.410420310. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Brambrink AM, Price AC, Kaiser A, Agnew DM, Ichord RN, Traystman RJ. Neuronal death in newborn striatum after hypoxia–ischemia is necrosis and evolves with oxidative stress. Neurobiol Dis. 2000;7:169–91. doi: 10.1006/nbdi.2000.0282. [DOI] [PubMed] [Google Scholar]
- Meng SZ, Isumi H, Takashima S. Neuropathological characteristics and alteration of the dopamine D2 receptor in hypoxic–ischemic basal ganglia necrosis. Brain Dev. 1998;20:98–104. doi: 10.1016/s0387-7604(98)00003-5. [DOI] [PubMed] [Google Scholar]
- Miller SP, Ramaswamy V, Michelson D, Barkovich AJ, Holshouser B, Wycliffe N, Glidden DV, Deming D, Partridge JC, Wu YW, Ashwal S, Ferriero DM. Patterns of brain injury in term neonatal encephalopathy. J Pediatr. 2005;146:453–60. doi: 10.1016/j.jpeds.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Nishi A, Fisone G, Snyder GL, Dulubova I, Aperia A, Nairn AC, Greengard P. Regulation of Na+, K+-ATPase isoforms in rat neostriatum by dopamine and protein kinase C. J Neurochem. 1999a;73:1492–501. doi: 10.1046/j.1471-4159.1999.0731492.x. [DOI] [PubMed] [Google Scholar]
- Nishi A, Snyder GL, Fienberg AA, Fisone G, Aperia A, Nairn AC, Greengard P. Requirement for DARPP-32 in mediating effect of dopamine D2 receptor activation. Eur J Neurosci. 1999b;11:2589–92. doi: 10.1046/j.1460-9568.1999.00724.x. [DOI] [PubMed] [Google Scholar]
- Nishi A, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J Neurosci. 1997;17:8147–55. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill MJ, Hicks CA, Ward MA, Cardwell GP, Reymann JM, Allain H, Bentue-Ferrer D. Dopamine D2 receptor agonists protect against ischaemia-induced hippocampal neurodegeneration in global cerebral ischaemia. Eur J Pharmacol. 1998;352:37–46. doi: 10.1016/s0014-2999(98)00333-1. [DOI] [PubMed] [Google Scholar]
- Pastuszko A, Saadat-Lajevardi N, Chen J, Tammela O, Wilson DF, Delivoria-Papadopoulos M. Effects of graded levels of tissue oxygen pressure on dopamine metabolism in the striatum of newborn piglets. J Neurochem. 1993;60:161–6. doi: 10.1111/j.1471-4159.1993.tb05834.x. [DOI] [PubMed] [Google Scholar]
- Qayyum I, Zubrow AB, Ashraf QM, Kubin J, Delivoria-Papadopoulos M, Mishra OP. Nitration as a mechanism of Na+, K+-ATPase modification during hypoxia in the cerebral cortex of the guinea pig fetus. Neurochem Res. 2001;26:1163–9. doi: 10.1023/a:1012331108641. [DOI] [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Ehlers MD. Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology. 2003;45:755–67. doi: 10.1016/s0028-3908(03)00250-8. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501–10. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine-and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18:10297–303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci. 1996;16:6579–91. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–96. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- Therien AG, Blostein R. Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol. 2000;279:C541–66. doi: 10.1152/ajpcell.2000.279.3.C541. [DOI] [PubMed] [Google Scholar]
- Yu XM, Salter MW. Gain control of NMDA-receptor currents by intracellular sodium. Nature. 1998;396:469–74. doi: 10.1038/24877. [DOI] [PubMed] [Google Scholar]
- Zhu C, Xu F, Wang X, Shibata M, Uchiyama Y, Blomgren K, Hagberg H. Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxia–ischaemia. J Neurochem. 2006;96:1016–27. doi: 10.1111/j.1471-4159.2005.03639.x. [DOI] [PubMed] [Google Scholar]