

Abstract
Translocations affecting the chromosomal region 15q11–13 and various other partners are recurrent in diffuse large-cell lymphomas (DLCL). To identify the putative gene, here named BCL8, involved in these translocations we have cloned the breakpoint region from a DLCL patient with t(14;15)(q32;q11–13) and the corresponding germ-line region from chromosome 15. The genomic locus on chromosome 15 is clonally rearranged in about 4% of DLCL in agreement with the frequency of 15q11–13 translocations. A probe derived from the BCL8 locus on chromosome 15 detected a transcript in human testis and prostate, whereas no expression was found in spleen, thymus, and blood leukocytes. Analysis of the BCL8 cDNA clones isolated from human testis cDNA library showed that the BCL8 gene generates a major transcript of 2.6 kb and a less prominent 4.5-kb species due to differential polyadenylylation. By reverse transcription–PCR analysis of RNA extracted from frozen DLCL samples and lymphoma cell lines, BCL8 expression was detected in all patients carrying 15q11–13 abnormalities and in a fraction of randomly selected DLCL patients. These results suggest that the BCL8 gene is not normally expressed in lymphoid tissues, but its expression can be activated by chromosomal translocation or by other mechanisms in DLCL. Ectopic expression of BCL8 in a significant proportion of DLCL suggests an important role for this gene in the molecular pathogenesis of B cell lymphoma.
Recurrent chromosomal translocations recognized in the majority of lymphomas provide clues to mechanisms of lymphomagenesis. Ig genes undergo specific rearrangements during differentiation of lymphoid cells, and errors in recombination lead to chromosome translocation and neoplastic transformation (1, 2). Diffuse large-cell lymphomas (DLCL) make up approximately 50% of non-Hodgkin lymphomas (3) and show significant variability in terms of their pathologic manifestation, response to therapy, and prognosis (4). DLCL are also highly heterogeneous with respect to karyotypic abnormalities and underlying molecular lesions. So far, three genes have been identified whose normal pattern of expression is altered by a specific chromosomal translocation in DLCL, namely, MYC, BCL2, and BCL6, occurring in approximately 10%, 20%, and 25% of DLCL, respectively, as detected by either cytogenetic or molecular methods (1, 2, 5–7). The remaining DLCL exhibit a number of small subsets each characterized by a specific site of recurrent chromosomal rearrangement. The molecular genetic analysis of these sites can contribute significantly to our understanding of the mechanisms of B cell lymphomagenesis.
One of the recurrent sites of rearrangement seen in 3–4% of DLCL cases is 15q11–13. A notable feature of this site is promiscuity of rearrangement. In addition to translocations involving the Ig gene sites 14q32 and 22q11, it exhibits translocations with multiple other sites, such as 9p13, 1p32, 7p13, 12q24, and 15q22 (R.S.K.C., unpublished observation). Translocations involving 15q11–13 also have been noted in nonlymphoid tumors (8). The 15q11–13 region has been implicated in genomic imprinting and contains putative genes for human imprinting-related disorders such as Prader–Willi and Angelman syndromes. Another unusual feature of the region is the presence of orphan, presumably nonfunctional, copies of V and D segments of the IGH gene (9, 10). Here we report the cloning of the t(14:15)(q32;q11–13) breakpoint from a DLCL patient and the isolation of a new gene, here named BCL8, located at 15q11–13 and potentially involved in the pathogenesis of DLCL.
MATERIALS AND METHODS
Molecular Cloning and Southern and Northern Blot Analysis.
A genomic library was constructed from case LL430 in λ GEM-11 phage vector (Promega) by partial Sau3A digestion of tumor DNA. Commercial λ DR2 cDNA library from human testis (CLONTECH) was used for cDNA cloning. Human IGH probes used for initial Southern blot analysis and genomic library screening were a 5.5-kb BamHI–HindIII fragment containing the entire JH locus (JH) and a 1.3-kb EcoRI fragment containing first two exons of the IGHM gene (Cμ). For somatic cell hybrid mapping and Northern analysis, probes from cloned chromosomal breakpoints were generated by subcloning of repeat-free fragments from recombinant phages into pBluescript (Stratagene).
