

Abstract
Lymphotoxin-α-deficient (LT-α−/−) mice manifest congenital absence of lymph nodes (LNs) and Peyer’s patches and disturbed spleen follicle structure. The splenic white pulp areas show loss of discrete T and B lymphocyte zones, of follicular dendritic cell (FDC) clusters, and of germinal centers (GCs). Tumor necrosis factor receptor I-deficient (TNFR-I−/−) mice show similar absence of FDC clusters and GCs but retain segregation of T and B cell zones. Rarely are mesenteric LNs found in LT-α−/− mice. These mesenteric LNs show segregation of T and B cell zones similar to wild-type mice. In contrast, mesenteric LNs in TNFR-I−/− mice manifest grossly disturbed organization of T and B cells. Both LT-α−/− and TNFR-I−/− mice lacked FDC clusters in LNs and spleen. Interestingly, although both LT-α−/− and TNFR-I−/− mice that had been immunized with sheep red blood cells failed to form GCs in the spleen, they both developed GC-like clusters of peanut agglutinin-positive (PNA+) cells in their LNs. Furthermore, when lethally irradiated recombination activating gene (RAG)-1-deficient (RAG-1−/−) mice that had received spleen cells from LT-α−/− mice were immunized with sheep red blood cells, they failed to generate PNA+ clusters in the reconstituted spleen but showed robust PNA+ clusters in the reconstituted LNs. These data demonstrate that the signals that regulate the development of distinct T and B cell zones as well as the signals that regulate B cell activation to produce clusters of PNA+ cells differ between the spleen and LNs.
Soluble lymphotoxin α (LT-α) and tumor necrosis factor α (TNF-α) are structurally related homotrimers that bind similarly to both the 55-kDa TNF receptor (TNFR-I) and the 75-kDa TNF receptor (TNFR-II) (1, 2). LT-α is also present as a heteromer with a structurally related, type II membrane protein, lymphotoxin-β (LT-β) (3), which is present in its most prevalent form with the stoichiometry LT-α1β2 (4). This heteromer has no measurable affinity for TNFR-I or TNFR-II, but interacts avidly with the LT-β receptor (3, 5). Mice with targeted disruption of the LT-α gene manifest congenital absence of lymph nodes (LNs) or Peyer’s patches (6, 7). Banks et al. (7) have reported that 4 of 14 examined LT-α−/− mice have a LN-like structure in the abdominal mesentery. In LT-α−/− mice, spleen structure is also disturbed, with small white pulp follicles that fail to segregate T and B cell zones, form a marginal zone of metallophilic macrophages, generate clusters of follicular dendritic cells (FDCs), or develop germinal centers (GCs) (6, 8). Administration of an LT-β receptor–Ig fusion protein to pregnant normal mice disrupted LN development and splenic microarchitecture of the progeny, indicating that these effects are mediated at least in part by the membrane LT-α–LT-β complex (9).
The organized structures of peripheral lymphoid tissues, including primary follicles and the GC-containing secondary follicles, are thought to support efficient regulated interaction of antigen-presenting cells and T and B lymphocytes. GCs appear in primary follicles after antigen challenge and have been identified as the sites of somatic hypermutation and antibody affinity maturation (10–12). The mechanisms of formation of both primary and secondary follicles are not well understood.
The present study was undertaken to define further the role of LT-α and TNFR-I in formation of peripheral lymphoid tissues in both LNs and spleen. We report that distinct signals regulate the formation of discrete B cell and T cell zones in the splenic white pulp and in mesenteric LNs. B/T cell segregation in the splenic white pulp requires expression of LT-α and is independent of TNFR-I. In contrast, B/T cell segregation in mesenteric LNs requires TNFR-I and is independent of LT-α. Furthermore, activation of B cells to form GC-like clusters of peanut agglutinin-positive (PNA+) cells can occur in the mesenteric LNs of LT-α−/− and TNFR-I−/− mice, but not in their spleens. But, strikingly, both LT-α−/− and TNFR-I−/− mice lack FDC clusters in both LNs and spleen. Thus, in LNs, FDC clusters are not required for the activation of PNA+ B cell clusters.
MATERIALS AND METHODS
Mice.
