

Abstract
Mutations are permanent DNA sequence changes that can be induced when replication occurs on a damaged DNA template. In Escherichia coli, the process of translesion synthesis past a lesion that hinders replication requires the induction of SOS-controlled gene products, among which are those of the umuDC operon. To study translesion synthesis in vivo, we have constructed single-stranded vectors containing single 2-acetylaminofluorene adducts located within −1 and −2 frameshift mutation hot spots formed by short repetitive sequences. These adducts strongly hinder DNA replication as only 2–5% of the molecules give rise to progeny under non-SOS-induced conditions. Induction of the SOS response lead to a 10-fold increase in survival. Adducts present within repetitive sequences trigger the formation of misaligned primer/template replication intermediates which, upon elongation, will result in the fixation of frameshift errors (mutagenic translesion synthesis). Surprisingly we find that elongation from the nonslipped intermediate depends upon functional umuDC+ gene products, whereas elongation from the slipped intermediate is umuDC+ independent but requires another, as yet biochemically uncharacterized, SOS function. These data are discussed in terms of the different steps involved during translesion synthesis through a replication-blocking lesion.
Keywords: replication-blocking lesions, 2-acetylaminofluorene adducts, slippage mutagenesis, frameshift mutation hot spots, UmuDC
Organisms have developed various strategies to protect their genomes from the damaging effects of endogenous and exogenous agents. Despite having robust excision repair systems that remove DNA lesions before replication, cells also possess efficient strategies for tolerating lesions persisting at the replication fork during DNA synthesis (1). Two basic strategies of lesion tolerance can be distinguished: (i) translesion synthesis (TLS) is a process during which the replication machinery reads through the lesion with an associated risk of fixing a mutation. If the lesion does not impede DNA synthesis, an unmodified replication complex can achieve TLS. However, a replication complex modified by accessory proteins encoded by genes associated with the SOS regulon is required for TLS of lesions that hinder the progression of replication; and (ii) damage avoidance designates a general strategy that facilitates replication of damaged DNA templates without the need for the polymerase to read through the lesion. This strategy takes advantage of the information contained in the complementary strand. Two models of damage avoidance have been proposed (1–3): (i) postreplication recombinational repair, in which the DNA polymerase, blocked at a lesion site, dissociates from the DNA and reinitiates replication downstream from the lesion, leaving a gap that is repaired by a recombination mechanism involving the sister chromatid; and (ii) polymerase strand switching, in which the DNA polymerase switches temporarily from the damaged parental template to the undamaged newly synthesized strand of the sister chromatid before returning to the parental template downstream from the lesion. Both damage avoidance models are believed to be efficient and error-free. The current level of understanding of the molecular mechanisms underlying these processes is rudimentary.
To study the processes of damage avoidance, we recently have developed an approach involving double-stranded DNA constructions with a genetic strand marker located across from a single 2-acetylaminofluorene guanine adduct (dG-AAF) (4). These adducts were previously shown, using primer-template extension reactions, to be strong blocks for various DNA polymerases in vitro. (5–7). However, double-stranded plasmids with single adducts exhibit the same colony-forming efficiency as nonmodified control molecules when propagated in vivo. The presence of a strand marker across from the adduct site allowed us to measure the fraction of colonies that effectively result from a TLS event. In fact, in the absence of SOS induction, only about 0.5% of the colonies were found to result from TLS events. The induction of the SOS response increased the proportion of TLS events to about 12%. Under both SOS− and SOS+ conditions the large majority of colonies contained only plasmids with the marker of the nonadducted strand, suggesting that the initial round of replication of the damaged plasmid molecule in these colonies occurred via a damage avoidance event (4).
The large predominance of damage avoidance events in double-stranded plasmids severely limits the study of the mechanism of TLS per se. Single-stranded DNA vectors containing single adducts offer a unique possibility to study TLS events because their single-stranded nature imposes TLS as the only process that can give rise to progeny in vivo. It is therefore possible to directly measure the toxicity, and the mutational efficiency and specificity of a given lesion. This approach was pioneered by C. Lawrence and colleagues who, using single-stranded M13 molecules with well defined UV lesions and abasic sites, demonstrated the critical role of the bacterial SOS response in promoting survival and base substitution mutagenesis (8, 9). Base substitution mutagenesis is known to strongly depend upon the expression of the SOS-controlled umuDC operon (10–13).
In contrast, we previously have described a frameshift mutation pathway that is SOS-inducible but umuDC independent (14–16). Frameshift mutations are efficiently induced by a large variety of chemical carcinogens that present an aromatic moiety. Most frameshift mutations occur within short mononucleotide or dinucleotide repeats and can be explained by slippage events during TLS (14, 17–19). Indeed, adducts formed by aromatic amines strongly stabilize misaligned primer/template replication intermediates [slipped mutagenic intermediates (SMI), (20–22)]. These slipped intermediates are in equilibrium with their nonslipped counterparts. Elongation from nonslipped and slipped intermediates will result in error-free and mutagenic TLS, respectively (20, 23). In this paper we have determined the efficiency of elongation from slipped and nonslipped replication intermediates at two different frameshift mutation hot spots in single-stranded DNA. The results show that while elongation from the nonslipped intermediate requires functional umuDC, elongation from the SMI is independent of umuDC but depends upon another SOS function. These results will be discussed in terms of the accessory factors that are potentially required when the replication machinery performs the different biochemical steps involved in replicating through a blocking lesion.