Fluorescence in Situ Hybridization Analysis.
Phage and plasmid probes were labeled with biotin-14-dUTP and hybridized to metaphase spreads from normal human lymphocytes as previously described (11). Hybridization signal and corresponding bands were visualized with fluorescein isothiocyanate-conjugated avidin (Oncor) following staining and counterstaining, respectively, with propidium iodide and 4′,6-diamidino-2-phenylindole.
Reverse Transcription–PCR (RT-PCR) Analysis.
Total RNA from frozen tumor specimen and cell lines was isolated by the guanidine isothiocyanate method using RNAgents kit (Promega). MessageClean (GenHunter, Nashville, TN) was used for further RNA purification resulting in DNA-free RNA preparations. The following primers were used for detection of BCL8 expression: GTTAAGTCCTAAAAGTCT (forward) and TATAGGAGTAAAGTCTAC (reverse). β-Actin specific primers were used as a positive control (12). Minus-RT controls were run for all samples and were negative.
RESULTS
Southern Blot Analysis of the Tumor DNA.
Patient 430, a DLCL, had a t(14;15)(q32;q11–13) translocation and did not express a clonal Ig heavy-chain phenotype. Southern blotting analysis of tumor DNA digested with BamHI and HindIII showed clonal rearrangements of both the JH and Cμ regions of the IGH gene (Fig. 1a–d). The JH and Cμ probes used in this analysis hybridized to the same germ-line BamHI fragment. The JH- and Cμ-rearranged bands in the tumor DNA did not comigrate, suggesting either that both alleles of IGH underwent nonproductive rearrangements or that the JH and Cμ regions of one allele were separated by a translocation breakpoint. In the latter case, the rearranged JH and Cμ fragments would be expected to represent the two reciprocal junctions (Fig. 1 a and b).
Figure 1.
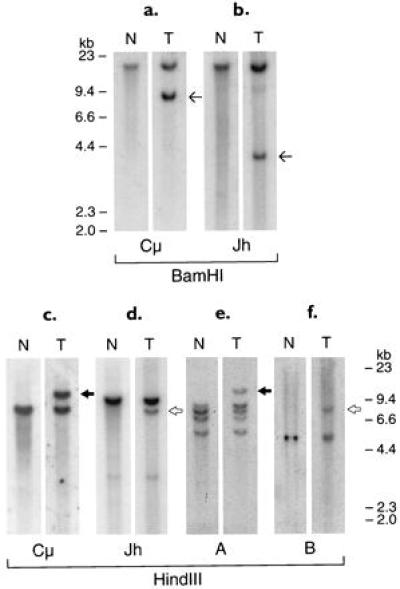
Southern blot analysis of tumor 430 DNA digested with BamHI and HindIII enzymes and hybridized with IGH probes (a–d) and probes located across the chromosomal breakpoints (e and f). Thin arrows indicate non-comigrating Cμ and JH BamHI rearrangement bands. Solid arrows indicate comigrating HindIII rearrangement bands with Cμ and probe A (Fig. 2I). Open arrows indicate comigrating HindIII rearrangement bands with JH and probe B (Fig. 2III). The same filter was successively rehybridized with the different probes.
Cloning of the t(14;15)(q32;q11–13) Translocation Breakpoint.
To isolate the chromosomal breakpoint, a genomic library of the tumor DNA partially digested with Sau3A was constructed and screened with the JH and Cμ probes. Clones positive with one probe but negative with the other were selected, mapped (Fig. 2), and partially sequenced. To further confirm that the clones selected represented the original rearranged bands in the tumor, probes from the cloned regions located across the breakpoints were hybridized to genomic Southern blots of tumor DNA. The same rearranged bands as those initially detected by the JH and Cμ probes were identified (Fig. 1 e and f).
Figure 2.