Recombination activating gene 1-deficient (RAG-1−/−) mice were obtained from The Jackson Laboratory. LT-α−/− mice (6) were maintained on a mixed 129Sv × C57BL/6 background and were bred under specific pathogen-free conditions. TNFR-I−/− mice were provided by J. Peschon (Immunex). Both TNFR-I−/− and RAG-1−/− mice are on the C57BL/6 background.
Immunohistology of Spleen and LNs.
Spleens or LNs were harvested 10 days after i.p. immunization with 108 sheep red blood cells (SRBCs), embedded in O.C.T. compound (Miles), and frozen in liquid nitrogen. Frozen sections (6–10 μm thick) were fixed in ice-cold acetone. Endogenous peroxidase was quenched with 0.2% H2O2 in methanol. After washing, the sections were stained by first incubating with fluorescein isothiocyanate (FITC)-conjugated B220 or biotinylated Thy1.2 (PharMingen), anti-CR1 (8C12, PharMingen), PNA (Vector Laboratories), or MOMA-1 (Serotec), all at a 1:100 dilution. Horseradish peroxidase (HRP)-conjugated rabbit anti-FITC (Dako; diluted 1:10) was added 1 h later. Sections were then incubated for 1 h with a 1:20 dilution of alkaline phosphatase (AP)-conjugated streptavidin (Zymed) and color development for bound AP and HRP was with an AP reaction kit (Vector Laboratories) and with diaminobenzidine.
Reconstitution of RAG-1−/− Mice.
Cell suspensions were prepared by mincing donor spleen with scissors and then disrupting by squeezing between sterile microscope slides. The cells from an entire spleen were injected i.v. into individual RAG-1−/− recipients that had been irradiated 3 h earlier with 5 Gy. SRBCs (108 cells) were injected i.v. with the spleen cell suspensions. Two weeks later, spleen and LN sections were prepared and stained with PNA-biotin and MOMA-1.
RESULTS AND DISCUSSION
LT-α Is Not Required for T Cell/B Cell Segregation in LNs.
The spleen is composed of many discrete white pulp lymphoid cell clusters, each with a central Thy1.2-staining T cell zone (the periarteriolar lymphoid sheath) and a peripheral B220-staining B cell zone (Fig. 1a). Within the B cell zone are clusters of FDCs that provide sites for the formation of antigen-driven GCs (Figs. 2a and 3a). LNs, in contrast, have a single large central T cell zone and a surrounding peripheral B cell zone (Fig. 1d). As in the spleen, clusters of FDCs are found in the B cell compartment of LNs where they provide sites for the formation of GCs (Figs. 2d and 3d). The signals that regulate the establishment of natural spleen and LNs follicular structure have not been defined; however, the recognition that the splenic follicles of LT-α−/− mice are severely disturbed, with defective segregation of T and B cell zones (Fig. 1c), demonstrates that LT-α provides important signals defining cellular compartments during follicle biogenesis. Although LT-α−/− mice manifest a profound failure to form peripheral LNs, a solitary mesenteric node can be found in a small fraction of these animals. Approximately 500 LT-α−/− mice have been evaluated in our study, and 15 showed a gross LN-like structure in the mesenteric fat. Ten of these structures showed histology typical of LNs, with the others being nonlymphoid tissue. Thus, we identified a mesenteric LNs in 2% of LT-α−/− mice. Banks et al. (7) identified a mesenteric LN-like structure in 4 of 14 LT-α−/− mice that they examined. The basis of the differences in frequency of mesenteric LNs in these two studies is not clear but could relate to the housing of our strain under specific pathogen-free conditions and the Banks strain in a conventional facility.
Figure 1.
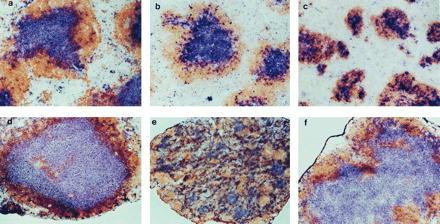
Disturbed follicular segregation of T cell and B cell zones in LNs of TNFR-I−/− mice and spleen of LT-α−/− mice. Wild-type (a and d), TNFR-I−/− (b and e), and LT-α−/− (c and f) mice were immunized i.p. with 108 SRBCs, and then 10 days later spleen (a–c) and LNs (d–f) were harvested and sections were prepared and stained with anti-Thy1.2 antibody (blue) and anti-B220 (brown) to visualize the T cell and B cell zones, respectively. (a–d and f, ×100; e, ×40.)