MATERIALS AND METHODS
Oligonucleotides.
Oligonucleotides with single 2-acetylaminofluorene (AAF) adducts located within the run of three Gs or the NarI site have been purified and characterized as described previously (20, 24). The oligonucleotides were phosphorylated using T4 polynucleotide kinase and [γ-32P]ATP (specific activity: 3,000 Ci/mmol). The kinased oligonucleotides were repurified by polyacrylamide gel electrophoresis (20%).
Construction and Quantification of Single-Adducted Single-Stranded Plasmids.
We developed a new strategy to produce high-quality single-stranded plasmids containing a single adduct (ref. 25; Fig. 1). Briefly, double-stranded molecules containing single AAF adducts were produced and purified as described previously (20, 24). The nonadducted strand of these constructs, which contains a few uracil residues, could be selectively degraded in vitro thus yielding the single-stranded mono-adducted vectors (25). The single-stranded vectors were quantified, with a PhosphorImager, from the radioactivity present in the corresponding band of a dried agarose gel knowing the specific activity of the 32P-labeled oligonucleotide (25).
Figure 1.
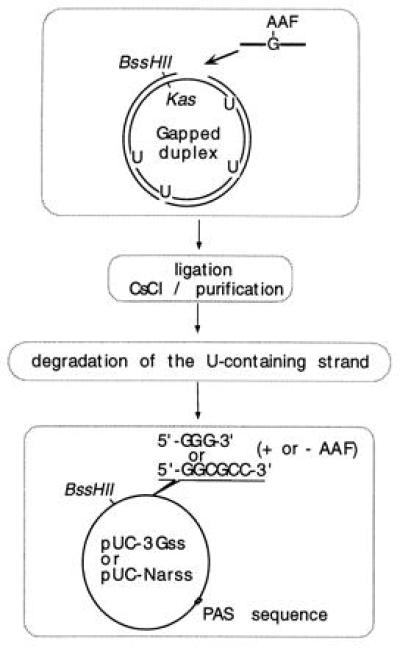
Strategy for the construction of single-stranded DNA. Gapped-duplex intermediates were produced as previously described (20, 24). The gapped strand of the duplex was provided by a plasmid that was grown in a wild-type strain, whereas the complementary strand contained several uracil residues as the corresponding plasmid was propagated in a dut, ung strain (CJ 236) (25). Single AAF-adducted oligonucleotides were ligated into the gap, and the covalently closed circular plasmid was purified from CsCl equilibrium sedimentation. The uracil-containing strand was then selectively degraded using an enzymatic mixture containing uracil DNA glycosylase, and the 3′ → 5′ exonuclease of exonucleaseIII and T7 DNA polymerase (25). Constructions containing the G-AAF adduct (G*) within the sequence 5′-GGG*- or 5′-GGCG*CC- were made along with the corresponding control vectors (no adduct).
Tranformation, Strains, and Mutant Selection.
The single-stranded plasmids were used to transform strain JM103 and its ΔumuDC derivative by the CaCl2 technique. When indicated, the SOS response was induced in both strains by UV irradiation at 50 J/m2 as previously described (4). After UV irradiation, the survival in strain JM103 and its ΔumuDC derivative was 20% and 15%, respectively.
Vectors pUC-3G.ss and pUC-Nar.ss carry a lacZ− allele that reverts to lacZ+ by −1 and −2 frameshift mutagenesis respectively (20, 26). Wild-type and mutant transformants were quantified on indicator plates containing ampicillin, 5-bromo-4-chloro-3-indolyl β-d-galactoside, and isopropyl β-d-thiogalactoside as white and blue colonies, respectively. In addition, −1 and −2 frameshift mutants were identified by direct sequencing or by differential colony hybridization (see below) using the following specific probes: −1 mutants, 5′-AGT CAT ACC CGG ACA TGG AC-3′; and −2 mutants, 5′-TCA TCA CCG GCC ACA GAC TA-5′.
To ascertain that transformants originate from the single-stranded vectors and not from a small amount of contaminating double-stranded DNA we performed colony hybridization experiments making use of a strand marker (BssHII/KasI, Fig. 1) that allows unambiguous identification of any contaminating double-stranded molecules (4, 25).
RESULTS
Nomenclature of the Single-Stranded Vectors.
The AAF adduct is located in two sequence contexts, a run of three Gs 5′-CCCG1G2G3-3′, or the NarI sequence 5′-G1G2CG3CC-3′, that were previously identified as hotspots for −1 and −2 frameshift mutations, respectively (14, 27). For both sequence contexts the AAF adduct is at G3, the most mutagenic position within each hot spot (20, 28). The single-stranded vectors with the run of three Gs were designated pUC-3G.0ss and pUC-3G.3ss for the unmodified control and the G3-AAF modified vector, respectively. Similarly, control and G3-AAF modified vectors at the NarI target sequence were called pUC-Nar.0ss and pUC-Nar.3ss, respectively.
Specificity of AAF-Induced Mutagenesis.