Restriction maps of the deleted IGH allele (I), the normal IGH locus (II), the der(14)t(14;15)(q32;q11–13) translocation allele (III), and the corresponding germ-line chromosome 15 region (IV). Lines under the maps indicate probes used for Southern and Northern analysis. Open arrow shows the direction of BCL8 transcription.
The Cμ-positive, JH-negative phage clone hybridized to both chromosomes 14 and 15 in fluorescence in situ hybridization analysis, suggesting that it may contain the translocation breakpoint (Fig. 3A). A smaller fragment derived from the cloned region (A in Fig. 2I) also recognized both chromosomes when used as a probe on a panel of somatic cell hybrids yielding four bands on chromosome 14 and a single band on chromosome 15 (Fig. 4 a–c). Partial sequencing of the 5′ end of the phage revealed 80% to 90% homology with the D regions of IGH. Previously, four D clusters (D1–D4) were shown to be tandemly located at 14q32, 23 kb telomeric to JH, and one (D5) was mapped to 15q11–13 (9, 10, 13). Because the only sequence data available for all five D clusters were those for Dxp1–Dxp5 segments, we located and sequenced the Dxp segment contained in the cloned region to determine its chromosomal origin. The sequence was identical to Dxp2. The order of the D segments in the cloned fragment was also consistent with that of D1–D4 clusters but not with that of D5 (data not shown). Based on these data, we concluded that the cloned phage contained parts of D2 and D3 clusters from chromosome 14, which crosshybridized with the D5 cluster sequences located on chromosome 15, and with other D clusters on chromosome 14. The Cμ-positive, JH-negative rearranged band, therefore, represented a microdeletion in one of the IGH alleles and was not involved in the t(14;15) translocation.
Figure 3.
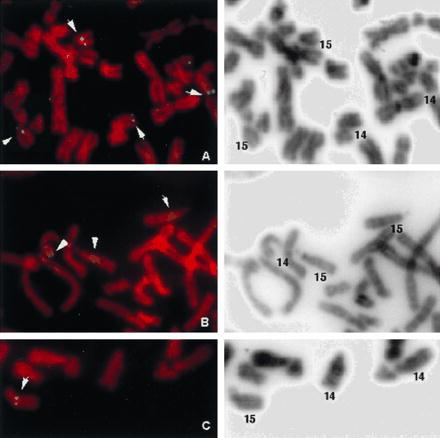
Fluorescence in situ hybridization mapping of the phage clones representing the deleted (A) and translocated (B) IGH alleles and the fragment C (see Fig. 2III) subcloned in pBluescript. (Left) Propidium iodide staining of hybridized metaphase. (Right) G-like banding of the same metaphase revealed by 4′,6-diamidino-2-phenylindole staining.
Figure 4.
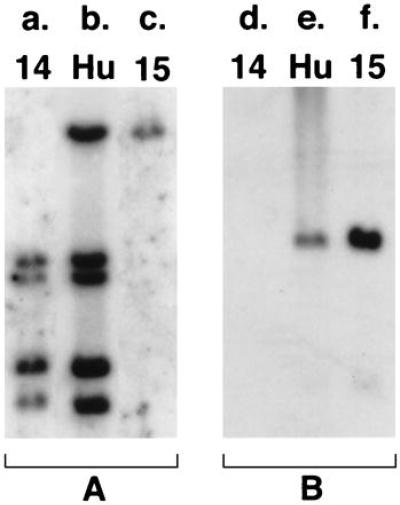
Somatic cell hybrid mapping of recombinant clones. (a–c) BamHI digestion of human/rodent somatic cell hybrids (Bios, New Haven, CT) hybridized to probe A (Fig. 2I). (d–f) BamHI digestion of monochromosomal cell hybrids containing human chromosomes 14 and 15 (Coriell Cell Repositories, Camden, NJ) hybridized to probe B (see Fig. 2III).