Figure 2.
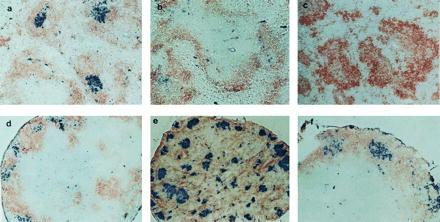
Clusters of PNA+ cells in LNs but not spleen of TNFR-I−/− and LT-α−/− mice. Sections of spleen and LNs were prepared as in Fig. 1 and stained with PNA-biotin (blue) and anti-IgD (brown). a–f are as in Fig. 1. (a–d and f, ×100; e, ×40.)
Figure 3.
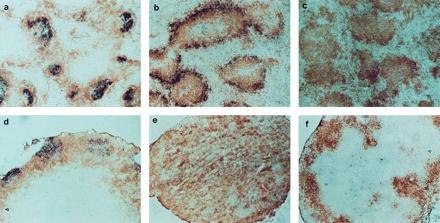
FDC clusters are absent from both spleen and LNs of TNFR-I−/− and LT-α−/− mice. Sections of spleen and LNs were prepared as in Fig. 1 and stained with biotinylated-8C12 (PharMingen) to detect FDCs (blue) and anti-B220 (brown). a–f are as in Fig. 1. (a–d and f, ×100; e, ×40.)
Interestingly, histological examination of the LT-α−/− mesenteric LNs revealed that, in contrast to spleen follicles, these mesenteric LNs showed structure very similar to the structure of mesenteric nodes in wild-type animals (Fig. 1 d and f), with a central T cell zone surrounded by a rim of B220+ B cells. Although Banks et al. (7) showed hematoxylin/eosin-stained sections suggesting that the corticomedullary structure of the mesenteric LNs they observed was disorganized and typical follicles were absent, no immunohistology was shown. Consequently, it is difficult to assess the nature of any possible discrepancies between their study and ours.
Our data demonstrate that although LT-α is absolutely required for the establishment of proper T cell and B cell compartmentalization in the white pulp areas of the spleen, this cytokine is not required for the establishment of segregated T cell and B cell zones in mesenteric LNs. Thus, the signals that regulate the development of proper follicle structure in the spleen differ from those in the mesenteric LNs. Additionally, the finding of a single mesenteric node in LT-α−/− mice and the finding by Rennert et al. (9) that soluble LT-β receptor–Ig fusion protein blocks, depending on the day of gestation it is administered, the development of all LNs except the mesenteric suggest that the signals governing LN biogenesis are not identical for all nodes. Furthermore, the fact that the mesenteric LN is present in only a minority of LT-α−/− mice suggests that LT plays a signaling role at this site as well but that the requirement for an LT signal can be bypassed by alternative signals that are occasionally expressed. The differences in signals required for LN development are underscored by the selective absence of inguinal LNs observed in BLR1-deficient mice (13).
TNFR-I Is Required for T Cell/B Cell Segregation in Mesenteric LNs.
In contrast to the small size of the spleen white pulp structure of LT-α−/− mice, these structures in TNF-α−/− and TNFR-I−/− mice are similar in size to those in wild-type mice (8, 14–16). The splenic white pulp areas in TNF-α−/− and TNFR-I−/− mice, like those in LT-α−/− animals, show absence of FDC clusters and fail to support formation of GCs in response to immunization with T cell-dependent antigens. This suggests that signaling through TNFR-I is not required for the establishment of proper T cell/B cell zones in spleen follicles, although TNFR-I is required for the development of FDC clusters and GCs. To investigate whether TNFR-I may play a role in the development of follicle structure in LNs, we examined the morphology of spleen and mesenteric LNs from TNFR-I−/− mice (Fig. 1 b and e). Strikingly, the structure of the mesenteric LNs in TNFR-I−/− mice was grossly disturbed, with diffuse loss of the normal T cell and B cell zones. Thus, the development of T cell and B cell compartmentalization in spleen and mesenteric LNs are independently controlled by different members of the TNF/LT gene family. LT-α-independent signaling through TNFR-I is required for the development of normal T and B cell zones in mesenteric LNs but not in the spleen. In contrast, LT-α is required for development of these zones in spleen follicles but not in the mesenteric LNs. Of interest, preliminary investigation of the structure of inguinal LNs in TNFR-I−/− mice shows less severe disturbance of the T/B organization (data not shown), again suggesting that all LNs are not created equal. We currently are systematically examining a variety of different LNs for potential changes in microarchitecture.