Previous studies using a forward mutation assay and randomly modified double-stranded DNA have shown that approximately 90% of AAF-induced mutations are frameshifts (14, 27). The specificity of AAF-induced mutations was reinvestigated using the single-stranded vectors. In our constructions the single adduct is located in either a SmaI or a NarI restriction site. We analyzed plasmid preparations from 112 independent colonies obtained by transformation of a SOS-induced wild-type strain with pUC-3G.3ss. Among the 112 plasmids, 13 were found to be resistant to SmaI digestion (SmaIR). Sequence analysis of the SmaIR plasmids revealed a distribution of 12 frameshift (−G) mutations (CCCGGG → CCCGG) and one G → T targeted transversion (CCCGGG → CCCGGT). Therefore, in agreement with our previous results, AAF adducts induce frameshift mutations at a much higher frequency than base substitutions (14, 27). For the sake of simplicity only −1 and −2 frameshift mutations were subsequently scored using the lacZ′ detection assay described below.
Nonslipped and Slipped TLS in Vivo.
The efficiency of TLS in a given bacterial cell is defined as the transformation efficiency of the single adducted vector relative to that of the corresponding nonmodified vector. The vectors have been engineered to allow direct determination of the induced frameshift mutation frequencies using the lacZ′ α-complementation assay. Vectors pUC-3G and pUC-Nar carry a lacZ− allele that reverts to lacZ+ by −1 and −2 frameshift mutations, respectively (20, 26). Using indicator plates containing 5-bromo-4-chloro-3-indolyl β-d-galactoside and isopropyl β-d-thiogalactoside, we define the efficiency of nonslipped and slipped TLS from the number of white and blue colonies, respectively (Table 1). The mutation frequency (i.e., the relative proportion of blue colonies divided by the total number of colonies) is a direct measurement of the polymerase error rate per TLS event in vivo because damage avoidance mechanisms cannot operate with single-stranded vectors.
Table 1.
Translesion synthesis within DNA sequences containing AAF adducts
| Measurement | Wild type
|
ΔumuDC
|
||
|---|---|---|---|---|
| SOS− | SOS+ | SOS− | SOS+ | |
| TLS at -GGGAAF- | ||||
| Nonslipped TLS* | 2.34 ± 1.2 | 22.8 ± 3.7 | 2.65 ± 1.48 | 2.07 ± 0.95 |
| (Number of white clones) | (454) | (1,668) | (1,555) | (189) |
| Slipped TLS† | 0.10 ± 0.07 | 3.08 ± 0.31 | 0.072 ± 0.003 | 1.37 ± 0.08 |
| (Number of blue clones) | (20) | (226) | (42) | (125) |
| Survival | 2.44 | 25.88 | 2.72 | 3.44 |
| Mutation frequency,‡ % | 4.1 | 11.9 | 2.6 | 40 |
| TLS at -GGCGAAFCC- | ||||
| Nonslipped TLS* | 3.06 ± 1.48 | 16.7 ± 1.0 | 2.19 ± 0.45 | 2.31 ± 0.78 |
| (Number of white clones) | (329) | (1,197) | (251) | (39) |
| Slipped TLS† | 1.03 ± 0.34 | 34.5 ± 0.84 | 2.70 ± 0.78 | 54.3 ± 1.2 |
| (Number of blue clones) | (111) | (2,464) | (309) | (913) |
| Survival | 4.09 | 51.2 | 4.89 | 56.6 |
| Mutation frequency,‡ % | 25.2 | 67.4 | 55.2 | 96 |
Nonmodified vectors form only white colonies. The total number of blue and white colonies that have been scored to obtain the TLS values are indicated in parenthesis. The average value of three independent experiments and the standard deviation are indicated. Survival is the sum of nonslipped and slipped TLS.
Nonslipped TLS is the number of white colonies per ng of modified vector divided by the number of (white) colonies per ng formed by the corresponding nonmodified vector, expressed as a percentage.
Slipped TLS is the number of blue colonies per ng of modified vector divided by the number of (white) colonies per ng formed by the corresponding nonmodified vector, expressed as a percentage.
The mutation frequency is the percent of slipped TLS divided by total TLS. It represents the elongation error-rate of the replication machinery per TLS event.
The single-stranded vectors were introduced into both wild-type (JM103) and the corresponding ΔumuDC mutant strain using the CaCl2 transformation procedure. The SOS system of these cells was either noninduced (SOS−) or was induced by previous irradiation with UV light at a dose of 50 J/m2 (SOS+). Experiments were generally repeated at least three times. Average values and standard deviations for nonslipped and slipped TLS are indicated in Table 1. Despite the relatively high toxicity of the AAF adduct under certain physiological conditions (i.e., SOS− conditions), reliable determinations of nonslipped and slipped TLS could be obtained by scoring relatively large numbers of white and blue colonies as reported in Table 1.
Survival and Polymerase Error Rate at the −1 Frameshift Mutation Hot Spot -GGG-.
In these experiments the dG-AAF adduct is located at the 3′ end of the run of three Gs (-GGGAAF-), the most mutagenic position for the induction of −1 frameshift mutations (20, 23). In a given strain, the toxicity conferred by the single adduct was measured by comparing the transformation efficiency of the modified vector (pUC-3G.3ss) with that of the corresponding nonmodified (pUC-3G.0ss) vector. In a wild-type strain, the survival rate increased from 2.4% to 25% when inducing the SOS response by UV irradiation. In contrast, in a ΔumuDC strain, the survival rate remained low, 2.7% and 3.4%, under UV− and UV+ conditions, respectively.