Because part of one IGH allele, including the JH region, was deleted, the JH-rearranged BamHI band identified in the Southern blot analysis of tumor DNA was not reciprocal to the Cμ-rearranged band. It was considered to originate from the second IGH allele, possibly linked to chromosome 15 by translocation. The fact that both IGH alleles were affected by nonproductive rearrangements was consistent with the lack of clonal Ig heavy-chain expression by the tumor. JH-positive, Cμ-negative clones obtained from the genomic library also hybridized to both chromosomes 14 and 15 by fluorescence in situ hybridization (Fig. 3B) and somatic cell hybrid mapping (Fig. 4 d–f). Restriction mapping and partial sequencing revealed IGH-related sequences both 3′ and immediately 5′ of JH (Fig. 2III). IGHE (Cɛ) and ɛ-switch sequences were identified 3′ of the JH region. V-related sequences were found 5′ of JH. Both JH and Cɛ regions contained multiple deletions and point mutations, the JH region showing only 80% homology with the germ-line sequence. The IGH enhancer region was retained in the cloned fragment as confirmed by sequencing. The cloned V-related fragment had 60–70% homology with a number of previously sequenced V regions of IGH, suggesting that it either originated from a germ-line V segment that has previously not been sequenced or it has been altered by deletions and mutations, as was the case with the adjacent JH. Because some V genes have been mapped to 15q11–13 (9, 10), this fragment may have originated either from chromosome 14 or 15. The fragment from the 5′ end of the cloned region (C in Fig. 2III) did not show significant homology to IGH or any other sequence in the database and hybridized to chromosome 15 alone in somatic cell hybrids (Fig. 4 d–f) and in fluorescence in situ hybridization analysis (Fig. 3C). These data thus demonstrated that this allele was involved in a translocation with a previously unknown sequence on chromosome 15.
BCL8 Transcriptional Unit.
The germ-line chromosome 15 region (Fig. 2IV) was cloned using probe B (Fig. 2III) to screen a genomic library. Overlapping phage clones spanning the breakpoint area and flanking regions were mapped and partially sequenced. To identify a possible transcriptional unit, a series of probes were derived from the region and hybridized to human multiple tissue Northern blots. Probe C (Fig. 2IV) detected a 4.5-kb transcript with abundant expression in prostate and testis but not in lymphoid tissues such as thymus, spleen, and blood leukocytes (data not shown).
Cloning of the BCL8 cDNA.
Probe C was used to screen a human testis cDNA library. Restriction mapping and sequence alignment of clones obtained showed that the sequence homologous to probe C was located at the 3′ end of the cDNA. cDNA clones with two different 3′ ends were found. This was confirmed by preliminary analysis of the cDNA sequence, which revealed two potential sites of polyadenylylation resulting in two possible transcripts, 2.6 kb and 4.5 kb in size. Using blast program no significant homology was found with any DNA sequence other than an expressed sequence tag cloned from human brain cDNA library. The direction of transcription on chromosome 15 was from telomere to centromere. The breakpoint in tumor 430 was located 3′ of the gene, bringing the intact gene to the proximity of the retained IGH enhancer, which is capable of activating it across the rearrangement junction (Fig. 2).
Northern Blot Analysis of BCL8.
To verify that both types of BCL8 transcripts with alternative polyadenylylation are expressed in human tissues, we used cDNA probes for Northern analysis. As expected from the cDNA map, the 3′ terminal probe C detected only the 4.5-kb transcript, whereas the 5′- terminal probe A and probe B located upstream of the internal polyadenylylation site recognized both RNA species. The 2.6-kb transcript appeared to be expressed more abundantly than the 4.5-kb species when detected with these probes. The sizes of the transcripts detected in prostate are consistently smaller than those in the testis. This may indicate alternative splicing and/or an alternative promoter in this tissue. Additional 3.7-kb- and 6-kb bands hybridizing to probe A are not consistent with the cDNA map and may indicate the existence of crosshybridizing, related genes expressed in testis only (Fig. 5). On a multiple species Southern blot, cDNA probes detected strong bands in human and monkey lanes. Detectable bands also were observed across the panel of species including yeast, indicating phylogenetic conservation of the cDNA sequence (data not shown).
Figure 5.