Different Requirements for Development of PNA+ GC-Like Clusters in Spleen and LNs.
PNA+ B lymphocytes, generally acknowledged as the defining feature of GCs, represent a special form of activated B cells found only in clusters within maturing lymphoid follicles at sites of somatic mutation, affinity maturation, and generation of memory B cells (10–12). Activated PNA+ B cell clusters can be detected in the B cell zones of lymphoid follicles in spleen and LNs of wild-type mice 7–10 days after immunization with SRBCs (Fig. 2 a and d). To determine whether GCs could form within the context of the altered follicles of both the TNFR-I−/− and LT-α−/− mice, animals were similarly immunized with SRBCs. As seen previously (8, 15), GC failed to develop in the spleens of TNFR-I−/− or LT-α−/− mice (Fig. 2 b and c). In dramatic contrast, there was strong development of GC-like PNA+ clusters within the mesenteric LN structures of both strains (Fig. 2 e and f). The distribution of PNA+ clusters was different in the TNFR-I−/− and the LT-α−/− mice, correlating with the structures of the primary follicles. In LT-α−/− LNs, which have T and B cell zones similar to wild type, the PNA+ clusters were distributed within the B cell area around the periphery of the node; whereas, in the TNFR-I−/− LNs, which manifest failure to segregate T and B cell zones, the PNA+ clusters were distributed apparently randomly throughout the node. These data demonstrate that the signals for formation of PNA+ clusters in the spleen and mesenteric LNs are different. Expression of both LT-α and TNFR-I are required for the formation of GCs in the spleen, whereas neither is required in the mesenteric LNs. These data also suggest that the signals required for activation of B cells in the spleen may be different from those in the LNs. This may be particularly relevant because in vitro studies of B cell activation and function have generally used spleen or LN B cells interchangeably and have assumed that peripheral lymphoid tissue B cells in the host are monomorphic.
Development of PNA+ GC-Like Cells Without FDC Clusters.
Clusters of FDCs are thought to be required to support formation and maturation of GCs (10–12). In support of this concept, both FDC clusters and GCs are absent from the spleens of immunized LT-α−/−, TNF-α−/−, and TNFR-I−/− mice (8, 15, 16). We investigated whether clusters of FDCs were present in the LNs of these SRBC-immunized mice by using immunohistochemical staining with the 8C12 anti-mouse CR1 monoclonal antibody (17). Clearly defined clusters of 8C12+ cells with dendritic morphology were detected within the B220-staining B cell zones in the spleen and LNs of wild-type mice (Fig. 3 a and d). Although the peripheral zone of B cells in TNFR-I−/− mice showed increased CR1 staining with the 8C12 monoclonal antibody (Fig. 3b), there were no clusters of 8C12-staining cells with dendritic morphology in either the spleen or LNs of TNFR-I−/− mice (Fig. 3 b and e). No 8C12+ cells were detected in the LT-α−/− mice (Fig. 3 c and f). Spleen and mesenteric LNs from TNFR-I−/− and LT-α−/− mice also showed no staining with the FDC-M1 antibody (data not shown), supporting the absence of FDCs in the lymphoid tissues of these two strains. Thus, LT-α and TNFR-I are each required for the formation of FDC clusters in the follicles of spleen and LNs, but in LNs the formation of foci of PNA+ cells is not dependent on either FDC clusters or signaling by LT-α or TNFR-I.
LT-α Is Required for Development of PNA+ GC-Like Clusters in Spleen but Not in Mesenteric LNs.