In the wild-type strain, the induced −1 frameshift mutation frequency increased from 4% to 12% when inducing the SOS response. Most surprisingly however, UV irradiation caused an increase of the mutation frequency from about 2.6% to 40% in the ΔumuDC strain.
Survival and Polymerase Error Rate at the −2 Frameshift Mutation Hot Spot -GGCGCC-.
In vector pUC-Nar.3ss, the dG-AAF adduct is located at the third G residue within the NarI site -GGCGAAFCC-. It was previously demonstrated using singly adducted double-stranded vectors that this position represents the only position in the NarI site that induces −2 frameshift mutations (26, 28). In a wild-type strain, the survival rate of pUC-Nar.3ss increased from 4.1% to 51% when inducing the SOS response by UV irradiation. Similarly, in a ΔumuDC strain, the survival rate also increased from 5% to 57% under UV− and UV+ conditions, respectively. In the wild-type strain, the induced −2 frameshift mutation frequency increased from 25% to 67% upon induction of the SOS response. However, UV irradiation of the ΔumuDC strain caused a further increase of the mutation frequency from ≈55% to 96%. It should be stressed that under these conditions virtually all TLS events are mutagenic.
DISCUSSION
Mechanism of DNA Replication of the Single-Stranded Vectors.
The single-stranded plasmids that we have constructed correspond to the template for lagging strand replication of the corresponding pUC double-stranded plasmid. Double-stranded pUC plasmids contain a unidirectional ColE1 origin of replication. Their lagging strand contains a DNA sequence named primosome assembly site (29, 30). This sequence is recognized by protein PriA, which directs the assembly of a φX174-like primosome involving several proteins including DNA primase (DnaG) (31). The primase synthesizes primers that are extended by PolIII holoenzyme.
We believe that the majority of mutations scored in our assay are fixed during the first round of replication. Indeed, after the first round of replication, the adduct will be located in double-stranded DNA, and UvrABC-mediated nucleotide excision repair can efficiently excise the lesion, thus preventing mutation fixation during subsequent rounds of replication. In fact, all mutant colonies that we have analyzed contained the mutant plasmid in a pure form and not as a mixture of mutant and wild-type sequences as would be expected if mutations were formed after the first round of replication (data not shown).
Survival and Polymerase Error Rate During TLS at Single AAF Adducts.
In this study, we investigated the effect on TLS of the covalent adduct formed by the strong chemical carcinogen AAF at the C8 position of guanine (dG-AAF). This lesion is known to be a strong block for in vitro DNA synthesis reactions (5–7). In vivo studies using a forward mutation assay have shown that dG-AAF induces frameshift mutations at high frequency when situated in certain DNA sequence contexts (14, 27). Studies with singly adducted double-stranded constructions have demonstrated that −1 and −2 frameshift mutations are induced at high efficiency when AAF is bound to position G3 within a run of three Gs (5′-G1G2G3-3′) and the NarI sequence (5′-G1G2CG3CC-3′), respectively (20, 23, 28).
Single-stranded vectors offer a unique opportunity to study the mechanisms of translesion synthesis in vivo because both the efficiency of TLS (survival rate) and the error rate per TLS event can be examined under various physiological conditions that are known to affect lesion tolerance. In Escherichia coli, the induction of the SOS response is known to promote lesion-induced mutagenesis, a key component in this process being the umuDC operon (12, 13).
Within both the 5′-GGGAAF-3′ and 5′-GGCGAAFCC-3′ sequence contexts, a single AAF adduct strongly impairs replication of the vector in vivo because only ≈2% to 5% survival was observed in the absence of SOS induction (Table 1). Because the same values are seen in SOS uninduced wild-type and ΔumuDC host cells, this low level of replication seems to reflect the basal level of TLS that can be mediated by the normal replication machinery, and is probably not TLS mediated by a replication complex in the presence of constitutive levels of UmuD/C proteins. Induction of the SOS response in the wild-type strain by UV irradiation caused a greater than 10-fold increase in survival in both sequence contexts (Table 1). Under SOS-induced conditions the survival of the modified vectors in the wild-type strain range between 25% and 50%, illustrating the extensive capacity of the SOS-modified replication complex to perform TLS through otherwise strongly blocking lesions. In a ΔumuDC strain, UV irradiation of the host did not result in an increase in survival of vectors in which the AAF adduct was situated within a run of three Gs. In contrast, survival of the pUC-Nar.3ss vector was enhanced by more than 10-fold in the UV-irradiated ΔumuDC strain, almost exclusively as a consequence of slipped TLS (Table 1; see below). The survival of single-stranded, M13-derived vectors containing UV lesions has previously been measured in E. coli. T-T cyclobutane dimers and T-T pyrimidine-pyrimidone (6–4) lesions exhibit similar survival values under SOS− conditions and undergo a similar increase in survival under SOS-induced conditions as reported here for AAF adducts (8, 9). However, in striking contrast with the results described here for AAF bound to the NarI site, no increase in TLS was observed for cyclobutane dimer containing single-stranded DNA in a UV-irradiated ΔumuDC strain (32).
Frameshift Mutagenesis Within Repetitive Sequences: Elongation from Nonslipped Versus Elongation from Slipped Replication Intermediates.