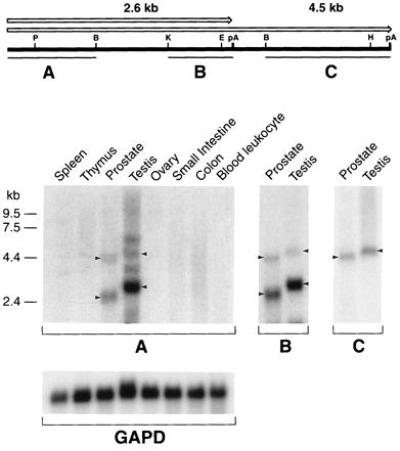
BCL8 cDNA map and Northern analysis of BCL8 expression in human tissues. Open arrows above the map show two alternative transcripts indicated by arrowheads on Northern blots. Probes used in the analysis are indicated by lines under the map. Multiple tissue Northern blot (CLONTECH) was successively rehybridized with all probes. GAPD, glyceraldehyde-3-phosphate dehydrogenase.
BCL8 Rearrangements in DLCL.
To determine whether translocation breakpoints in other DLCL cases cluster within the same region on chromosome 15, a panel of DLCL DNAs was screened by Southern blotting for clonal rearrangements using probe B (Fig. 2III). Three of 71 tumor DNAs showed clonal rearrangement within 5 kb of BCL8 locus (Fig. 6). The frequency of BCL8 DNA rearrangement was in agreement with that of chromosomal translocations involving 15q11–13 (R.S.K.C., unpublished observations).
Figure 6.
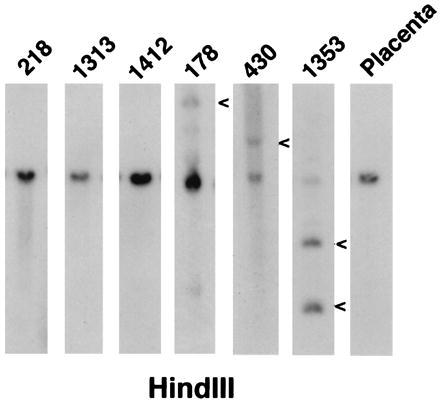
Southern blot analysis of DLCL DNA using probe B (see Fig. 2III). Rearrangement bands are indicated by arrowheads.
RT-PCR Analysis of BCL8 Expression in Lymphomas.
To detect BCL8 expression in RNA extracted from frozen tumor specimen by RT-PCR, we designed primers within the 2.6-kb transcript upstream of the internal polyadenylylation site. The primers specifically amplified a 390-nt fragment from human testis RNA. In all tested tumors carrying 15q11–13 abnormalities, an expected 390-bp band was detected. BCL8 expression was also detectable in 4 of 9 randomly selected DLCL but in none of the 3 tested hyperplastic lymph nodes (Fig. 7a). To test the possibility that the recognized bands originated from contamination of the tumor tissues with nonlymphoid cells that may normally express BCL8, we also studied a panel of DLCL cell lines. Six of 15 cell lines without 15q11–13 rearrangements were also positive for BCL8 expression, whereas a B lymphoblastoid cell line was negative (Fig. 7b). These data clearly demonstrate ectopic activation in BCL8 in DLCL.
Figure 7.
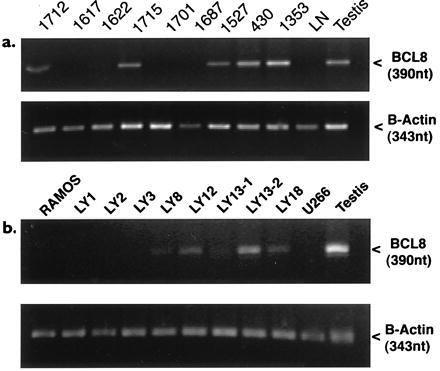
RT-PCR analysis of BCL8 expression in DLCL (a) and lymphoma cell lines (b). Arrowheads indicate bands amplified using BCL8 specific primers (Upper) and β-actin primers (Lower). Tumors 1527, 430, and 1353 had translocations affecting 15q11–13. LY numbers represent DLCL cell lines. RAMOS is a Burkitt lymphoma cell line, U226 is a plasmacytoma cell line, and LN is a hyperplastic lymph node with nonmalignant proliferation of T and B lymphocytes.