The mesenteric LN is detected in too small a fraction of LT-α−/− mice to permit a comprehensive analysis of determinants of LN structure. Consequently, the role of LT-α in the formation of spleen and LN follicle structure was examined further by transfer of splenocytes from either wild-type or LT-α−/− mice to irradiated RAG-1-deficient (RAG-1−/−) recipients. These recipients have both spleen and LNs but have no mature B or T lymphocytes. Thus, they provide an environment in which the actions of LT-α on both spleen and LNs can be investigated. When RAG-1−/− mice were treated with spleen cells from wild-type mice and then immunized with SRBCs, clusters of PNA+ cells developed in both spleen and mesenteric LNs (Fig. 4 a and c). In contrast, when RAG-1−/− mice were treated with splenocytes from LT-α−/− donors (Fig. 4 b and d), clusters of PNA+ cells developed in the mesenteric LNs, but not in spleen. These clusters of PNA+ cells in the mesenteric LNs formed without detectable FDCs (data not shown). These data confirm that LT-α is required for the formation of GCs in spleen but not in mesenteric LNs. Furthermore, FDCs are not essential to support this activation. We speculate that FDC clusters act, rather, to maintain GCs in the course of an immune response and/or to retain antigen within GCs to support antigen-driven affinity maturation. Relevant to this, when RAG-1−/− mice reconstituted with LT-α−/− splenocytes or LT-α−/− mice reconstituted with wild-type splenocytes were immunized at the time of spleen cell transfer with SRBCs, no IgG anti-SRBC response was detected (unpublished data). This demonstrates that the PNA+ clusters without FDCs are not fully functional GCs. These observations have significant implications in relation to previous studies in which we demonstrated that immunization of LT-α−/− mice with (4-hydroxy-3-nitrophenyl)acetyl (NP)-haptenated ovalbumin adsorbed to alum elicited high-affinity IgG anti-NP antibodies with sequence changes characteristic of somatic hypermutation (14). These experiments were performed with more than 12 individual mice, none of which showed evidence of the mesenteric LN that is observed in a small fraction of these mice. Thus, a contribution of FDC-deficient undetected LNs to the observed IgG response is unlikely. Furthermore, additional experiments comparing immunization of LT-α−/− mice with SRBCs without adjuvant to immunization with adjuvant demonstrated that IgG responses were dependent on the presence of adjuvant (unpublished data).
Figure 4.
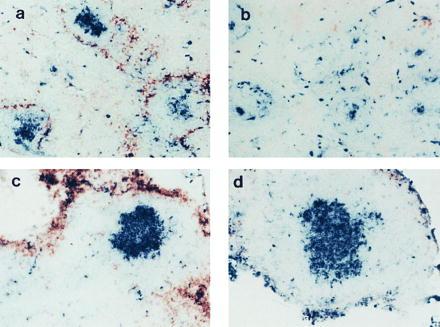
LT-α-expressing cells are required for formation of GCs in spleen but not in LNs. RAG-1−/− mice were irradiated and reconstituted with spleen cells from either wild-type (a and c) or LT-α−/− (b and d) mice. At the same time, they were immunized i.v. with SRBCs, and then 10 days later sections of spleen (a and b) and LNs (c and d) were stained with PNA-biotin (blue) and anti-MOMA-1 (brown). (×100.)
In prior studies, ectopic development of lymphoid follicles and GCs had been detected within the thymus of patients with myasthenia gravis or within the synovial tissues of patients with rheumatoid arthritis (18–21). Overexpression of LT-α or TNF-α by cells within these inflammatory lesions may provide the initial signals for biogenesis of the GCs. This concept is supported by the observation that transgenic mice expressing LT-α under control of the rat insulin promoter show ectopic follicles with GC-like structures in the pancreas and kidneys (22). Transgenic mice overexpressing TNF-α developed similar structures in the thymus and lungs (23). Thus with the present results, these data support the hypothesis that LT-α and TNF-α independently regulate the formation of lymphoid follicles. Furthermore, the maturation of lymphoid follicles in spleen and LNs is regulated differently. This can be seen by their differential dependence on LT-α for the development of clusters of PNA+ cells in response to antigen challenge. Future studies will determine whether this represents an intrinsic difference in the lymphoid cells in these two organs or perhaps indicates the presence of an additional regulatory cell type present in one organ and absent from the other.
Acknowledgments
We thank J. Peschon for providing the TNFR-I−/− mouse strain and G. Kelsoe and M. Kosco-Vilbois for anti-FDC antibodies. D.D.C. is an investigator of the Howard Hughes Medical Institute. These studies were supported in part by National Institutes of Health Grant AI34580.