Frameshift mutations induced by AAF adducts within both runs of Gs and NarI sites can be best explained by a slippage mechanism (20, 33). We recently have shown that the NarI site belongs to a family of −2 frameshift mutation hot spots whose consensus sequence is 5′-GCGCX-3′ (33). The key feature of the consensus sequence is a repetition of the GpC dinucleotide, the nucleotide X on the 3′ side determining the relative “hotness” of the sequence. Therefore, both runs of Gs and NarI sequence contexts can be viewed as short repetitions of single Gs and GpC dinucleotides, respectively.
Based on experiments with singly adducted plasmids, a two-step model for frameshift mutagenesis has been proposed (20, 23, 33). The first step involves incorporation of cytosine across from the dG-AAF adduct, followed by a slippage step during which the primer template forms one or two “correct” GC base pairs at the primer terminus (Fig. 2). These SMI (20, 21, 33) extrude the adduct in a bulge containing one base for the run of Gs, or two bases for the NarI site (Fig. 2). The presence of the AAF adduct within the bulge has been shown by thermal stability measurements, chemical probes, and NMR to strongly stabilize the SMIs (21, 22, 34). Elongation from the SMI will fix the frameshift event (−1 and −2) in the newly synthesized strand. Provided a cytosine residue is incorporated opposite the AAF adduct, TLS within these sequences can be either error-free if it proceeds from the nonslipped intermediate or mutagenic—i.e., leading to a frameshift mutation—if it proceeds from the SMI (Fig. 2). It should be stressed that frameshift mutations do not result from a polymerase misincorporation error as do base pair substitutions, but from a polymerase miselongation step from a slipped primer/template terminus (Fig. 2).
Figure 2.
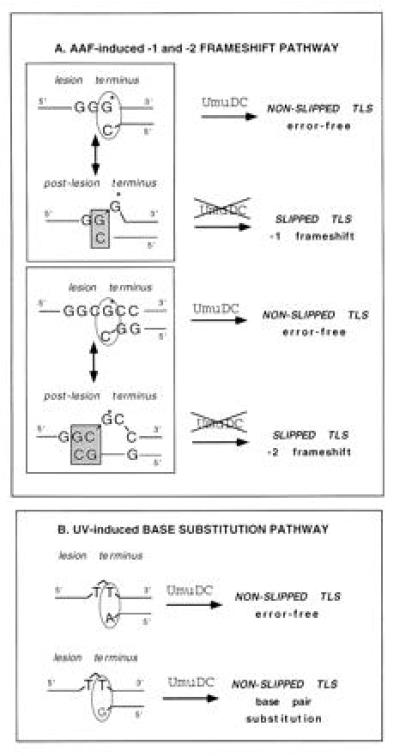
Structure of replication intermediates and involvement of SOS factors. (A) AAF-induced frameshift mutagenesis. When AAF adducts are situated in repetitive sequence contexts associated with both −1 and −2 frameshifts, the TLS intermediates can assume two forms: a nonslipped intermediate or a slipped intermediate. Elongation from the nonslipped intermediate, i.e., error-free elongation, proceeds from a lesion terminus at which the terminal nucleotide of the nascent strand is situated across from the AAF lesion (circled). Elongation from the lesion terminus appears to be stimulated by UmuDC. In contrast, frameshift mutations result from the elongation of the postlesion terminus of the slipped intermediate, at which the terminal nucleotide of the nascent strand is correctly paired with residues situated 5′ to the adduct on the template strand (shown in the square). G* is the AAF adducted guanine residue. (B) UV-induced base substitution mutagenesis. Both error-free and mutagenic TLS across a photoproduct involve elongation from a lesion terminus because, in the intermediate, the terminal nucleotide in the nascent strand is situated across from the lesion (circled). Both elongation events are strongly stimulated by the presence of UmuDC.
TLS Within Repetitive Sequences: Involvement of SOS Factors.
As discussed above, at repetitive sequences TLS can be error-free if it occurs from the nonslipped intermediate, or mutagenic if it proceeds from the SMI. We have previously described an SOS function that is essential for the efficient induction of −2 frameshift mutations within the NarI sequence context (15, 16, 35). Although the corresponding gene has not yet been isolated it was tentatively named npf (NarI processing factor) (16). Analysis of the present data in terms of elongation from slipped versus nonslipped replication intermediates allow us to define more precisely the role of both umuDC and npf factors during TLS in vivo.
Elongation from nonslipped replication intermediates is UmuDC dependent.
Error-free elongation is achieved at both repetitive G and NarI sequence contexts with the same low efficiency in SOS-uninduced wild-type cells, and in SOS-induced or uninduced ΔumuDC cells (Table 1). At both sequence contexts, a low level of about 2.4% of TLS is measured under all these conditions, suggesting that a basal level of nonslipped TLS is achieved by the replication machinery in the absence of UmuD/C+ proteins. However, in the wild-type strain, the induction of the SOS response stimulated TLS (by 5- to 10-fold) at both sequence contexts indicating that UmuD/C proteins strongly promote elongation from the nonslipped intermediate. A posteriori, it is not surprising that the efficiency of elongation from the nonslipped primer template is sequence context independent in view of the similarities of the primer template structure (Fig. 2). Indeed, both have a dG-AAF:C “mispair” at the terminus.
Elongation from the SMI is UmuDC independent.