DISCUSSION
DLCL is a clinically important subset of non-Hodgkin’s lymphoma that is highly heterogeneous both at the cytogenetic and molecular levels. The most frequent recurrent translocations in DLCL, so far identified are t(3;14), t(14;18), and t(8;14), which together are seen in approximately 55% of cases (5–7). Our cytogenetic analysis over the past 10 years has revealed several new recurrent chromosomal sites that translocate with the Ig gene sites in DLCL, suggesting deregulation of hitherto unidentified genes in the etiology of this disorder. Several of these sites, including 15q11–13, exhibit promiscuity in rearrangement; i.e., they participate in recurring translocations involving IG genes and other chromosomal sites, suggesting that the candidate genes at these sites may be deregulated by formation of chimeric gene products, utilization of unrelated promoters, or other mechanisms.
Orphan V and D segments with unknown function have previously been mapped to 15q11–13 (9–10). It is possible that the V/D segments on chromosome 15 can rearrange with D/J segments on chromosome 14, resulting in interchromosomally rearranged IGH genes. Such a rearrangement would be detectable cytogenetically as t(14;15)(q32;q11–13). In tumor 430, although the chromosomal origin of the V-related sequence found in the translocated allele is not clear, it is not located immediately downstream of BCL8 on the germ-line chromosome 15. We therefore consider it more likely to be of chromosome 14 origin; thus both V-D and D-J recombinations in this tumor may have involved only partners from chromosome 14.
We have identified and cloned the genomic region of BCL8 located at 15q11–13, which is a recurrent site of rearrangements in a fraction of DLCL. The results of Southern blot analysis of randomly selected DLCLs indicated that BCL8 rearrangements may account for a significant part of all cytogenetically detectable 15q11–13 abnormalities.
The BCL8 region contains a transcriptional unit that is not normally expressed in lymphoid tissues. The sequence of the gene has been conserved throughout evolution, suggesting its functional significance. So far, there is no direct evidence pointing to the possible function of BCL8 such as a sequence homology with a gene coding a known protein. Our RT-PCR analysis of the gene expression in lymphomas, however, showed that BCL8 is frequently expressed in DLCL, being the only known gene so far to be ectopically activated in lymphomas. These data suggest that BCL8 may play an important role in the pathogenesis of DLCL.
In tumor 430 the most likely mechanism of BCL8 deregulation is through its juxtaposition to the IGH enhancer located immediately downstream of JH and retained in the translocated allele. Similar mechanisms of activation by a regulatory element across the translocation breakpoint have previously been reported for t(8;14) and t(14;18) (1, 2). The same mechanism may apply to other cases with 15q11–13 translocations involving sites for the Ig genes 14q32 and 22q11 and possibly some of the unknown rearrangement partners. However, the RT-PCR analysis showed BCL8 expression in a significant proportion of DLCL, including those without cytogenetically detectable 15q11–13 abnormalities. Therefore mechanisms of activation other than translocations such as mutations in regulatory region or deregulation by a trans-acting factor also have to be considered (14). One of the possible mechanisms includes gene deregulation by changes in the imprinting pattern of the entire chromosomal region, which has been shown to contain multiple imprinted genes (15, 16).
In summary, we report here the cloning of a new gene, BCL8, which is activated by chromosome rearrangement in B cell DLCL, thereby enhancing our understanding of the molecular mechanisms of this genetically heterogenous and clinically important group of human B cell lymphomas. Although its function is yet unknown, it is the first known frequently activated gene in DLCL whose normal expression is not constitutive to normal B cells.
Acknowledgments
This investigation was supported by National Institutes of Health/National Cancer Institute Grants CA-34775 and CA-66999 (R.S.K.C.) and CA-44029 (R.D.F.).