ABBREVIATIONS
- LT
lymphotoxin
- TNF
tumor necrosis factor
- R
receptor
- LN
lymph node
- FDC
follicular dendritic cell
- GC
germinal center
- SRBC
sheep red blood cell
- PNA
peanut agglutinin
- RAG-1
recombination activating gene 1
References
- 1.Ruddle N H. Curr Opin Immunol. 1992;4:327–332. doi: 10.1016/0952-7915(92)90084-r. [DOI] [PubMed] [Google Scholar]
- 2.Ware C F, VanArsdale T L, Crowe P D, Browning J L. Curr Top Microbiol Immunol. 1995;198:175–218. doi: 10.1007/978-3-642-79414-8_11. [DOI] [PubMed] [Google Scholar]
- 3.Browning J L, Ngam-ek A, Lawton P, DeMarinis J, Tizard R, Chow E P, Hession C, O’Brine-Greco B, Foley S F, Ware C F. Cell. 1993;72:847–856. doi: 10.1016/0092-8674(93)90574-a. [DOI] [PubMed] [Google Scholar]
- 4.Browning J L, Dougas I, Ngam-ek A, Bourdon P R, Ehrenfels B N, Miatkowski K, Zafari M, Yampaglia A M, Lawton P, Meier W, Benjamin C P, Hession C. J Immunol. 1995;154:33–46. [PubMed] [Google Scholar]
- 5.Crowe P D, VanArsdale T L, Walter B N, Ware C F, Hession C, Ehrenfels B, Browning J L, Din W S, Goodwin R G, Smith C A. Science. 1994;264:707–710. [PubMed] [Google Scholar]
- 6.De Togni P, Goellner J, Ruddle N H, Streeter P R, Fick A, Mariathasan S, Smith S C, Carlson R, Shornick L P, Strauss-Schoenberger J, Russell J H, Karr R, Chaplin D D. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 7.Banks T A, Rouse B T, Kerley M K, Blair P J, Godfrey V L, Kuklin N A, Bouley D M, Thomas J, Kanangat S, Mucenski M L. J Immunol. 1995;155:1685–1693. [PubMed] [Google Scholar]
- 8.Matsumoto M, Mariathasan S, Nahm M H, Baranyay F, Peschon J J, Chaplin D D. Science. 1996;271:1289–1291. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- 9.Rennert P D, Browning J L, Mebius R, Mackay F, Hochman P S. J Exp Med. 1996;184:1999–2006. doi: 10.1084/jem.184.5.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacLennan I C M. Annu Rev Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 11.Kelsoe G. Immunity. 1996;4:107–111. doi: 10.1016/s1074-7613(00)80675-5. [DOI] [PubMed] [Google Scholar]
- 12.Rajewsky K. Nature (London) 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- 13.Forster R, Mattis A E, Kremmer E, Wolf E, Brem G, Lipp M. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto M, Lo S F, Carruthers C J L, Min J, Mariathasan S, Huang G, Plas D R, Martin S M, Geha R S, Nahm M H, Chaplin D D. Nature (London) 1996;382:462–466. doi: 10.1038/382462a0. [DOI] [PubMed] [Google Scholar]
- 15.Le Hir M, Bluethmann H, Kosco-Vilbois M H, Muller M, di Padova F, Moore M, Ryffel B, Eugster H-P. J Exp Med. 1996;183:2367–2372. doi: 10.1084/jem.183.5.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. J Exp Med. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinoshita T, Takeda J, Hong K, Kozono H, Sakai H, Inoue K. J Immunol. 1988;140:3066–3072. [PubMed] [Google Scholar]
- 18.Sloan H E J. Surgery. 1943;13:154–174. [Google Scholar]
- 19.Young C L, Adamson T C, Vaughan J H, Fox R I. Arthritis Rheum. 1984;27:32–39. doi: 10.1002/art.1780270106. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y-J, Banchereau J. J Exp Med. 1996;184:1207–1211. doi: 10.1084/jem.184.4.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroder A E, Greiner A, Seyfert C, Berek C. Proc Natl Acad Sci USA. 1996;93:221–225. doi: 10.1073/pnas.93.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kratz A, Campos-Neto A, Hanson M S, Ruddle N H. J Exp Med. 1996;183:1461–1472. doi: 10.1084/jem.183.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Douni E, Grell M, Pfizenmaier K, Kollias G. Eur Cytokine Network. 1996;7:164. (abstr.). [Google Scholar]