Mutagenic TLS within the run of three Gs and the NarI site result from elongation of SMIs that have one and two “correct” GC base pairs, respectively, at the primer template terminus (Fig. 2). In contrast to nonslipped TLS, mutagenic elongation of both −1 and −2 SMIs appears to be largely UmuDC+ independent but is strongly dependent upon another UV-inducible function (Table 1). Indeed, at both sequence contexts, the induction of the SOS response increases the extent of mutagenic TLS by about 25-fold (Table 1) in both the wild-type and ΔumuD/C cells. Moreover, mutagenic TLS appears to be ≈20-fold more frequent at the NarI sequence as compared with the run of three Gs under both SOS− and SOS+ conditions (Table 1). This observation suggests that the sequence context in the vicinity of the adduct, which determines the structure and/or the stability of the SMI, modulates the elongation efficiency of the slipped intermediate.
Frameshift Mutagenesis: Comparison Between Double-Stranded and Single-Stranded Vectors.
Using double-stranded vectors, we have previously characterized AAF-induced −1 and −2 frameshift mutagenesis as being umuDC dependent and umuDC independent, respectively (14–16). In the present study, using single-stranded vectors, −1 frameshift mutagenesis appears to be umuDC independent as well. The reason for this apparent discrepancy stems from the fact that when double-stranded vectors are introduced in a ΔumuDC strain, TLS is completely abolished (i.e., lesion tolerance occurs almost entirely through damage avoidance mechanisms) when the adduct is located within the run of Gs (4) while it is maintained at the same level as in the wild-type strain when the adduct is located at the NarI site (T. Broschard and R.P.P.F., unpublished results). Under SOS-induced conditions, we note that all TLS events are less frequent in double-stranded versus single-stranded templates (ref. 4, T. Broschard and R.P.P.F., unpublished results; present work). In the absence of UmuDC, this phenomenon is more pronounced except for −2 mutagenesis. Therefore, it appears that the coordinated synthesis of both leading and lagging strands, a distinct feature of double-stranded DNA replication, favors the dissociation of the polymerase from the lesion site in all cases except from the slipped intermediate at the NarI site. Consequently, in contrast to the −2 pathway that is clearly umuDC independent, the complete loss of TLS (and the consequent loss of frameshifts) in the −1 pathway makes it appear to be umuDC dependent. This example clearly illustrates the fact that (i) mutation frequencies determined with double-stranded DNA can by no means be taken as measurements of DNA polymerase fidelity; and (ii) the genetic control of a mutation pathway determined with double-stranded vectors does not necessarily reflect the genetic control of the TLS pathway itself. Therefore, single-stranded vectors provide an unambiguous analysis of the TLS pathway itself whereas double-stranded vectors enable the study of recovery mechanisms other than TLS. In conclusion, both −1 and −2 frameshift mutagenesis induced as a consequence of AAF-mediated slippage occurs through a SOS-dependent but umuDC-independent pathway.
Structure of Replication Intermediates: A Comparison Between UV-Induced Base Substitution and AAF-Induced Frameshift Mutagenesis.
AAF adducts and UV-induced pyrimidine dimers (cyclobutane or 6–4 photoproducts) are both strong replication blocking lesions. However, they differ in that UV lesions essentially induce base pair substitutions with little sequence context effect (for a review see ref. 1) whereas AAF adducts induce frameshift mutations with a strong effect of DNA context (14, 27). In addition, UV-light-induced base substitutions are umuDC dependent (1, 10) whereas AAF-induced frameshifts are not but depend upon a different SOS-controlled gene (this work, ref. 16). Despite these differences both mutation pathways can be understood in similar terms when considering the steps involved in TLS.
Let us analyze TLS in terms of the structure of the replication intermediates (Fig. 2): a replication intermediate in which the last nucleotide of the primer is located opposite the lesion in the template will be referred to as a “lesion terminus.” In contrast when the last nucleotide of the primer is paired with a (nondamaged) nucleotide located 5′ from the lesion in the template (such as in the SMIs) we will call it a “postlesion terminus.” Elongation from a “lesion terminus” is strongly promoted by UmuDC proteins, whereas elongation from a postlesion terminus is UmuDC independent. For UV lesions, incorporation of either a correct or an incorrect nucleotide across from the lesion will yield lesion termini, the elongation of which are stimulated by UmuDC proteins (32). As a result UV mutagenesis is umuDC dependent.
For AAF lesion, cytosine (i.e., the “correct nucleotide”) is almost always inserted across from the adduct, thus accounting for the poor capacity of AAF adducts to induce base substitutions (this work, refs. 7 and 14). Elongation from this lesion terminus is, as with the UV-lesions, umuDC dependent and is error-free (Table 1). However, in repetitive sequences the lesion terminus can be converted into a postlesion terminus in the absence of a nucleotide addition step, simply by a slippage event (Fig. 2). Elongation from postlesion termini that are present in the SMI generate frameshift mutations. Although elongation from a postlesion terminus is umuDC independent, the whole process of TLS requires another SOS function (Table 1). Therefore, the differences between UV and AAF mutagenesis in (i) mutation specificity (base substitutions vs. frameshifts), (ii) DNA sequence context effects (no strong sequence effect vs. hot spots), and (iii) genetic requirements all can be understood within the same model of TLS.
Replication Intermediates in TLS and Genetic Requirements.