ABBREVIATIONS
- DLCL
diffuse large-cell lymphoma
- RT-PCR
reverse transcription–PCR
References
- 1.Chaganti R S K, Klein E A. In: Molecular Genetics in Cancer Diagnosis. Cossman J, editor. New York: Elsevier; 1991. pp. 73–101. [Google Scholar]
- 2.Rabbits T H. Nature (London) 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- 3.Simon R, Durrleman S, Hoppe R T, Bonadonna G, Bloomfield C D, Rudders R A, Cheson B D, Berard C W. Ann Intern Med. 1988;109:939–945. doi: 10.7326/0003-4819-109-12-939. [DOI] [PubMed] [Google Scholar]
- 4.Magrath I. In: The Non-Hodgkin Lymphoma. Magrath I, editor. Baltimore: Williams & Wilkins; 1989. pp. 29–48. [Google Scholar]
- 5.Ladanyi M, Offit K, Jhanwar S C, Filippa D A, Chaganti R S K. Blood. 1991;77:1057–1063. [PubMed] [Google Scholar]
- 6.Offit K, Chaganti R S K. In: Hematology/Oncology Clinics of North America: Non-Hodgkin’s Lymphoma. Armitage J O, editor. Philadelphia: Saunders; 1991. pp. 853–869. [PubMed] [Google Scholar]
- 7.Offit K, Lo Coco F, Louie D C, Parsa N Z, Leung D, Portlock C, Ye B M, Lista F, Filippa D A, Rosenbaum A, Ladanyi M, Jhanwar S, Dalla-Favera R, Chaganti R S K. N Engl J Med. 1994;331:74–80. doi: 10.1056/NEJM199407143310202. [DOI] [PubMed] [Google Scholar]
- 8.Mittelman F. Catalog of Chromosomal Aberrations in Cancer. New York: Wiley–Liss; 1994. [Google Scholar]
- 9.Tomlinson I M, Cook G P, Carter N P, Elaswarapu R, Smith S, Walter G, Buluwela l, Rabbits T H, Winter G. Hum Mol Genet. 1994;3:853–860. doi: 10.1093/hmg/3.6.853. [DOI] [PubMed] [Google Scholar]
- 10.Wintle R F, Cox D W. Genomics. 1994;23:151–157. doi: 10.1006/geno.1994.1471. [DOI] [PubMed] [Google Scholar]
- 11.Rao P H, Murty V V V S, Gaidano G, Hauptschein R, Dalla-Favera R, Chaganti R S K. Genomics. 1993;16:426–430. doi: 10.1006/geno.1993.1206. [DOI] [PubMed] [Google Scholar]
- 12.Ladanyi M, Wang S. Diag Mol Path. 1992;1:31–35. doi: 10.1097/00019606-199203000-00005. [DOI] [PubMed] [Google Scholar]
- 13.Matsuda F, Lee K H, Nakai S, Sato T, Kodaira M, Zong S Q, Ohno H, Fukuhara S, Honjo T. EMBO J. 1988;7:1047–1051. doi: 10.1002/j.1460-2075.1988.tb02912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dalla-Favera R, Ye B H, Lo Coco F, Chang C-C, Cechova K, Zhang J, Miglazza H, Mellado W, Niu H, Chaganti S, Chen W, Rao P H, Pazsa N, Louie D C, Offit K, Chaganti R S K. Cold Spring Harbor Symp Quant Biol. 1994;59:117–123. doi: 10.1101/sqb.1994.059.01.015. [DOI] [PubMed] [Google Scholar]
- 15.Ledbetter D H, Engel E. Hum Mol Genet. 1995;4:1757–1764. doi: 10.1093/hmg/4.suppl_1.1757. [DOI] [PubMed] [Google Scholar]
- 16.Dittrich B, Buiting K, Korn B, Rickard S, Buxton J, Saitoh S, Nichols R D, Poustka A, Winterpacht A, Zabel B, Horsthemke B. Nat Genet. 1996;14:163–170. doi: 10.1038/ng1096-163. [DOI] [PubMed] [Google Scholar]