Although, the core replication machinery is capable of performing a low basal level of TLS and mutagenesis, proteins encoded by the SOS regulon strongly increase the capacity of the replication machinery to achieve TLS (this work, refs. 4, 36, and 37). For UV lesions and apurinic sites, genetic and biochemical data have shown that the SOS gene products RecA, UmuD′, and UmuC facilitate TLS (32, 38–41).
The first step in TLS, i.e., incorporation, appears to be efficiently performed by the polymerase itself as suggested by dNTP turnover measurements when a polymerase idles across from a variety of blocking lesions (42–44). However, to achieve complete TLS requires additional steps to be overcome: proofreading avoidance, stimulation of postlesion elongation, and/or avoidance of polymerase dissociation. It is not clear which of these steps are facilitated by RecA and UmuD/C, respectively (43, 45, 46). These proteins may only be required for elongation of lesion termini. On the other hand, during frameshift mutagenesis, the RecA and UmuD′/UmuC functions appear to be dispensable for the elongation of postlesion termini that are formed by slippage (Fig. 2). Yet an additional SOS-controlled factor was found to be involved in the whole process of TLS. Its precise role in TLS will await its isolation.
Acknowledgments
We thank S. Turban for excellent technical assistance. R.L.N. was supported by a fellowship from the Conselho Nacional de Desenvolvimento Cientifico e Technologico, Brazil. This project was supported in part by a North Atlantic Treaty Organization Collaborative Grant. Work in the laboratory of I.B.L is supported by the National Scientific and Engineering Research Council of Canada.
ABBREVIATIONS
- TLS
translesion synthesis
- dG-AAF
2-acetylaminofluorene guanine adduct
- SMI
slipped mutagenic intermediate
- AAF
2-acetylaminofluorene
References
- 1.Livneh Z, Cohen-Fix O, Skaliter R, Elizur T. Crit Rev Biochem Mol Biol. 1993;28:465–513. doi: 10.3109/10409239309085136. [DOI] [PubMed] [Google Scholar]
- 2.Higgins N P, Kato K, Strauss B. J Mol Biol. 1976;101:417–425. doi: 10.1016/0022-2836(76)90156-x. [DOI] [PubMed] [Google Scholar]
- 3.Rupp W D, Wilde C E, III, Reno D L, Howard-Flanders P. J Mol Biol. 1971;61:25–44. doi: 10.1016/0022-2836(71)90204-x. [DOI] [PubMed] [Google Scholar]
- 4.Koffel-Schwartz N, Coin F, Veaute X, Fuchs R P P. Proc Natl Acad Sci USA. 1996;93:7805–7810. doi: 10.1073/pnas.93.15.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belguise-Valladier P, Maki H, Sekiguchi M, Fuchs R P P. J Mol Biol. 1994;236:151–164. doi: 10.1006/jmbi.1994.1125. [DOI] [PubMed] [Google Scholar]
- 6.Belguise-Valladier P, Fuchs R P P. J Mol Biol. 1995;249:903–913. doi: 10.1006/jmbi.1995.0347. [DOI] [PubMed] [Google Scholar]
- 7.Lindsley J E, Fuchs R P P. Biochemistry. 1994;33:764–772. doi: 10.1021/bi00169a018. [DOI] [PubMed] [Google Scholar]
- 8.Banerjee S K, Christensen R B, Lawrence C W, LeClerc J E. Proc Natl Acad Sci USA. 1988;85:8141–8145. doi: 10.1073/pnas.85.21.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LeClerc J E, Borden A, Lawrence C W. Proc Natl Acad Sci USA. 1991;88:9685–9689. doi: 10.1073/pnas.88.21.9685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato T, Shinoura Y. Mol Gen Genet. 1977;156:121–131. doi: 10.1007/BF00283484. [DOI] [PubMed] [Google Scholar]
- 11.Steinborn G. Mol Gen Genet. 1978;165:87–93. doi: 10.1007/BF00270380. [DOI] [PubMed] [Google Scholar]
- 12.Woodgate R, Sedgwick S. Mol Microbiol. 1992;6:2213–2218. doi: 10.1111/j.1365-2958.1992.tb01397.x. [DOI] [PubMed] [Google Scholar]
- 13.Murli S, Walker G C. Curr Opin Genet Dev. 1993;3:719–725. doi: 10.1016/s0959-437x(05)80089-9. [DOI] [PubMed] [Google Scholar]
- 14.Koffel-Schwartz N, Verdier J M, Bichara M, Freund A M, Daune M P, Fuchs R P P. J Mol Biol. 1984;177:33–51. doi: 10.1016/0022-2836(84)90056-1. [DOI] [PubMed] [Google Scholar]
- 15.Koffel-Schwartz N, Fuchs R P P. Mol Gen Genet. 1989;215:306–311. doi: 10.1007/BF00339733. [DOI] [PubMed] [Google Scholar]
- 16.Maenhaut-Michel G, Janel-Bintz R, Fuchs R P P. Mol Gen Genet. 1992;235:373–380. doi: 10.1007/BF00279383. [DOI] [PubMed] [Google Scholar]
- 17.Refolo L M, Bennett C B, Humayun M Z. J Mol Biol. 1987;193:609–636. doi: 10.1016/0022-2836(87)90344-5. [DOI] [PubMed] [Google Scholar]
- 18.Schaaper R M, Koffel-Schwartz N, Fuchs R P P. Carcinogenesis. 1990;11:1087–1095. doi: 10.1093/carcin/11.7.1087. [DOI] [PubMed] [Google Scholar]
- 19.Lambert I B, Gordon A J E, Glickman B W, McCalla D R. Genetics. 1992;132:911–927. doi: 10.1093/genetics/132.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert I B, Napolitano R L, Fuchs R P P. Proc Natl Acad Sci USA. 1992;89:1310–1314. doi: 10.1073/pnas.89.4.1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia A, Lambert I B, Fuchs R P P. Proc Natl Acad Sci USA. 1993;90:5989–5993. doi: 10.1073/pnas.90.13.5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milhe C, Dhalluin C, Fuchs R P P, Lefevre J-F. Nucleic Acids Res. 1994;22:4646–4652. doi: 10.1093/nar/22.22.4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Napolitano R L, Lambert I B, Fuchs R P P. Biochemistry. 1994;33:1311–1315. doi: 10.1021/bi00172a004. [DOI] [PubMed] [Google Scholar]
- 24.Koehl P, Burnouf D, Fuchs R P P. J Mol Biol. 1989;207:355–364. doi: 10.1016/0022-2836(89)90259-3. [DOI] [PubMed] [Google Scholar]
- 25.Napolitano, R. L. & Fuchs, R. P. P. (1997) Chem. Res. Toxicol., in press. [DOI] [PubMed]
- 26.Veaute X, Fuchs R P P. Science. 1993;261:598–600. doi: 10.1126/science.8342022. [DOI] [PubMed] [Google Scholar]
- 27.Fuchs R P P, Schwartz N, Daune M P. Nature (London) 1981;294:657–659. doi: 10.1038/294657a0. [DOI] [PubMed] [Google Scholar]
- 28.Burnouf D, Koehl P, Fuchs R P P. Proc Natl Acad Sci USA. 1989;86:4147–4151. doi: 10.1073/pnas.86.11.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zypursky S L, Marians K J. Proc Natl Acad Sci USA. 1980;77:6521–6525. doi: 10.1073/pnas.77.11.6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zypursky S L, Marians K J. Proc Natl Acad Sci USA. 1981;78:6111–6115. doi: 10.1073/pnas.78.10.6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kornberg A, Baker T. DNA Replication. 2nd Ed. New York: Freeman; 1991. pp. 284–286. [Google Scholar]
- 32.Szekeres E S, Woodgate R, Lawrence C W. J Bacteriol. 1996;178:2559–2563. doi: 10.1128/jb.178.9.2559-2563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koffel-Schwartz N, Fuchs R P P. J Mol Biol. 1995;252:507–513. doi: 10.1006/jmbi.1995.0515. [DOI] [PubMed] [Google Scholar]
- 34.Milhe C, Fuchs R P P, Lefevre J-F. Eur J Biochem. 1996;235:120–127. doi: 10.1111/j.1432-1033.1996.00120.x. [DOI] [PubMed] [Google Scholar]
- 35.Burnouf D, Fuchs R P P. Biochimie. 1985;67:385–392. doi: 10.1016/s0300-9084(85)80085-7. [DOI] [PubMed] [Google Scholar]
- 36.Christensen J R, LeClerc J E, Valone Tata P, Christensen R B, Lawrence C W. J Mol Biol. 1988;203:635–641. doi: 10.1016/0022-2836(88)90198-2. [DOI] [PubMed] [Google Scholar]
- 37.Tessman I. Proc Natl Acad Sci USA. 1985;82:6614–6618. doi: 10.1073/pnas.82.19.6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sommer S, Bailone A, Devoret R. Mol Microbiol. 1993;10:963–971. doi: 10.1111/j.1365-2958.1993.tb00968.x. [DOI] [PubMed] [Google Scholar]
- 39.Frank E G, Hauser J, Levine A S, Woodgate R. Proc Natl Acad Sci USA. 1993;90:8169–8173. doi: 10.1073/pnas.90.17.8169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rajagopalan M, Lu C, Woodgate R, O’Donnell M, Goodman M F, Echols H. Proc Natl Acad Sci USA. 1992;89:10777–10781. doi: 10.1073/pnas.89.22.10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawrence C L, Borden A, Woodgate R. Mol Gen Genet. 1996;251:493–498. doi: 10.1007/BF02172378. [DOI] [PubMed] [Google Scholar]
- 42.Villani G, Boiteux S, Radman M. Proc Natl Acad Sci USA. 1978;75:3037–3041. doi: 10.1073/pnas.75.7.3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shwartz H, Shavitt O, Livneh Z. J Biol Chem. 1988;263:18277–18285. [PubMed] [Google Scholar]
- 44.Hevroni D, Livneh Z. Proc Natl Acad Sci USA. 1988;85:5046–5050. doi: 10.1073/pnas.85.14.5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fersht A R, Knill-Jones J W. J Mol Biol. 1983;165:669–682. doi: 10.1016/s0022-2836(83)80273-3. [DOI] [PubMed] [Google Scholar]
- 46.Bridges B A, Woodgate R. Mutat Res. 1985;150:133–139. doi: 10.1016/0027-5107(85)90110-1. [DOI] [PubMed] [Google Scholar]